
Tissue Barrier-on-Chip: A Technology for Reproducible Practice in Drug Testing

 , ,
, , 
Abstract
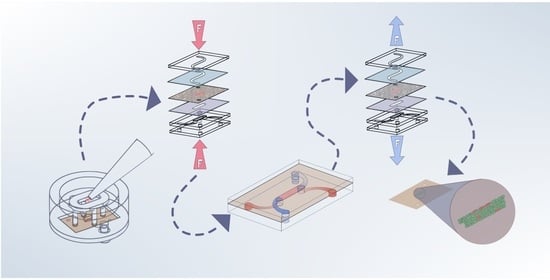
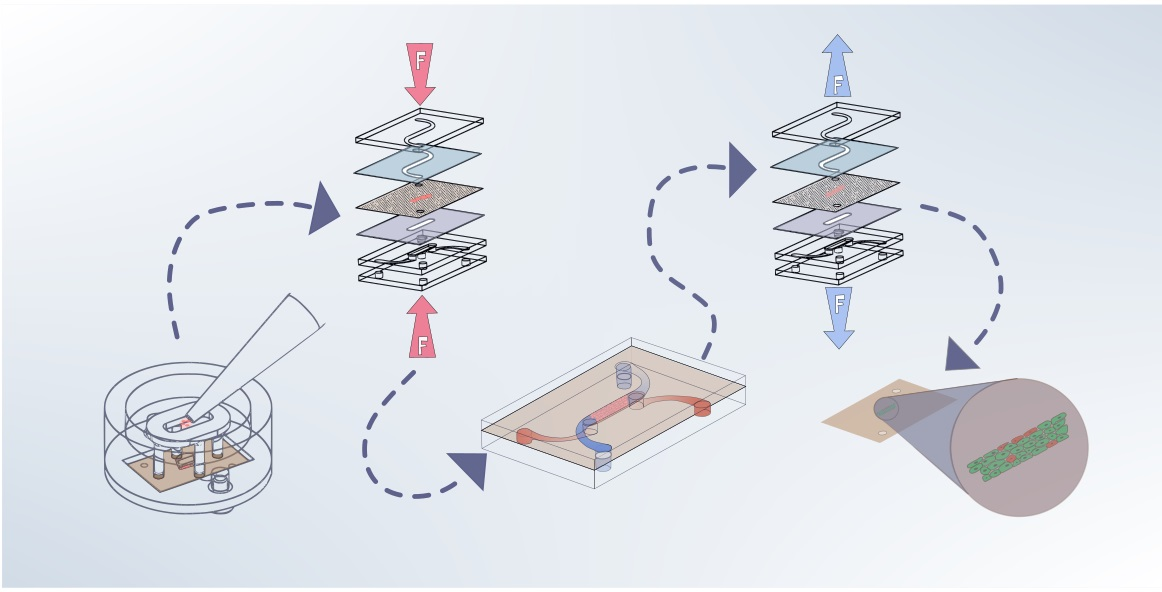
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
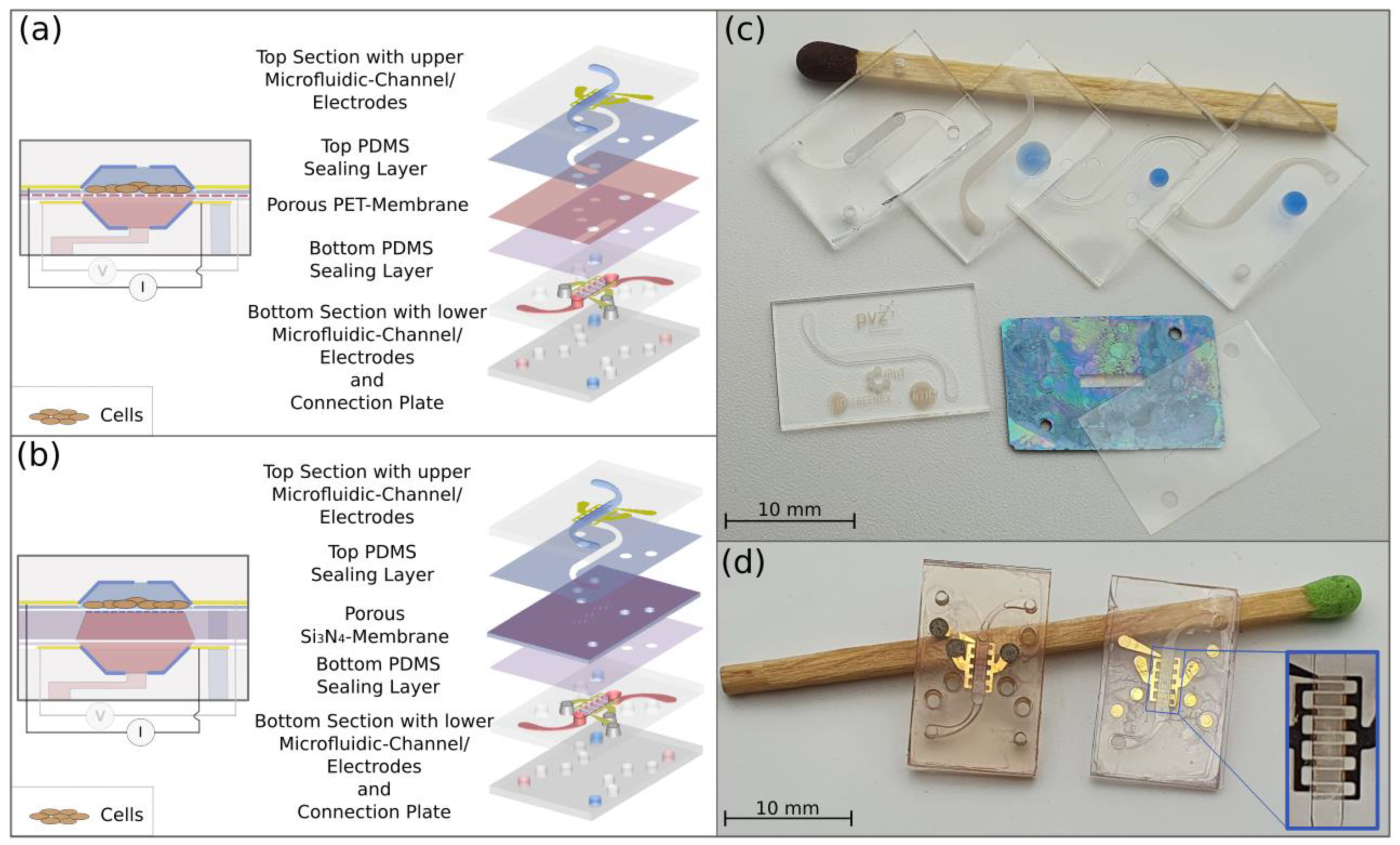
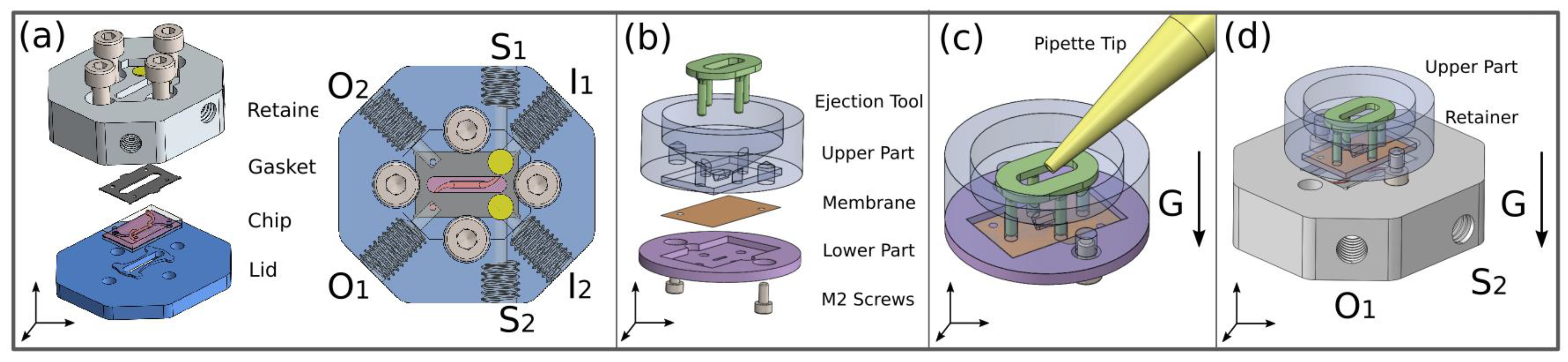
2.1. Microfluidic Chip Design
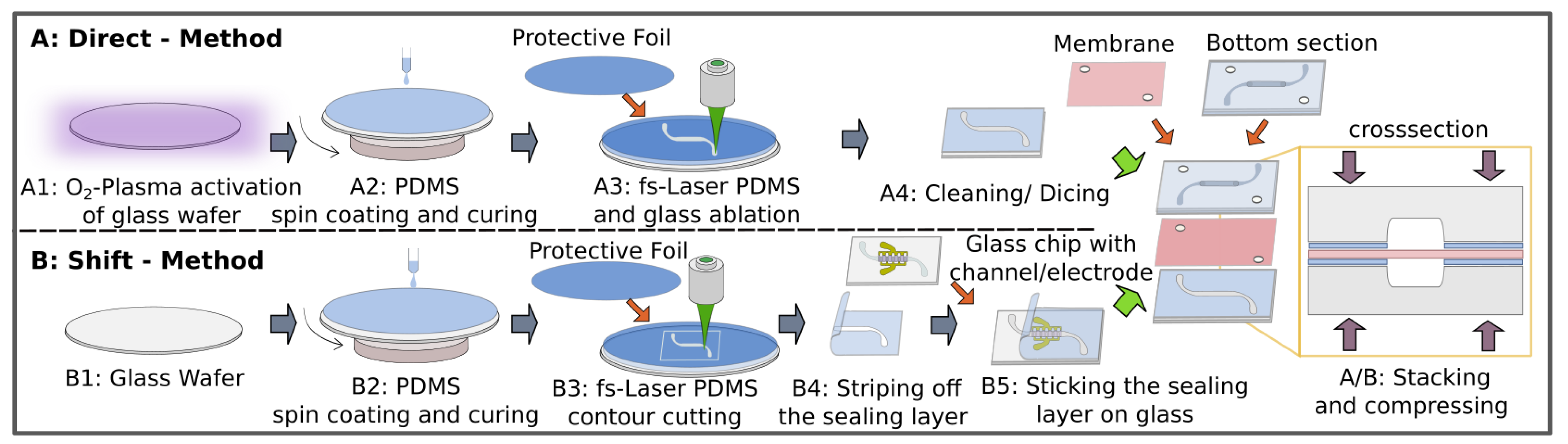
2.2. Fabrication of the Chip
2.3. Fabrication of the Sealing Layer
2.4. Microfluidic Chip Holder
2.5. Transfer Holder
2.6. Cell Cultivation
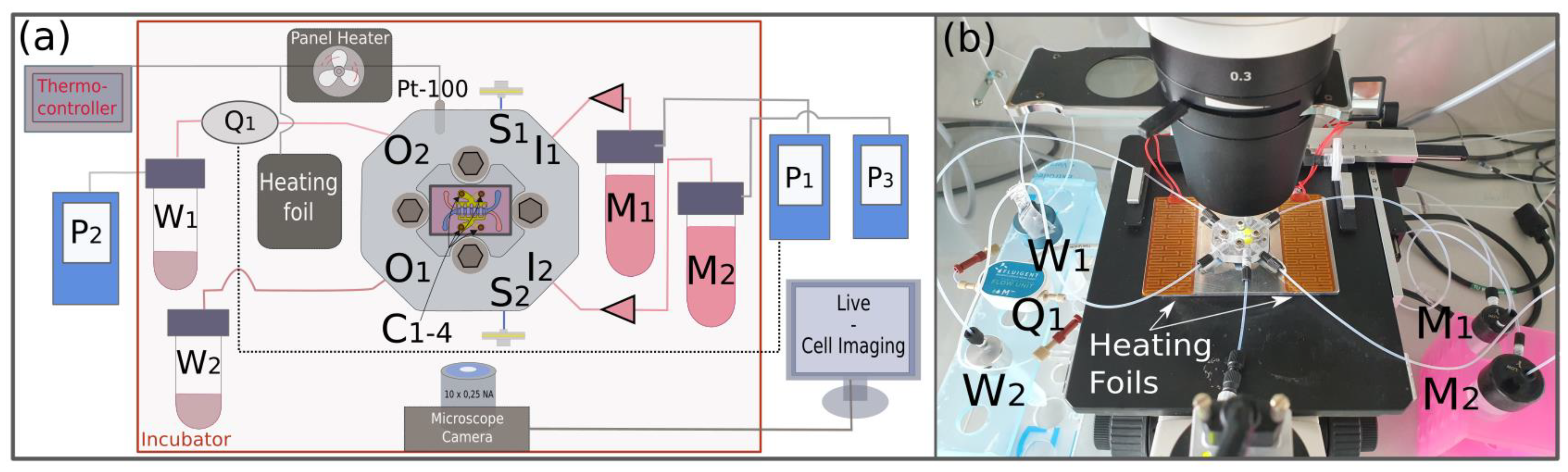
2.7. Experimental Setup
2.8. LIF Measurements
2.9. Gold-Etching Leak Test
2.10. TEER Measurements
3. Results and Discussion
3.1. Gas Trapping
3.2. Material Cell Compatibility Tests
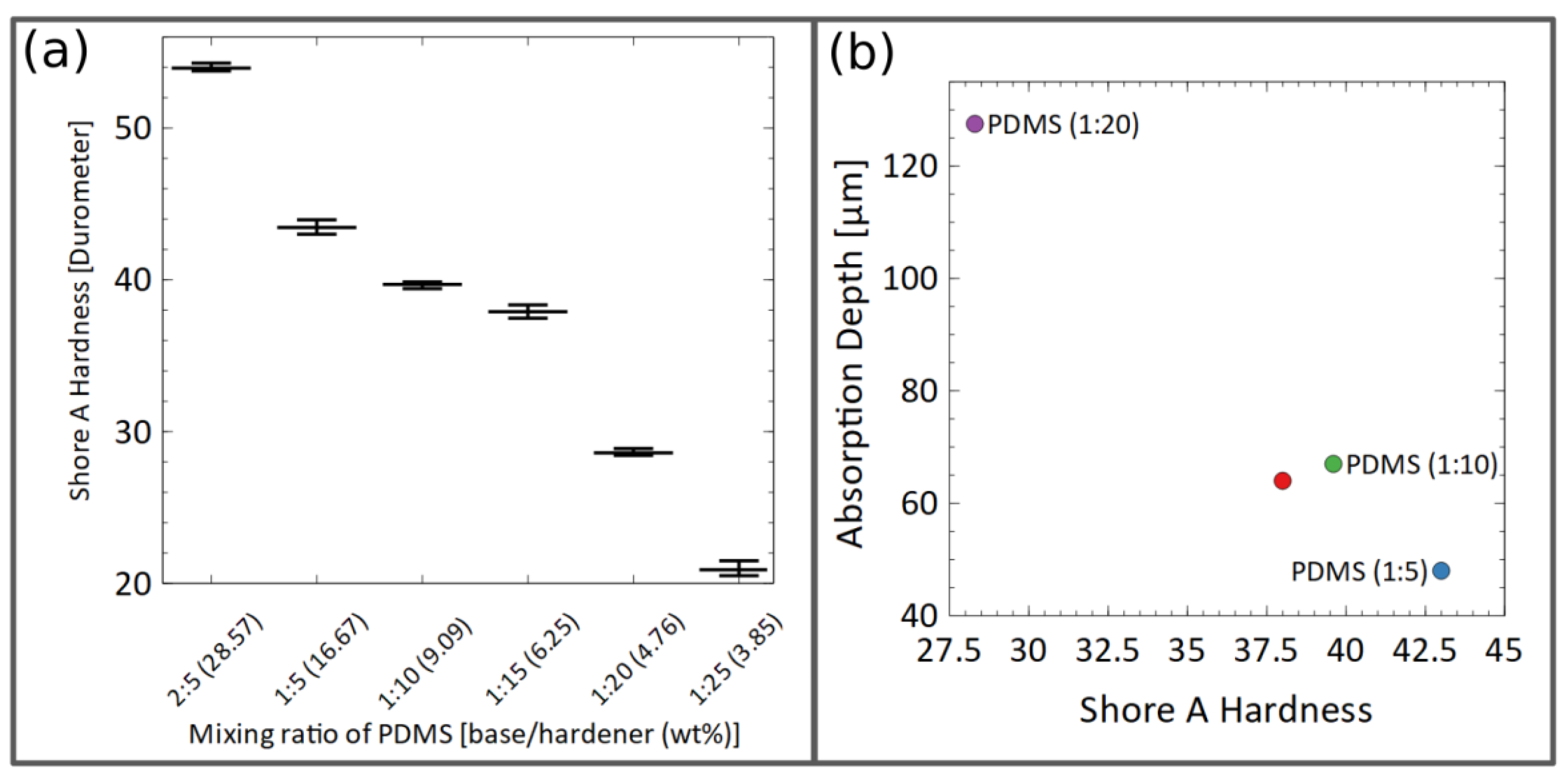
3.3. Sealing-Layer Parameters
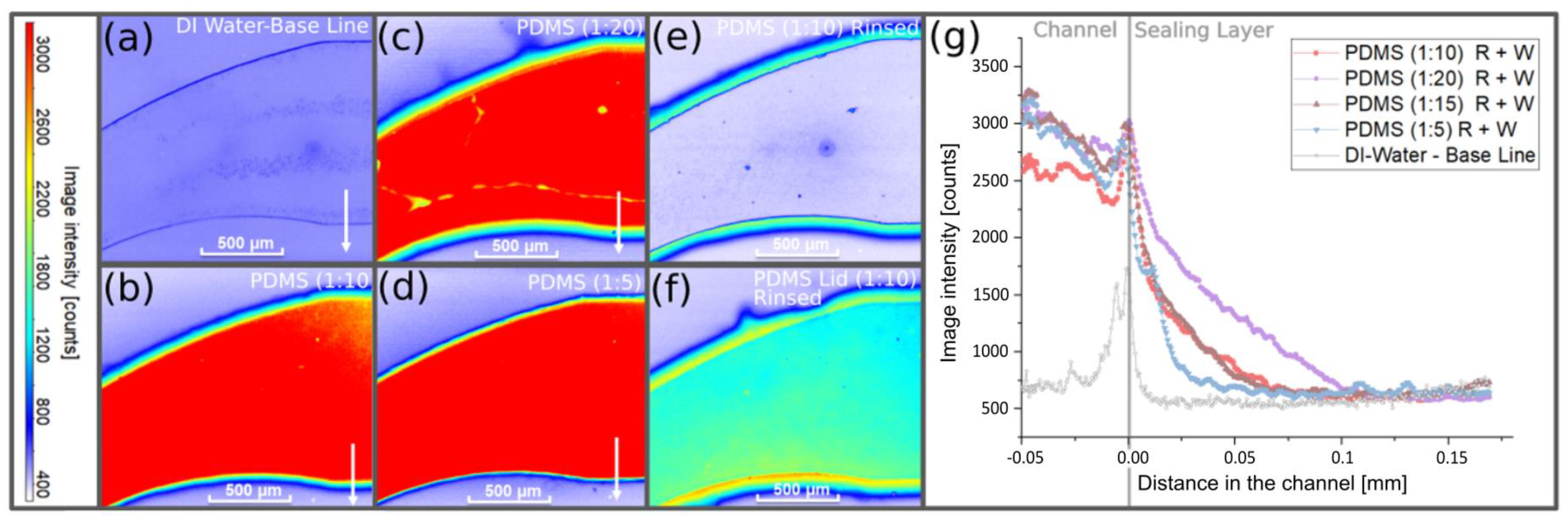
3.4. LIF Measurements
3.5. Leak Test
3.6. Transfer Method
3.7. TEER Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jin, I.S.; Yoon, M.S.; Park, C.-W.; Hong, J.T.; Chung, Y.B.; Kim, J.-S.; Shin, D.H. Replacement techniques to reduce animal experiments in drug and nanoparticle development. J. Pharm. Investig. 2020, 50, 327–335. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.I.; Sei, Y.J.; Park, H.-J.; Kim, J.; Ryu, Y.; Choi, J.J.; Sung, H.