
Biotinylated Polymer-Ruthenium Conjugates: In Vitro and In Vivo Studies in a Triple-Negative Breast Cancer Model

 ,
,  ,
,  , ,
, ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Synthesis
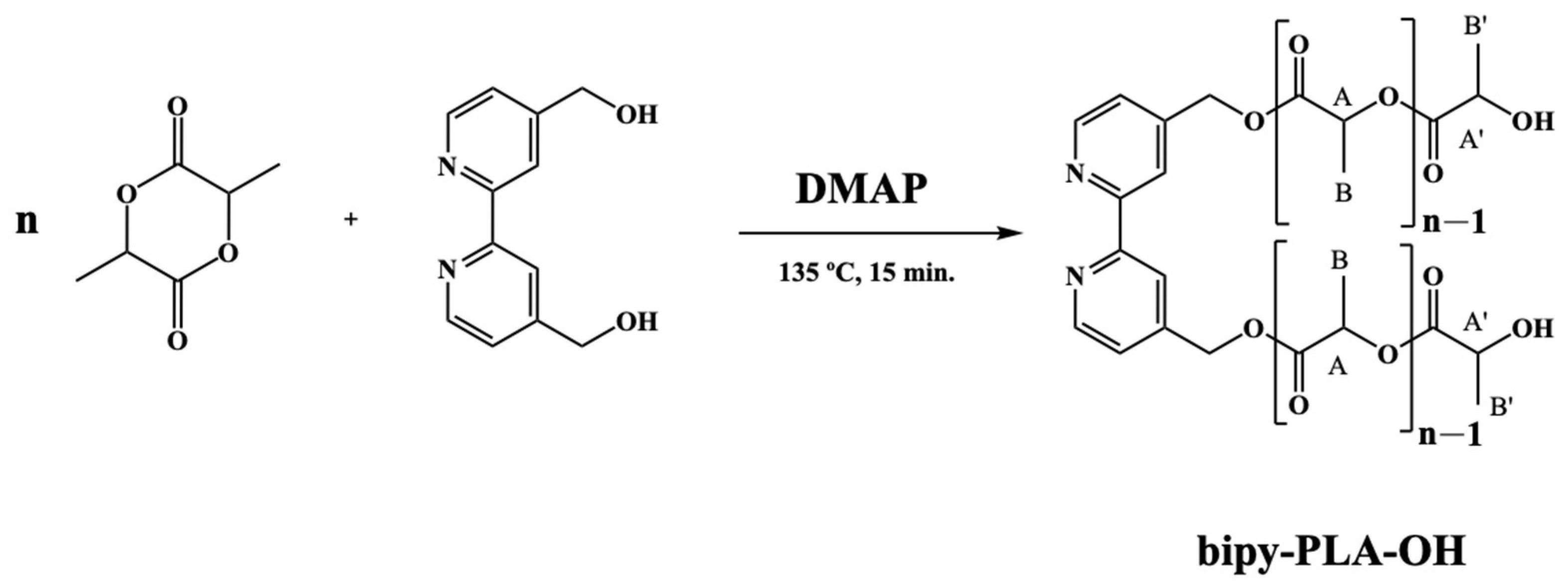
