
Nanoparticles-Based Strategies to Improve the Delivery of Therapeutic Small Interfering RNA in Precision Oncology


Abstract
:1. Introduction
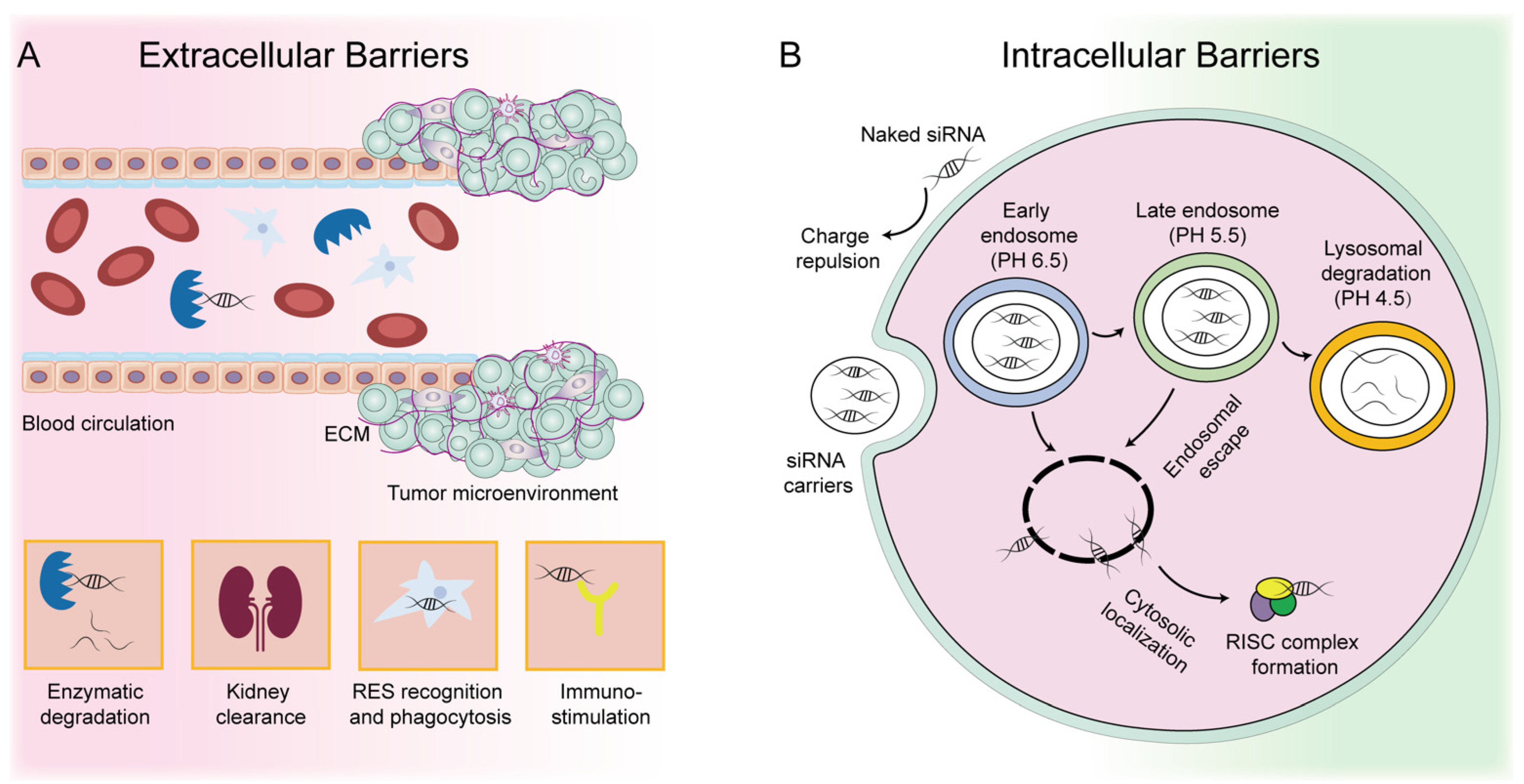
2. Development and Challenges of siRNA-Based Therapeutics
3. Strategies to Improve siRNA Delivery in Cancer Therapy
3.1. Chemical Modification
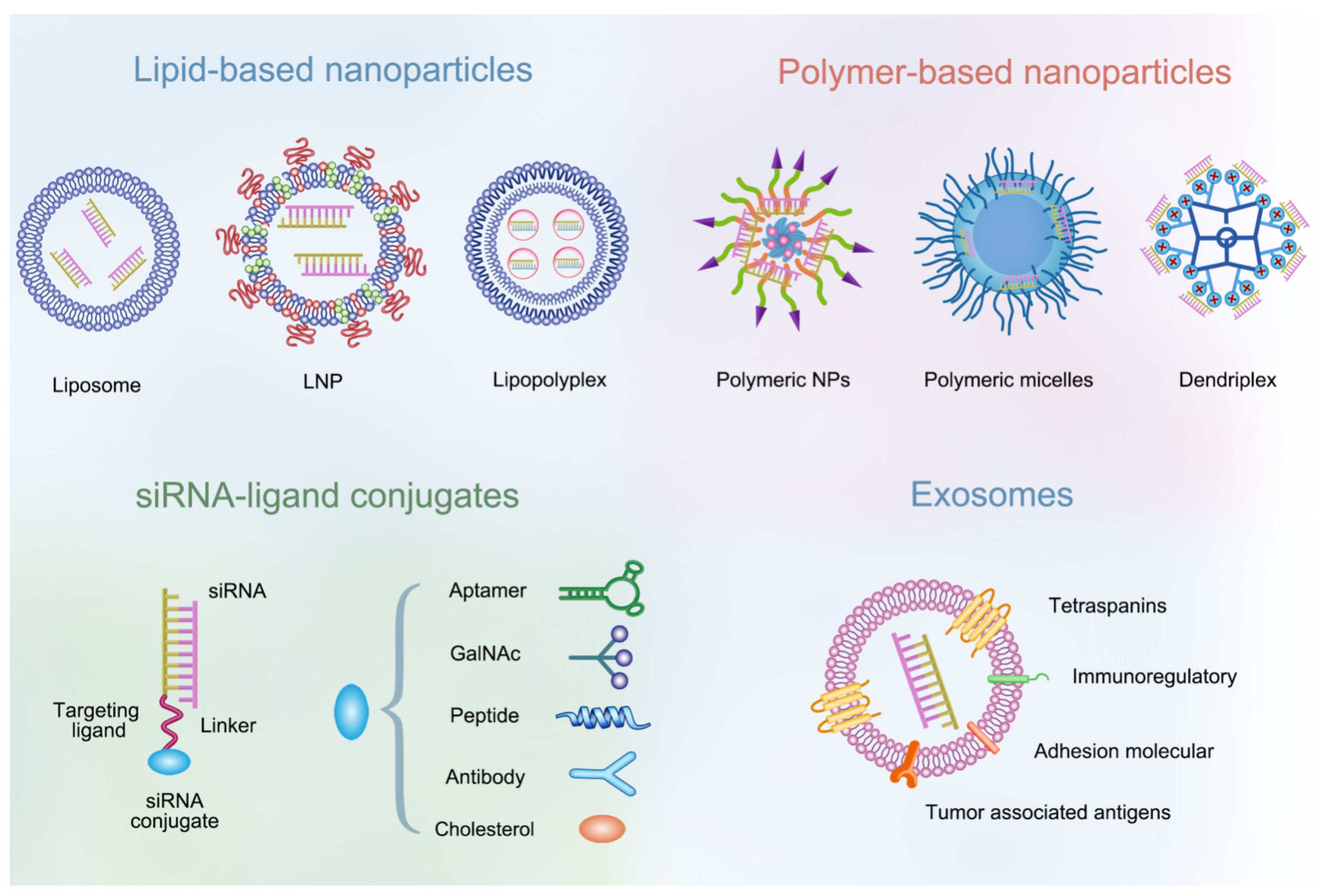
3.2. NP-Based Delivery Systems
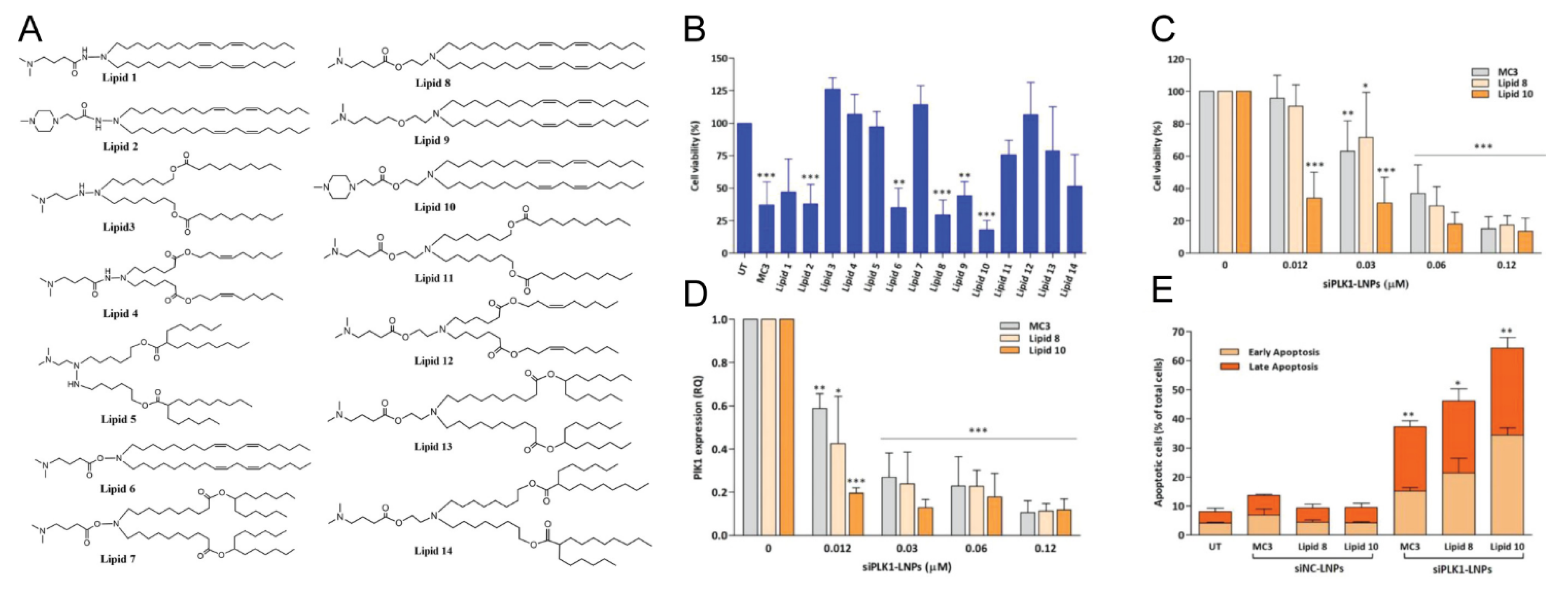
3.2.1. Lipid-Based NPs
3.2.2. Polymer-Based NPs
3.2.3. siRNA-Ligand Conjugates
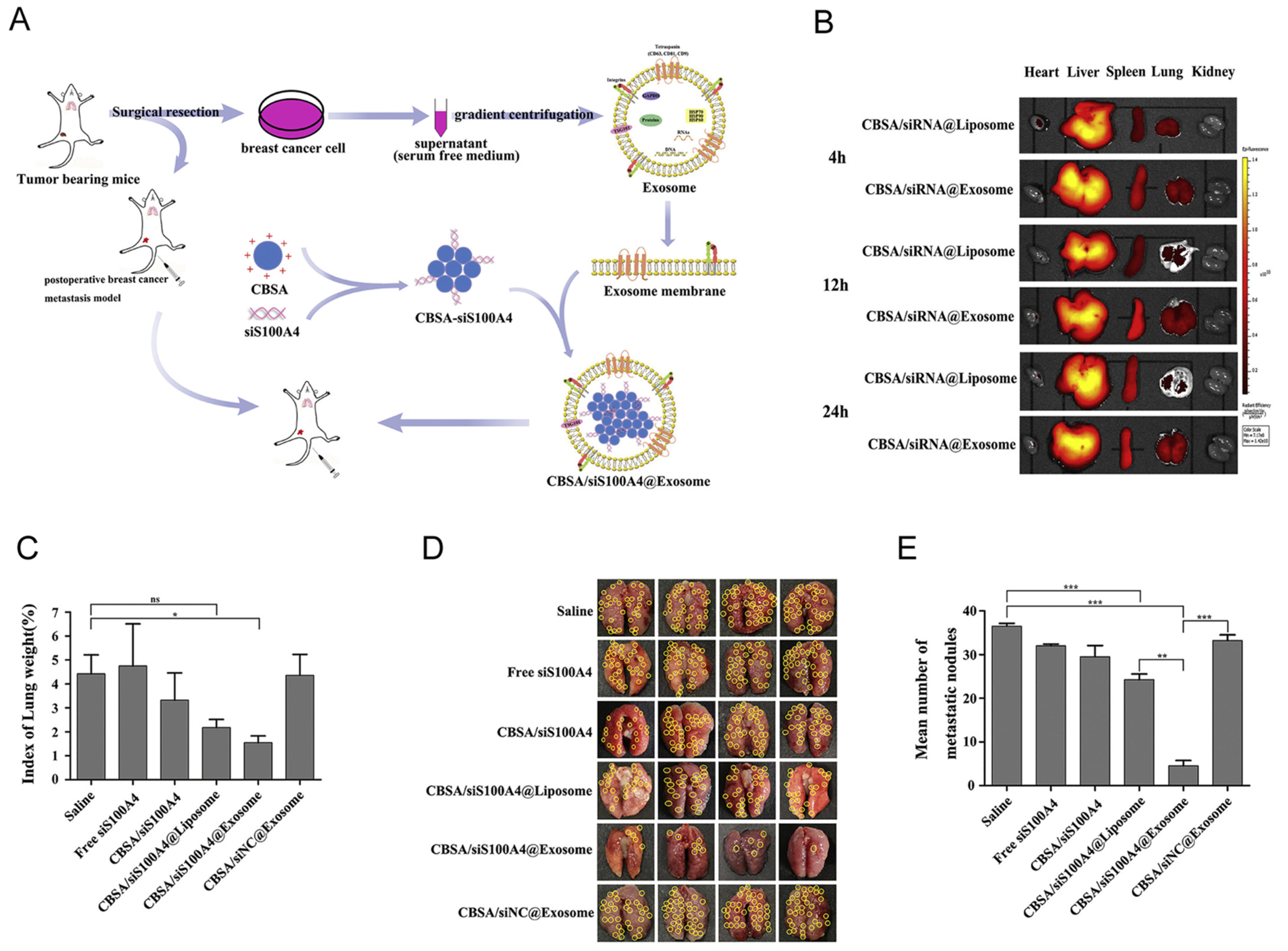
3.2.4. Exosomes
4. Potential Targets for siRNA-Based Cancer Therapeutics
4.1. Genes Promoting Tumor Growth
4.2. Genes Supplying the Tumor Nutrients
4.3. Genes Promoting Tumor Drug Resistance or Metastasis
4.4. Genes Modulating the TME
4.4.1. Targeting Cancer-Associated Fibroblasts
4.4.2. Targeting Immunosuppressive Cells or Immune Checkpoints
5. Combined Strategies with Other Therapeutic Modalities
5.1. Combined Gene Therapy
5.2. SiRNA Therapeutics Combined with Chemotherapy
5.3. SiRNA Therapeutics Combined with Radiotherapy
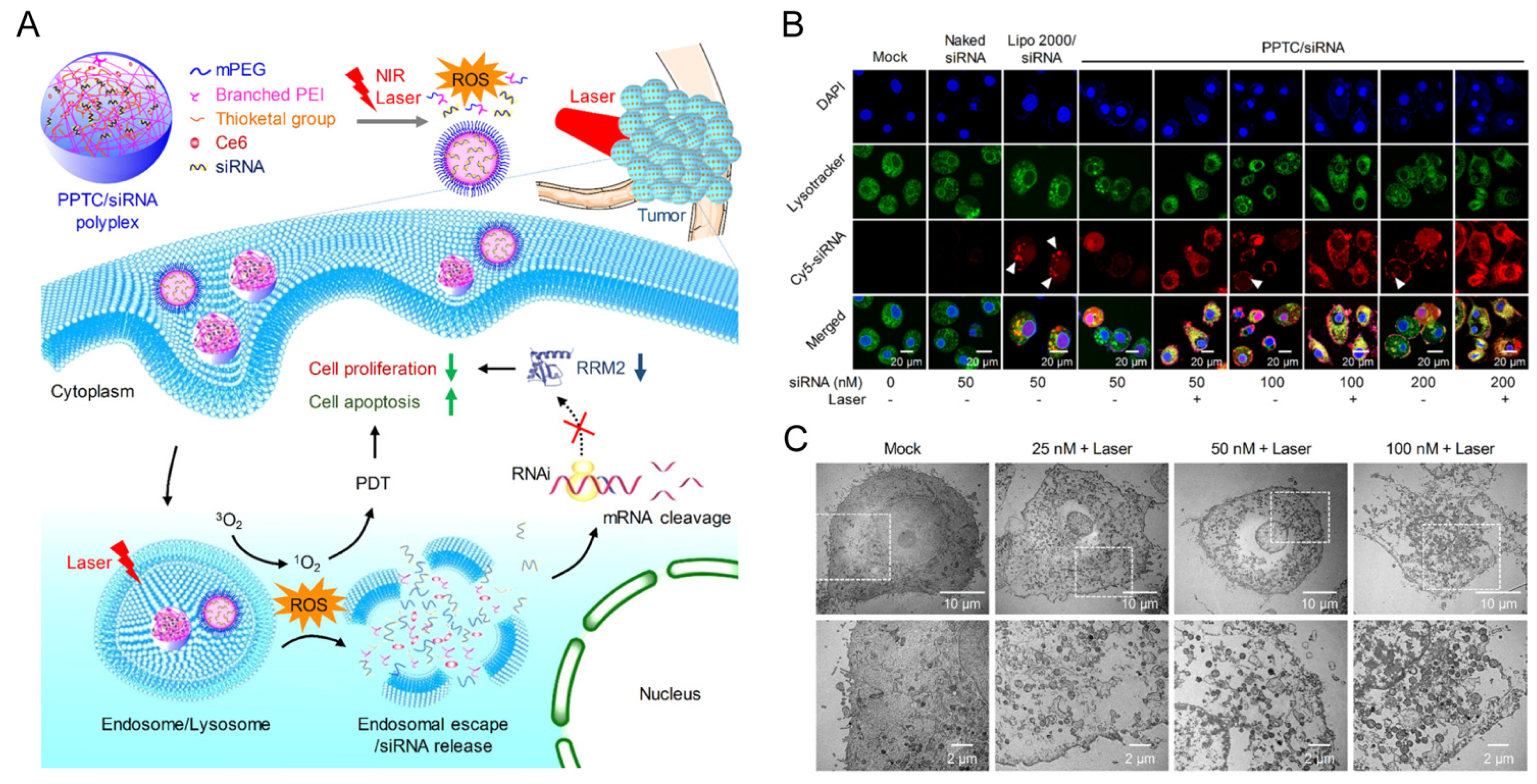
5.4. SiRNA Therapeutics Combined with Photodynamic/Photothermal Therapy
6. Safety and Toxicity
7. Outlook
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ting, L.Y.; Tan, Y.J.; Ein, O.C. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Bholakant, R.; Qian, H.; Zhang, J.; Huang, X.; Huang, D.; Feijen, J.; Zhong, Y.; Chen, W. Recent Advances of Polycationic siRNA Vectors for Cancer Therapy. Biomacromolecules 2020, 21, 2966–2982. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, W.; Qi, R.; Mao, Z.W.; Shen, H. Engineering functional inorganic-organic hybrid systems: Advances in siRNA therapeutics. Chem. Soc. Rev. 2018, 47, 1969–1995. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Hamilton, A.J. A Species of Small Antisense RNA in Posttranscriptional Gene Silencing in Plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L. Delivery of siRNA therapeutics: Barriers and carriers. Aaps. J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Kang, L.; Wang, R.F.; Yan, P.; Liu, M.; Zhang, C.L.; Yu, M.M.; Cui, Y.G.; Xu, X.J. Noninvasive visualization of RNA delivery with 99mTc-radiolabeled small-interference RNA in tumor xenografts. J. Nucl. Med. 2010, 51, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Braasch, D.A.; Paroo, Z.; Constantinescu, A.; Ren, G.; Oz, O.K.; Mason, R.P.; Corey, D.R. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg. Med. Chem. Lett. 2004, 14, 1139–1143. [Google Scholar] [CrossRef]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.A.; Nam, K.T.; Lee, K.; et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting-strategies and applications. J. Control Release 2015, 203, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Zatsepin, T.S.; Kotelevtsev, Y.V.; Koteliansky, V. Lipid nanoparticles for targeted siRNA delivery-going from bench to bedside. Int. J. Nanomed. 2016, 11, 3077–3086. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [Green Version]
- El Dika, I.; Lim, H.Y.; Yong, W.P.; Lin, C.C.; Yoon, J.H.; Modiano, M.; Freilich, B.; Choi, H.J.; Chao, T.Y.; Kelley, R.K.; et al. An Open-Label, Multicenter, Phase I, Dose Escalation Study with Phase II Expansion Cohort to Determine the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Intravenous TKM-080301 in Subjects with Advanced Hepatocellular Carcinoma. Oncologist 2019, 24, 747-e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Röder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef]
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Feist, M.; Gebhardt, F.; Khan, M.; et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers 2020, 12, 3130. [Google Scholar] [CrossRef]