-J.; MacDonald, T.J.; Levey, A.I.; Kim, Y. Microengineered human blood-brain barrier platform for understanding nanoparticle transport mechanisms. Nat. Commun. 2020, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Leong, K.W. Advances in microphysiological blood-brain barrier (BBB) models towards drug delivery. Curr. Opin. Biotechnol. 2020, 66, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Hudecz, D.; Khire, T.; Chung, H.L.; Adumeau, L.; Glavin, D.; Luke, E.; Nielsen, M.S.; Dawson, K.A.; McGrath, J.L.; Yan, Y. Ultrathin Silicon Membranes for in Situ Optical Analysis of Nanoparticle Translocation across a Human Blood-Brain Barrier Model. ACS Nano 2020, 14, 1111–1122. [Google Scholar] [CrossRef]
- Coluccio, M.L.; Perozziello, G.; Malara, N.; Parrotta, E.; Zhang, P.; Gentile, F.; Limongi, T.; Raj, P.M.; Cuda, G.; Candeloro, P.; et al. Microfluidic platforms for cell cultures and investigations. Microelectron. Eng. 2019, 208, 14–28. [Google Scholar] [CrossRef]
- Volpe, D.A.; Faustino, P.J.; Ciavarella, A.B.; Asafu-Adjaye, E.B.; Ellison, C.D.; Yu, L.X.; Hussain, A.S. Classification of Drug Permeability with a Caco-2 Cell Monolayer Assay. Clin. Res. Regul. Aff. 2008, 24, 39–47. [Google Scholar] [CrossRef]