2.1.1. General Procedure for the Polymerization of D,L-Lactide (Bipy-PLA-OH)
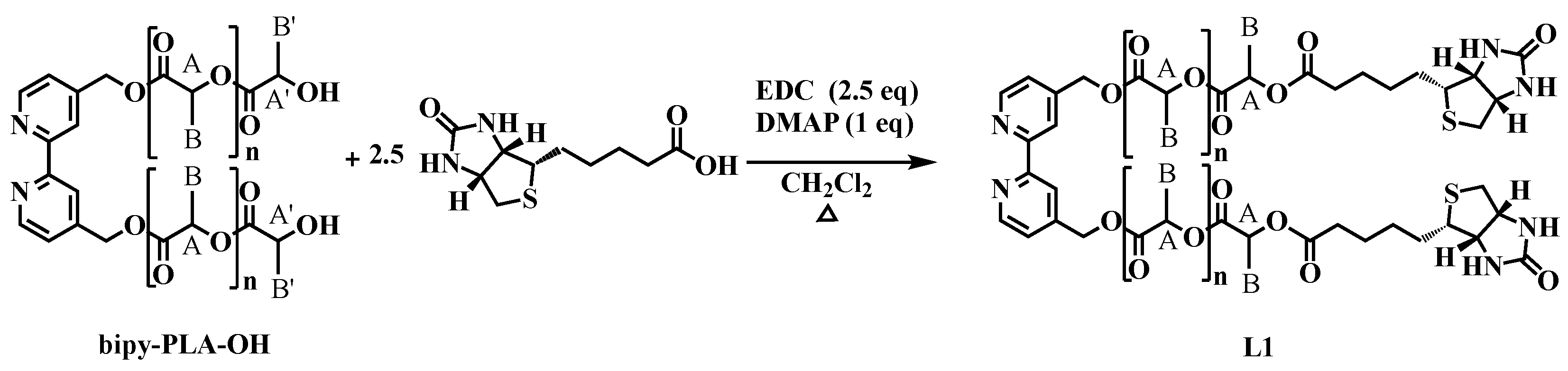
2.1.2. General Procedure for the Synthesis of 2,2′-Bipyridine-4,4′-PLA-Biotin (L1)
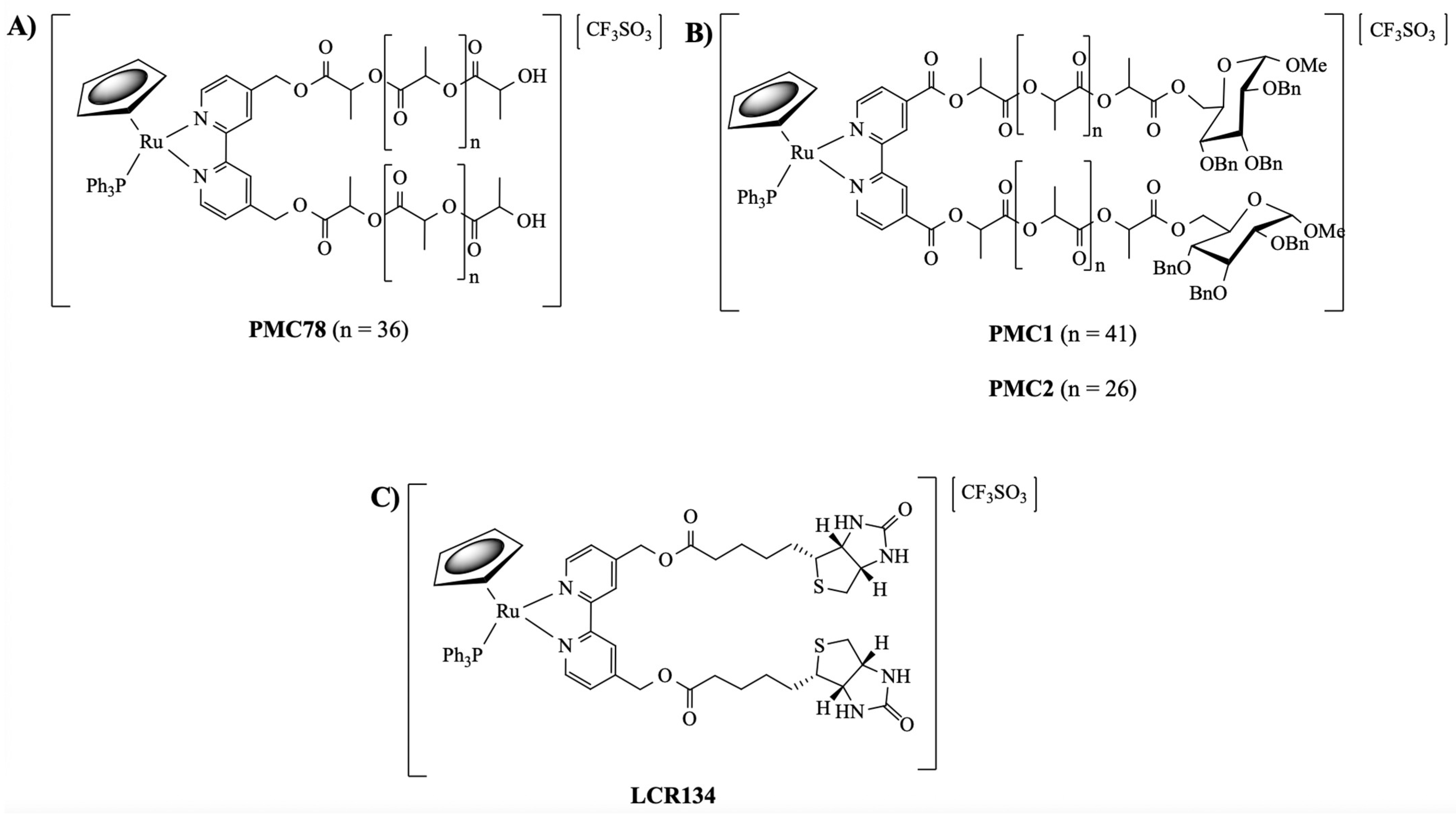
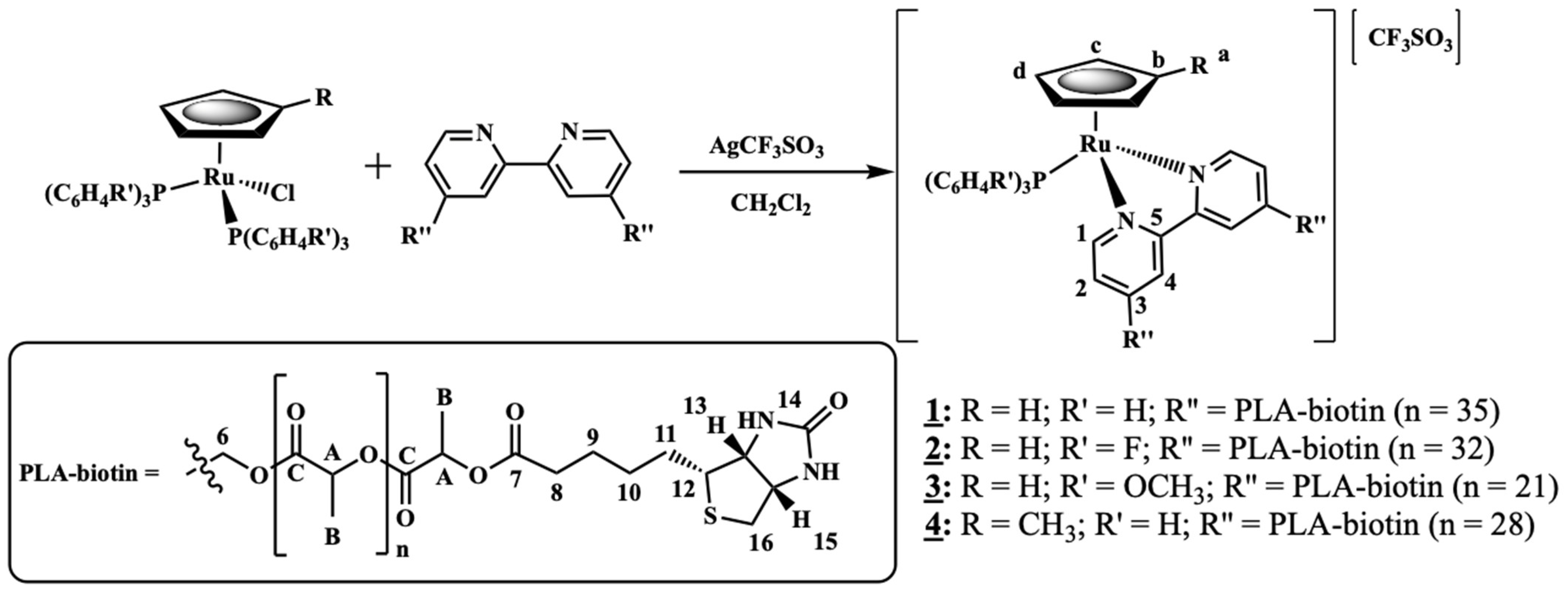