- Schlee, M.; Hornung, V.; Hartmann, G. siRNA and isRNA: Two edges of one sword. Mol. Ther. 2006, 14, 463–470. [Google Scholar] [CrossRef]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.; Yamasaki, S.; Itaya, M.; Pan, Y. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Terrazas, M.; Kool, E.T. RNA major groove modifications improve siRNA stability and biological activity. Nucleic Acids Res. 2009, 37, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Amarzguioui, M.; Holen, T.; Babaie, E.; Prydz, H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003, 31, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, M. RNA interference and chemically modified small interfering RNAs. Curr. Opin. Chem. Biol. 2004, 8, 570–579. [Google Scholar] [CrossRef]
- Judge, A.D.; Bola, G.; Lee, A.C.; MacLachlan, I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 2006, 13, 494–505. [Google Scholar] [CrossRef]
- Sioud, M. Single-stranded small interfering RNA are more immunostimulatory than their double-stranded counterparts: A central role for 2’-hydroxyl uridines in immune responses. Eur. J. Immunol. 2006, 36, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman , J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heide, J.D.; Ribas , A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941. [Google Scholar] [CrossRef]
- Martin, D.T.; Steinbach, J.M.; Liu, J.; Shimizu, S.; Kaimakliotis, H.Z.; Wheeler, M.A.; Hittelman, A.B.; Saltzman, W.M.; Weiss, R.M. Surface-modified nanoparticles enhance transurothelial penetration and delivery of survivin siRNA in treating bladder cancer. Mol. Cancer Ther. 2014, 13, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, M.S.; Nicolas, V.; Wilhelm, C.; Ménager, C.; Barratt, G.; Lesieur, S. The in vitro kinetics of the interactions between PEG-ylated magnetic-fluid-loaded liposomes and macrophages. Biomaterials 2007, 28, 4143–4153. [Google Scholar] [CrossRef]
- Ngoune, R.; Peters, A.; von Elverfeldt, D.; Winkler, K.; Pütz, G. Accumulating nanoparticles by EPR: A route of no return. J. Control. Release 2016, 238, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.; Tavares, A.J.; Qin, D.; Ohta, S.; Chan, W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Alipour, M.; Baneshi, M.; Hosseinkhani, S.; Mahmoudi, R.; Arabzadeh, A.J.; Akrami, M.; Mehrzad, J.; Bardania, H. Recent progress in biomedical applications of RGD-based ligand: From precise cancer theranostics to biomaterial engineering: A systematic review. J. Biomed. Mater. Res. Part A 2020, 108, 839–850. [Google Scholar] [CrossRef]
- Zwicke, G.L.; Mansoori, G.A.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, H. Application of hyaluronic acid as carriers in drug delivery. Drug Deliv. 2018, 25, 766–772. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharm. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Wang, H.-X.; Zuo, Z.-Q.; Du, J.-Z.; Wang, Y.-C.; Sun, R.; Cao, Z.-T.; Ye, X.-D.; Wang, J.-L.; Leong, K.W.; Wang, J. Surface charge critically affects tumor penetration and therapeutic efficacy of cancer nanomedicines. Nano Today 2016, 11, 133–144. [Google Scholar] [CrossRef]
- Wang, K.; Tu, Y.; Yao, W.; Zong, Q.; Xiao, X.; Yang, R.M.; Jiang, X.Q.; Yuan, Y. Size-Switchable Nanoparticles with Self-Destructive and Tumor Penetration Characteristics for Site-Specific Phototherapy of Cancer. ACS Appl. Mater. Interfaces 2020, 12, 6933–6943. [Google Scholar] [CrossRef]
- Ma, T.; Chen, R.; Lv, N.; Chen, Y.; Qin, H.; Jiang, H.; Zhu, J. Size-Transformable Bicomponent Peptide Nanoparticles for Deep Tumor Penetration and Photo-Chemo Combined Antitumor Therapy. Small 2022, 18, e2106291. [Google Scholar] [CrossRef] [PubMed]
- Canton, I.; Battaglia, G. Endocytosis at the nanoscale. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Su, I.K.; Akaike, T.; Chong, S.C. Synergistic effect of poly(ethylenimine) on the transfection efficiency of galactosylated chitosan/DNA complexes. J. Control. Release Off. J. Control. Release Soc. 2005, 105, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.; Merkel, O.; Fink, L.; Czubayko, F.; Kissel, T.; Aigner, A. In vivo pharmacokinetics, tissue distribution and underlying mechanisms of various PEI(-PEG)/siRNA complexes. Toxicol. Appl. Pharm. 2009, 236, 97–108. [Google Scholar] [CrossRef]