- Sakolish, C.M.; Esch, M.B.; Hickman, J.J.; Shuler, M.L.; Mahler, G.J. Modeling Barrier Tissues In Vitro: Methods, Achievements, and Challenges. EBioMedicine 2016, 5, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Azizipour, N.; Avazpour, R.; Rosenzweig, D.H.; Sawan, M.; Ajji, A. Evolution of Biochip Technology: A Review from Lab-on-a-Chip to Organ-on-a-Chip. Micromachines 2020, 11, 599. [Google Scholar] [CrossRef]
- Piergiovanni, M.; Leite, S.B.; Corvi, R.; Whelan, M. Standardisation needs for organ on chip devices. Lab Chip 2021, 21, 2857–2868. [Google Scholar] [CrossRef] [PubMed]
- Bunge, F.; van den Driesche, S.; Vellekoop, M.J. PDMS-free microfluidic cell culture with integrated gas supply through a porous membrane of anodized aluminum oxide. Biomed. Microdevices 2018, 20, 98. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.I.; Amaral, M.H.; Costa, P.C.; Lopes, C.M.; Lamprou, D.A. Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies. Pharmaceutics 2020, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Beiβner, N.; Mattern, K.; Dietzel, A.; Reichl, S. DynaMiTES—A dynamic cell culture platform for in vitro drug testing PART 2—Ocular DynaMiTES for drug absorption studies of the anterior eye. Eur. J. Pharm. Biopharm. 2018, 126, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Hinkel, S.; Mattern, K.; Dietzel, A.; Reichl, S.; Müller-Goymann, C.C. Parametric investigation of static and dynamic cell culture conditions and their impact on hCMEC/D3 barrier properties. Int. J. Pharm. 2019, 566, 434–444. [Google Scholar] [CrossRef]