2.1.3. General Procedure for the Synthesis of [Ru(CpR)(P(C6H4R’)3)(2,2′-Bipyridine-4,4′-PLA-Biotin][CF3SO3] R = H; R’ = H (1) or R = H; R’ = F (2) or R = H; R’ = OCH3 (3) or R = CH3; R’ = H (4)
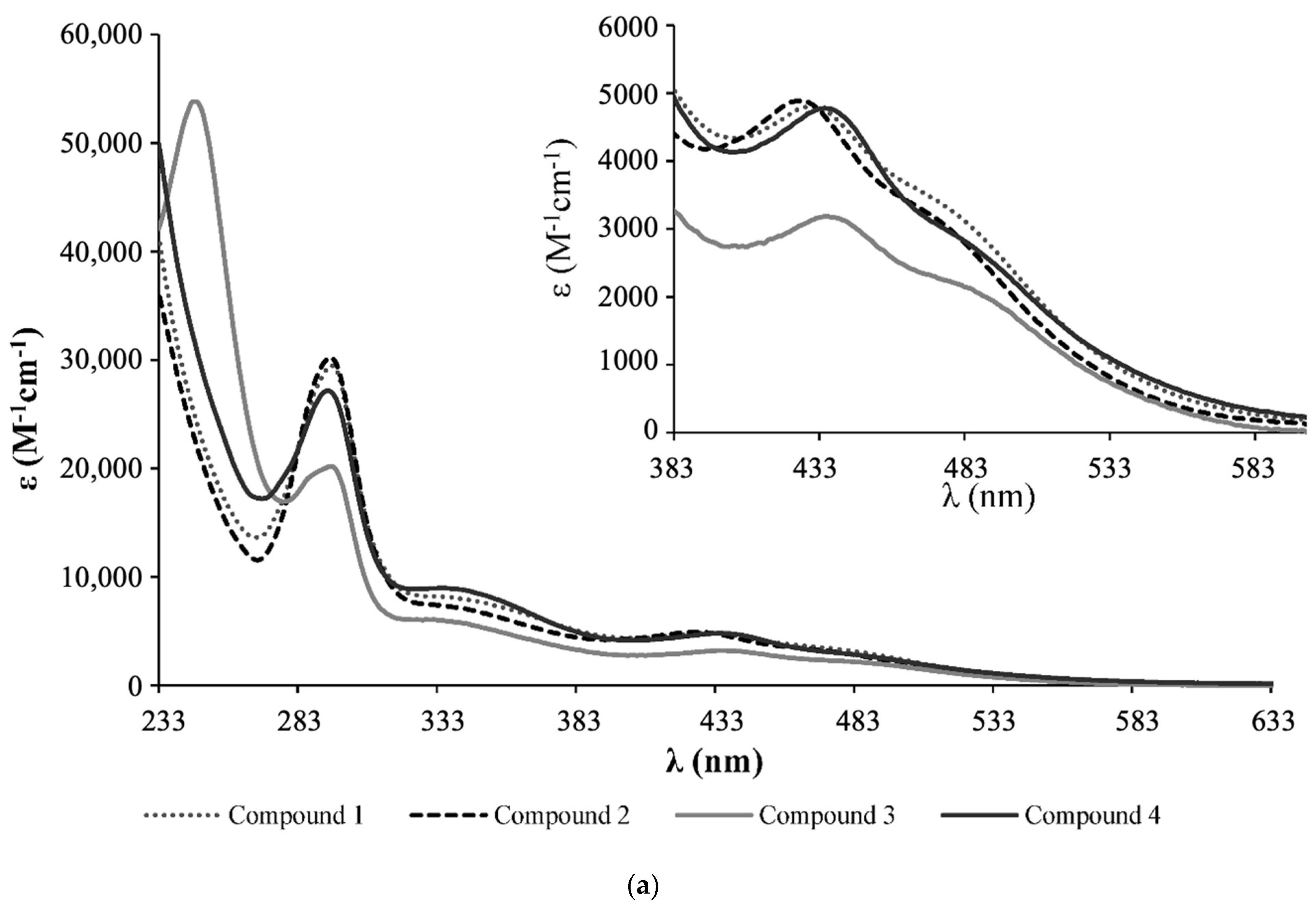