- Li, C.; Zhou, J.; Wu, Y.; Dong, Y.; Du, L.; Yang, T.; Wang, Y.; Guo, S.; Zhang, M.; Hussain, A.; et al. Core Role of Hydrophobic Core of Polymeric Nanomicelle in Endosomal Escape of siRNA. Nano Lett. 2021, 21, 3680–3689. [Google Scholar] [CrossRef]
- Peeler, D.J.; Thai, S.N.; Cheng, Y.; Horner, P.J.; Sellers, D.L.; Pun, S.H. pH-sensitive polymer micelles provide selective and potentiated lytic capacity to venom peptides for effective intracellular delivery. Biomaterials 2019, 192, 235–244. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi, D.K.; Shin, J.Y.; Shin, S.M.; Park, S.W.; Cho, H.S.; Kim, Y.S. Endosomal acidic pH-induced conformational changes of a cytosol-penetrating antibody mediate endosomal escape. J. Control Release 2016, 235, 165–175. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zheng, N.; Song, Z.; Kim, K.H.; Yao, C.; Zhang, R.; Zhang, C.; Huang, Y.; Uckun, F.M.; Cheng, J.; et al. Suppression of Hepatic Inflammation via Systemic siRNA Delivery by Membrane-Disruptive and Endosomolytic Helical Polypeptide Hybrid Nanoparticles. ACS Nano 2016, 10, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cheng, Q.; Wei, T.; Yu, X.; Johnson, L.T.; Farbiak, L.; Siegwart, D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat. Mater. 2021, 20, 701–710. [Google Scholar] [CrossRef]
- Li, S.; Yuan, H.; Hui, C.; Wang, X.; Shu, W. Cationic Poly(p-phenylene vinylene) Materials as A Multifunctional Platform for Light-Enhanced siRNA Delivery. Chem.-Asian J. 2016, 11, 2686–2689. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, W.; Duan, Y.; Xu, L.; Xu, Y.; Hou, L.; Meng, X.; Zhu, X.; Liu, B. Light-Induced Self-Escape of Spherical Nucleic Acid from Endo/Lysosome for Efficient Non-Cationic Gene Delivery. Angew. Chem. Int. Ed. Engl. 2020, 59, 19168–19174. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.E.; Drummond, D.C.; Hong, K.; Park, J.W.; Kirpotin, D.B. Assembly of nucleic acid-lipid nanoparticles from aqueous-organic monophases. BBA-Biomembr. 2006, 1758, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Sakurai, N.; Okamoto, A.; Koide, H.; Asai, T. Design of a Novel PEGylated Liposomal Vector for Systemic Delivery of siRNA to Solid Tumors. Biol. Pharm. Bull. 2019, 42, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.; Szebeni, J. Complement Activation: A Potential Threat on the Safety of Poly(ethylene glycol)-Coated Nanomedicines. ACS Nano 2020, 14, 7682–7688. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Gunther, M.; Gu, Z.; Wagner, E. Pyridylhydrazone-based PEGylation for pH-reversible lipopolyplex shielding. Biomaterials 2011, 32, 858–869. [Google Scholar] [CrossRef]
- Chi, Y.; Yin, X.; Sun, K.; Feng, S.; Liu, J.; Chen, D.; Guo, C.; Wu, Z. Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J. Control. Release 2017, 261, 113–125. [Google Scholar] [CrossRef]
- Kamada, H.; Taki, S.; Nagano, K.; Inoue, M.; Ando, D.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S. Generation and characterization of a bispecific diabody targeting both EPH receptor A10 and CD3. Biochem. Biophys. Res. Commun. 2015, 456, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, Z.; Pan, S.; Shi, M.; Li, J.; Yang, C.; Hu, H.; Qiao, M.; Chen, D.; Zhao, X. Overcoming Multidrug Resistance by Codelivery of MDR1-Targeting siRNA and Doxorubicin Using EphA10-Mediated pH-Sensitive Lipoplexes: In Vitro and In Vivo Evaluation. ACS Appl. Mater. Interfaces 2018, 10, 21590–21600. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Tam, Y.Y.; Cullis, P.R. Lipid nanoparticles for short interfering RNA delivery. Adv. Genet. 