- Yang, Q.; Ju, D.; Liu, Y.; Lv, X.; Xiao, Z.; Gao, B.; Song, F.; Xu, F. Design of organ-on-a-chip to improve cell capture efficiency. Int. J. Mech. Sci. 2021, 209, 106705. [Google Scholar] [CrossRef]
- Mattern, K.; Beißner, N.; Reichl, S.; Dietzel, A. DynaMiTES—A dynamic cell culture platform for in vitro drug testing PART 1—Engineering of microfluidic system and technical simulations. Eur. J. Pharm. Biopharm. 2018, 126, 159–165. [Google Scholar] [CrossRef]
- Dehne, E.-M.; Marx, U. The universal physiological template—A system to advance medicines. Curr. Opin. Toxicol. 2020, 23–24, 1–5. [Google Scholar] [CrossRef]
- Wagner, I.; Materne, E.-M.; Brincker, S.; Süssbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [Green Version]
- Yahyazadeh Shourabi, A.; Kashaninejad, N.; Saidi, M.S. An integrated microfluidic concentration gradient generator for mechanical stimulation and drug delivery. J. Sci. Adv. Mater. Devices 2021, 6, 280–290. [Google Scholar] [CrossRef]
- Wang, J.D.; Douville, N.J.; Takayama, S.; ElSayed, M. Quantitative analysis of molecular absorption into PDMS microfluidic channels. Ann. Biomed. Eng. 2012, 40, 1862–1873. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Huang, B.; Zare, R.N. Construction of microfluidic chips using polydimethylsiloxane for adhesive bonding. Lab Chip 2005, 5, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- van Meer, B.J.; de Vries, H.; Firth, K.S.A.; van Weerd, J.; Tertoolen, L.G.J.; Karperien, H.B.J.; Jonkheijm, P.; Denning, C.; IJzerman, A.P.; Mummery, C.L. Small molecule absorption by PDMS in the context of drug response bioassays. Biochem. Biophys. Res. Commun. 2017, 482, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regehr, K.J.; Domenech, M.; Koepsel, J.T.; Carver, K.C.; Ellison-Zelski, S.J.; Murphy, W.L.; Schuler, L.A.; Alarid, E.T.; Beebe, D.J. Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 2009, 9, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Akther, F.; Yakob, S.B.; Nguyen, N.-T.; Ta, H.T. Surface Modification Techniques for Endothelial Cell Seeding in PDMS Microfluidic Devices. Biosensors 2020, 10, 182. [Google Scholar] [CrossRef]
- Hirama, H.; Satoh, T.; Sugiura, S.; Shin, K.; Onuki-Nagasaki, R.; Kanamori, T.; Inoue, T. Glass-based organ-on-a-chip device for restricting small molecular absorption. J. Biosci. Bioeng. 2019, 127, 641–646. [Google Scholar] [CrossRef]
- Vera, D.; García-Díaz, M.; Torras, N.; Álvarez, M.; Villa, R.; Martinez, E. Engineering Tissue Barrier Models on Hydrogel Microfluidic Platforms. ACS Appl. Mater. Interfaces 2021, 13, 13920–13933. [Google Scholar] [CrossRef]