2014, 88, 71–110. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.J.; Kulkarni, M.M.; Mercer , J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.T.; Suys, E.; Lee, J.S.; Dai, H.N.; Truong, N.P. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Suhr, O.B. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Benson, M.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.; Sekijima, Y.; Sipe, J.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2019, 25, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Kinami, Y.; Hashiba, K.; Harashima, H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J. Control. Release 2020, 322, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Harvie, P.; Wong, F.M.; Bally, M.B. Use of poly(ethylene glycol)–lipid conjugates to regulate the surface attributes and transfection activity of lipid–DNA particles. J. Pharm. Sci. 2000, 89, 652–663. [Google Scholar] [CrossRef]
- Fz, A.; Ms, A.; Hv, B.; Zm, C.; Bb, D. Liposome and immune system interplay: Challenges and potentials. J. Control. Release 2019, 305, 194–209. [Google Scholar] [CrossRef]
- Ramishetti, S.; Hazan-Halevy, I.; Palakuri, R.; Chatterjee, S.; Naidu Gonna, S.; Dammes, N.; Freilich, I.; Kolik Shmuel, L.; Danino, D.; Peer, D. A Combinatorial Library of Lipid Nanoparticles for RNA Delivery to Leukocytes. Adv. Mater. 2020, 32, e1906128. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Kato, A.; Hida, Y.; Hamada, J.; Maishi, N.; Hida, K.; Harashima, H. Synergistic Enhancement of Cellular Uptake With CD44-Expressing Malignant Pleural Mesothelioma by Combining Cationic Liposome and Hyaluronic Acid–Lipid Conjugate. J. Pharm. Sci. 2019, 108, 3218–3224. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ishihara, H. Difference in the lipid nanoparticle technology employed in three approved siRNA (Patisiran) and mRNA (COVID-19 vaccine) drugs. Drug Metab Pharm. 2021, 41, 100424. [Google Scholar] [CrossRef]
- Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of Lipid Nanoparticles for RNA Delivery. Acc. Chem. Res. 2022, 55, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Ferraresso, F.; Strilchuk, A.W.; Juang, L.J.; Poole, L.G.; Luyendyk, J.P.; Kastrup, C.J. Comparison of DLin-MC3-DMA and ALC-0315 for siRNA Delivery to Hepatocytes and Hepatic Stellate Cells. Mol. Pharm. 2022, 19, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wagner, E. Polymeric Carriers for Nucleic Acid Delivery: Current Designs and Future Directions. Biomacromolecules 2019, 20, 3613–3626. [Google Scholar] [CrossRef]
- Cavallaro, G.; Sardo, C.; Craparo, E.F.; Giammona, B. Polymeric nanoparticles for siRNA delivery: Production and applications. Int. J. Pharm. 2017, 525, 313–333. [Google Scholar] [CrossRef]
- Gary, D.J.; Puri, N.; Won, Y.Y. Polymer-based siRNA delivery: Perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J. Control. Release 2007, 121, 64–73. [Google Scholar] [CrossRef]
- Lv, P.; Zhou, C.; Zhao, Y.; Liao, X.; Yang, B. Modified-epsilon-polylysine-grafted-PEI-β-cyclodextrin supramolecular carrier for gene delivery. Carbohydr. Polym. 2017, 168, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Park, K.; Kim, J.; Kim, K.S.; Hahn, S.K. Target specific intracellular delivery of siRNA/PEI-HA complex by receptor mediated endocytosis. Mol. Pharm. 2009, 6, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, T.L.; Lu, Z.R. Targeted intracellular codelivery of chemotherapeutics and nucleic acid with a well-defined dendrimer-based nanoglobular carrier. Biomaterials 2009, 30, 5660–5666. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Lin, L.; Jiao, Z.; Guo, Z.; Chen, J.; Gao, S.; Zhu, X.; Chen, X. Polylysine-modified polyethylenimine inducing tumor apoptosis as an efficient gene carrier. J. Control. Release 2013, 172, 410–418. [Google Scholar] [CrossRef]