- Ferro, M.P.; Heilshorn, S.C.; Owens, R.M. Materials for blood brain barrier modeling in vitro. Mater. Sci. Eng. R Rep. 2020, 140, 100522. [Google Scholar] [CrossRef]
- Yeste, J.; Illa, X.; Gutiérrez, C.; Solé, M.; Guimerà, A.; Villa, R. Geometric correction factor for transepithelial electrical resistance measurements in transwell and microfluidic cell cultures. J. Phys. D Appl. Phys. 2016, 49, 375401. [Google Scholar] [CrossRef] [Green Version]
- Erfle, P.; Riewe, J.; Bunjes, H.; Dietzel, A. Stabilized Production of Lipid Nanoparticles of Tunable Size in Taylor Flow Glass Devices with High-Surface-Quality 3D Microchannels. Micromachines 2019, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.H.; Lepak, L.A.; Hussain, R.J.; Shain, W.; Shuler, M.L. An endothelial and astrocyte co-culture model of the blood-brain barrier utilizing an ultra-thin, nanofabricated silicon nitride membrane. Lab Chip 2005, 5, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh-Barforoushi, A.; Law, A.M.K.; Hejri, A.; Asadnia, M.; Ormandy, C.J.; Gallego-Ortega, D.; Ebrahimi Warkiani, M. Static droplet array for culturing single live adherent cells in an isolated chemical microenvironment. Lab Chip 2018, 18, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, Z.; Zhang, W.; Jiang, X. A simple PDMS-based microfluidic channel design that removes bubbles for long-term on-chip culture of mammalian cells. Lab Chip 2010, 10, 2906–2910. [Google Scholar] [CrossRef]
- Kratz, S.R.A.; Eilenberger, C.; Schuller, P.; Bachmann, B.; Spitz, S.; Ertl, P.; Rothbauer, M. Characterization of four functional biocompatible pressure-sensitive adhesives for rapid prototyping of cell-based lab-on-a-chip and organ-on-a-chip systems. Sci. Rep. 2019, 9, 9287. [Google Scholar] [CrossRef] [PubMed]
- Johnston, I.D.; McCluskey, D.K.; Tan, C.K.L.; Tracey, M.C. Mechanical characterization of bulk Sylgard 184 for microfluidics and microengineering. J. Micromech. Microeng. 2014, 24, 35017. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, E.V.; Ledwig, V.; Bendas, S.; Reichl, S.; Dietzel, A. Tissue Barrier-on-Chip: A Technology for Reproducible Practice in Drug Testing. Pharmaceutics 2022, 14, 1451. https://doi.org/10.3390/pharmaceutics14071451
Koch EV, Ledwig V, Bendas S, Reichl S, Dietzel A. Tissue Barrier-on-Chip: A Technology for Reproducible Practice in Drug Testing. Pharmaceutics. 2022; 14(7):1451. https://doi.org/10.3390/pharmaceutics14071451
Chicago/Turabian StyleKoch, Eugen V., Verena Ledwig, Sebastian Bendas, Stephan Reichl, and Andreas Dietzel. 2022. "Tissue Barrier-on-Chip: A Technology for Reproducible Practice in Drug Testing" Pharmaceutics 14, no. 7: 1451. https://doi.org/10.3390/pharmaceutics14071451