- Patil, M.L.; Zhang, M.; Minko, T. Multifunctional Triblock Nanocarrier (PAMAM-PEG-PLL) for the Efficient Intracellular siRNA Delivery and Gene Silencing. Acs Nano 2011, 5, 1877–1887. [Google Scholar] [CrossRef]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Gao, Y.; Zhou, T.; Zhao, X.L.; Hu, H.Y.; Chen, D.W.; Qiao, M.X. Enhanced response to PD-L1 silencing by modulation of TME via balancing glucose metabolism and robust co-delivery of siRNA/Resveratrol with dual-responsive polyplexes. Biomaterials 2021, 271, 120711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, H.; Li, Y.; Chen, W.; Li, H.; Peng, K.; Zhang, Z.; Sun, X. Targeting NF-kB signaling with polymeric hybrid micelles that co-deliver siRNA and dexamethasone for arthritis therapy. Biomaterials 2017, 122, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Lindstrom, A.; Li, Y. Porphyrin-Based Nanomedicines for Cancer Treatment. Bioconjug. Chem. 2019, 30, 1585–1603. [Google Scholar] [CrossRef]
- Zhang, M.; Weng, Y.; Cao, Z.; Guo, S.; Hu, B.; Lu, M.; Guo, W.; Yang, T.; Li, C.; Yang, X.; et al. ROS-Activatable siRNA-Engineered Polyplex for NIR-Triggered Synergistic Cancer Treatment. ACS Appl. Mater. Interfaces 2020, 12, 32289–32300. [Google Scholar] [CrossRef]
- Jeong, J.H.; Mok, H.; Oh, Y.K.; Park, T.G. siRNA Conjugate Delivery Systems. Bioconjugate Chem. 2009, 20, 5–14. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173. [Google Scholar] [CrossRef] [PubMed]
- Parsi, M.; Desai, M.H.; Desai, D.; Singhal, S.; Khandwala, P.M.; Potdar, R.R. PSMA: A game changer in the diagnosis and treatment of advanced prostate cancer. Med. Oncol. 2021, 38, 89. [Google Scholar] [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Khorev, O.; Stokmaier, D.; Schwardt, O.; Cutting, B.; Ernst, B. Trivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptor. Bioorganic Med. Chem. 2008, 16, 5216–5231. [Google Scholar] [CrossRef]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; Lieske, J.C. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inclisiran: First Approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- Brown, C.R.; Gupta, S.; Qin, J.; Racie, T.; He, G.; Lentini, S.; Malone, R.; Yu, M.; Matsuda, S.; Shulga-Morskaya, S.; et al. Investigating the pharmacodynamic durability of GalNAc-siRNA conjugates. Nucleic Acids Res. 2020, 48, 11827–11844. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Lunavat, T.R.; Jang, S.C.; Nilsson, L.; Park, H.T.; Repiska, G.; L?Sser, C.; Nilsson, J.A.; Gho, Y.S.; L?Tvall, J. RNAi delivery by exosome-mimetic nanovesicles–Implications for targeting c-Myc in cancer. Biomaterials 2016, 102, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Stevens, M.M. Strategic design of extracellular vesicle drug delivery systems. Adv. Drug Deliv. Rev. 2018, 130, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Xu, L.; Faruqu, F.N.; Lim, Y.M.; Lim, K.Y.; Liam-Or, R.; Walters, A.A.; Lavender, P.; Fear, D.; Wells, C.M.; Tzu-Wen Wang, J.; et al. Exosome-mediated RNAi of PAK4 prolongs survival of pancreatic cancer mouse model after loco-regional treatment. Biomaterials 2021, 264, 120369. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef]
- Jeyaram, A.; Lamichhane, T.N.; Wang, S.; Zou, L.; Dahal, E.; Kronstadt, S.M.; Levy, D.; Parajuli, B.; Knudsen, D.R.; Chao, W.; et al. Enhanced Loading of Functional miRNA Cargo via pH Gradient Modification of Extracellular Vesicles. Mol. Ther. 2020, 28, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. J. Control Release 2020, 318, 1–15. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [Green Version]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, F.; Ye, T.; Wang, J.; Lyu, C.; Qing, S.; Ding, Z.; Gao, X.; Jia, R.; Yu, D.; et al. Macrophage-tumor chimeric exosomes accumulate in lymph node and tumor to activate the immune response and the tumor microenvironment. Sci. Transl. Med. 2021, 13, eabb6981. [Google Scholar] [CrossRef] [PubMed]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Li, Z.; Xu, C.; Guo, B.; Guo, P. Folate-displaying exosome mediated cytosolic delivery of siRNA avoiding endosome trapping. J. Control. Release 2019, 311–312, 43–49. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajac-Kaye, M. Myc oncogene: A key component in cell cycle regulation and its implication for lung cancer. Lung Cancer 2001, 34 (Suppl. 2), S43–S46. [Google Scholar] [CrossRef]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31, 1203–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussey, K.J.; Bapat, A.; Linnehan, C.; Wandoloski, M.; Dastrup, E.; Rogers, E.; Gonzales, P.; Demeure, M.J. Targeting polo-like kinase 1, a regulator of p53, in the treatment of adrenocortical carcinoma. Clin. Transl. Med. 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, M.; Bergemann, A.D. Diverse roles for the Eph family of receptor tyrosine kinases in carcinogenesis. Microsc. Res. Tech 2002, 59, 58–67. [Google Scholar] [CrossRef]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeure, M.J.; Armaghany, T.; Ejadi, S.; Ramanathan, R.K.; Kowalski, M.M. A phase I/II study of TKM-080301, a PLK1-targeted RNAi in patients with adrenocortical cancer (ACC). J. Clin. Oncol. 2016, 34, 2547. [Google Scholar] [CrossRef]
- Wu, D.; Yang, J.; Xing, Z.; Han, H.; Wang, T.; Zhang, A.; Yang, Y.; Li, Q. Phenylboronic acid-functionalized polyamidoamine-mediated Bcl-2 siRNA delivery for inhibiting the cell proliferation. Colloids Surf. B Biointerfaces 2016, 146, 318–325. [Google Scholar] [CrossRef]
- Kreuger, J.; Claesson-Welsh, L.; Olsson, A.K.; Dimberg, A. VEGF receptor signalling-in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Wang, G.; Gao, X.; Guojun, G.U.; Shao, Z.; Li, M.; Wang, P.; Yang, J.; Cai, X.; Li, Y. Polyethylene glycol–poly(ε-benzyloxycarbonyl-L-lysine)-conjugated VEGF siRNA for antiangiogenic gene therapy in hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E.I. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2015, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Keller, K.; Doctor, Z.; Dwyer, Z.; Lee, Y.S. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol. Cell 2014, 53, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Dang, J.; Ye, H.; Li, Y.; Liang, Q.; Li, X.; Yin, L. Multivalency-assisted membrane-penetrating siRNA delivery sensitizes photothermal ablation via inhibition of tumor glycolysis metabolism. Biomaterials 2019, 223, 119463. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Sigdel, K.P.; Schaefer, K.G.; Mensah, G.A.K.; King, G.M.; Roberts, A.G. The effects of anthracycline drugs on the conformational distribution of mouse P-glycoprotein explains their transport rate differences. Biochem. Pharm. 2020, 174, 113813. [Google Scholar] [CrossRef]
- Shi, B.; Zheng, M.; Jiang, T.; Yang, W.; Zou, Y.; Wu, H.; Liu, X.; Mcdonald, K.; Ling, D.; Shi, J. The siRNAsome: A Cation-Free and Versatile Nanostructure for siRNA and Drug Co-delivery. Angew. Chem. Int. Ed. 2019, 58, 4938–4942. [Google Scholar] [CrossRef]
- Gao, J.; Li, H.R.; Jin, C.; Jiang, J.H.; Ding, J.Y. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin. Transl. Oncol. 2019, 21, 1287–1301. [Google Scholar] [CrossRef]
- Hayakawa, D.; Takahashi, F.; Mitsuishi, Y.; Tajima, K.; Takahashi, K. Activation of insulin-like growth factor-1 receptor confers acquired resistance to osimertinib in non-small cell lung cancer with EGFR T790M mutation. Thorac. Cancer 2019, 11, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Yin, Q.; Su, J.; Sun, H.; Meng, Q.; Chen, Y.; Chen, L.; Huang, Y.; Gu, W.; Xu, M.; et al. Inhibition of metastasis and growth of breast cancer by pH-sensitive poly (β-amino ester) nanoparticles co-delivering two siRNA and paclitaxel. Biomaterials 2015, 48, 1–15. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.; Zhao, X.; Qi, Y.; Zhang, Y.; Han, X.; Ding, Y.; Guan, J.; Ji, T.; Zhao, Y.; Nie, G. Reshaping Prostate Tumor Microenvironment To Suppress Metastasis via Cancer-Associated Fibroblast Inactivation with Peptide-Assembly-Based Nanosystem. ACS Nano 2019, 13, 12357–12371. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Melo, F.; Vermeulen, L.; Fessler, E.; Medema, J.P. Cancer heterogeneity—A multifaceted view. Embo Rep. 2013, 14, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Hattab, D.; Gazzali, A.M.; Bakhtiar, A. Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment. Pharmaceutics 2021, 13, 1009. [Google Scholar] [CrossRef]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.W.; Hsu, F.F.; Qiu, J.T.; Chern, G.J.; Chen, Y. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci. Adv. 2020, 6, eaax5032. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.B.; Swaminathan, S.K.; Sadhukha, T.; Ma, L.; Panyam, J. The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials 2010, 31, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Wang, K.; Kievit, F.M.; Chiarelli, P.A.; Stephen, Z.R.; Lin, G.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. siRNA nanoparticle suppresses drug-resistant gene and prolongs survival in an orthotopic glioblastoma xenograft mouse model. Adv. Funct. Mater. 2021, 31, 2007166. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, A.; Qian, J.; Xu, M.; Xu, W.; Zhang, Y.; Yao, Y. Folate-decorated PEGylated triblock copolymer as a pH/reduction dual-responsive nanovehicle for targeted intracellular co-delivery of doxorubicin and Bcl-2 siRNA. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 76, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Meijer, T.; Kaanders, J.; Span, P.N.; Bussink, J. Targeting Hypoxia, HIF-1, and Tumor Glucose Metabolism to Improve Radiotherapy Efficacy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, Y.; Zhang, C.; Gu, Z.; Du, J.; Guo, Z.; Dong, X.; Xie, J.; Zhang, G.; Liu, X.; Zhao, Y. Polyoxometalate-Based Radiosensitization Platform for Treating Hypoxic Tumors by Attenuating Radioresistance and Enhancing Radiation Response. ACS Nano 2017, 11, 7164–7176. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA A Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liang, X.; Zhao, B.; Chen, M.; Liu, R.; Sun, S.; Yue, X.; Wang, S. Ultrasound assisted gene and photodynamic synergistic therapy with multifunctional FOXA1-siRNA loaded porphyrin microbubbles for enhancing therapeutic efficacy for breast cancer. Biomaterials 2018, 173, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.M.; Moon, E.K.; Cengel, K.A.; Busch, T.M. Photodynamic Therapy and Immune Checkpoint Blockade. Photochem. Photobiol. 2020, 96, 954–961. [Google Scholar] [CrossRef]
- Wang, D.; Wang, T.; Liu, J.; Yu, H.; Li, Y. Acid-Activatable Versatile Micelleplexes for PD-L1 Blockade-Enhanced Cancer Photodynamic Immunotherapy. Nano Lett. 2017, 16, 5503–5513. [Google Scholar] [CrossRef]
- Huang, J.; Zhuang, C.; Chen, J.; Chen, X.; Li, X.; Zhang, T.; Wang, B.; Feng, Q.; Zheng, X.; Gong, M.; et al. Targeted drug/gene/photodynamic therapy via a stimuli-responsive dendritic polymer-based nano-cocktail for treatment of EGFR-TKI-resistant non-small cell lung cancer. Adv Mater 2022, 34, e2201516. [Google Scholar] [CrossRef]
- Han, H.S.; Choi, K.Y. Advances in Nanomaterial-Mediated Photothermal Cancer Therapies: Toward Clinical Applications. Biomedicines 2021, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.; Sharma, P.K. Clinical, Prognostic and Therapeutic Significance of Heat Shock Proteins in Cancer. Curr Drug Targets 2018, 19, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Gao, X.; Huang, X.; Ge, H.; Xie, M.; Qian, J.; Song, J.; Li, Y.; Zhu, X.; Zhang, C. Polydopamine-coated nucleic acid nanogel for siRNA-mediated low-temperature photothermal therapy. Biomaterials 2020, 245, 119976. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: Prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Stessl, M.; Amartey, J.; Noe, C.R. Off-target effects related to the phosphorothioate modification of nucleic acids. ChemMedChem 2010, 5, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Janas, M.M.; Schlegel, M.K.; Harbison, C.E.; Yilmaz, V.O.; Jiang, Y.; Parmar, R.; Zlatev, I.; Castoreno, A.; Xu, H.; Shulga-Morskaya, S. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun. 2018, 9, 723. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.A.; Dreicer, R.; Anderson, J.; Garcia, J.A.; Alva, A.; Hart, L.L.; Milowsky, M.I.; Posadas, E.M.; Ryan, C.J.; Graf, R.P.; et al. Safety and Efficacy of BIND-014, a Docetaxel Nanoparticle Targeting Prostate-Specific Membrane Antigen for Patients With Metastatic Castration-Resistant Prostate Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 1344–1351. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (Market Name) | Company | Gene Targets/Indications | Organ | Route of Administration | Delivery System | Approval |
|---|---|---|---|---|---|---|
| Patisiran (Onpattro) | Alnylam | TTR/hereditary transthyretin amyloidosis (hATTR) | Liver | Intravenous | LNP | August 2018 (FDA) |
| Givosiran (Givlaari) | Alnylam | ALAS1/acute hepatic porphyria (AHP) | Liver | Subcutaneous | GalNAc conjugate | November 2019 (FDA) |
| Lumasiran (Oxlumo) | Alnylam | HAO1/primary hyperoxaluria type 1 (PH1) | Liver | Subcutaneous | ESC-GalNAc conjugate | November 2020 (FDA, EMA) |
| Inclisiran (Leqvio) | Novartis | PCSK9/hyperlipidemia | Liver | Subcutaneous | ESC-GalNAc conjugate | 2020 (EMA) |
| Name | Indications (Tumor Types) | Delivery System | Gene Targets | Sponsor | Phase | Status | NCT ID |
|---|---|---|---|---|---|---|---|
| SiRNA-EphA2 | Advanced Malignant Solid Neoplasm | Neutral liposome | EphA2 | M.D. Anderson Cancer Center | I | Active, not recruiting | NCT01591356 |
| iExosomes | Pancreatic Cancer | Exosomes | KRAS G12D | M.D. Anderson Cancer Center | I | Recruiting | NCT03608631 |
| Atu027 Atu027 +Gemcitabine | Advanced Solid Tumors Advanced/metastatic pancreatic cancer | Lipople xLipoplex | PKN3 PKN3 | Silence Therapeutics GmbH Silence Therapeutics GmbH | I II | Completed Completed | NCT00938574 NCT01808638 |
| CALAA-01 | Solid Tumors | Transferrin receptor-targeted cyclodextrin nanoparticle | RRM2 | Calando Pharmaceuticals | I | Terminated | NCT00689065 |
| ALN-VSP02 | Solid Tumors | lipid nanoparticle | VEGF, KSP | Alnylam Pharmaceuticals | I | Completed | NCT00882180 |
| siG12D-LODER | Pancreatic Ductal Adenocarcinoma | Miniature biodegradable polymeric matrix | KRAS G12D | Silenseed Ltd. | II | Unknown | NCT01676259 |
| TKM-080301 | Advanced Solid Tumors | SNALP | PLK1 | Arbutus Biopharma Corporation | II | Completed | NCT01262235 NCT02191878 |
| STP705 | Squamous Cell Carcinoma in Situ | Peptide-Nano particle | TGF-β1, COX-2 | Sirnaomics | II | Recruiting | NCT04844983 |
| NBF-006 | Non-Small Cell Lung Cancer Pancreatic Cancer Colorectal Cancer | LNP | GSTP | Nitto BioPharma | I | Recruiting | NCT03819387 |
| DCR-MYC | Hepatocellular Carcinoma | EnCore LNP | MYC | Dicerna pharmaceuticals | 1b/2 | Terminated | NCT02314052 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Xiao, K. Nanoparticles-Based Strategies to Improve the Delivery of Therapeutic Small Interfering RNA in Precision Oncology. Pharmaceutics 2022, 14, 1586. https://doi.org/10.3390/pharmaceutics14081586
Huang J, Xiao K. Nanoparticles-Based Strategies to Improve the Delivery of Therapeutic Small Interfering RNA in Precision Oncology. Pharmaceutics. 2022; 14(8):1586. https://doi.org/10.3390/pharmaceutics14081586
Chicago/Turabian StyleHuang, Jinxing, and Kai Xiao. 2022. "Nanoparticles-Based Strategies to Improve the Delivery of Therapeutic Small Interfering RNA in Precision Oncology" Pharmaceutics 14, no. 8: 1586. https://doi.org/10.3390/pharmaceutics14081586