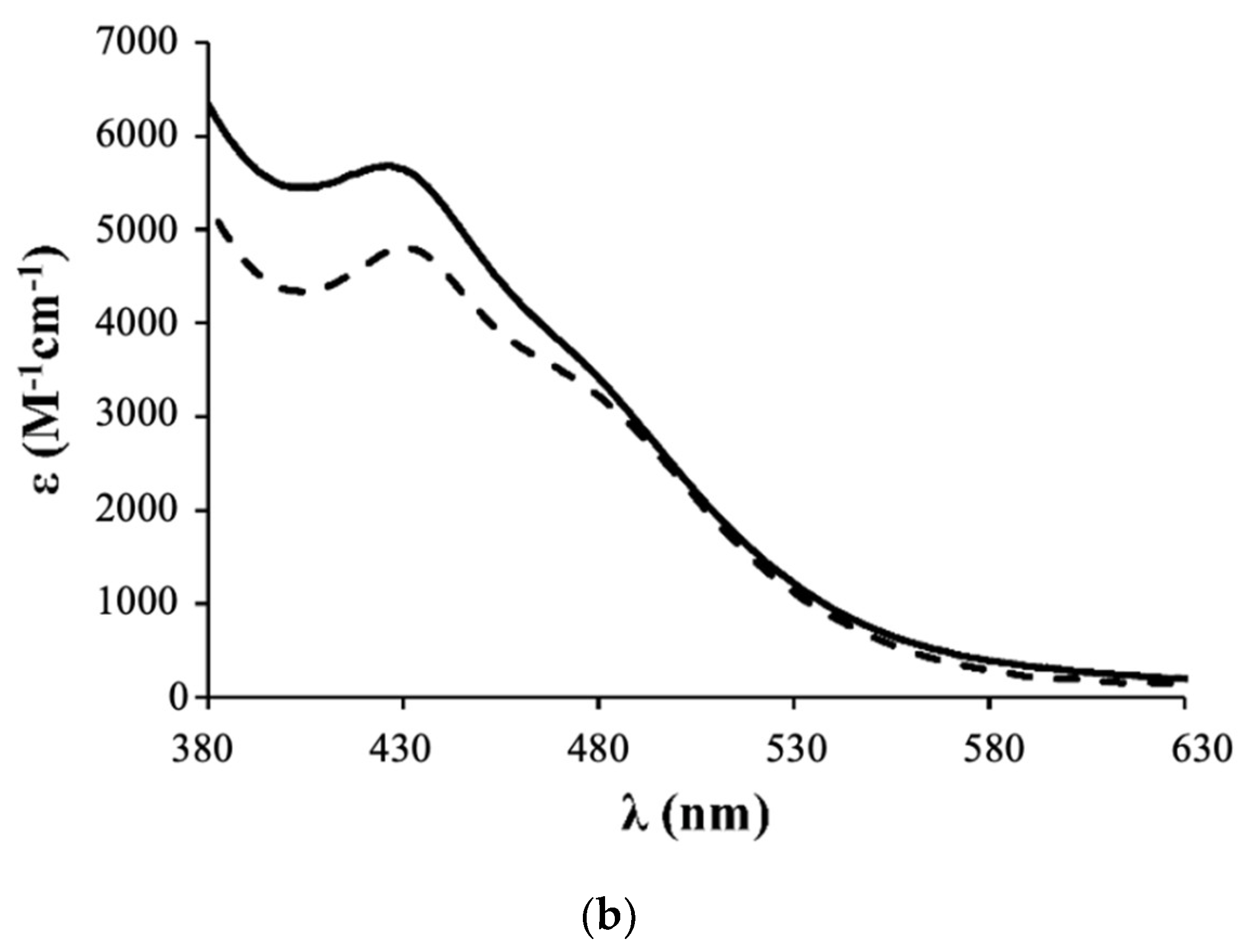
- Complex 1: 1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.52 (m, 2, H1); 8.09 (m, 2, H4); 7.45 (m, 3, HparaPPh3); 7.34 (m, 8, H2+HmetaPPh3); 7.12 (m, 6, HorthoPPh3); 5.35 (m, 4, H6); 5.20 (m, 99, HA, PLA); 4.95 (s, 5, η5-C5H5); 4.77 (m, 2, H15); 4.69 (m, 2, H13); 3.36 (m, 2, H12); 2.86 (m, under de signal of the water of solvent, H16); 2.42 (m, 4, H8); 1.79 (m, 4, H9); 1.55 (m, 179, H10+H11+HB, PLA). 13C NMR [(CD3)2CO, δ/ppm]: 173.25 (C7); 170.10 (CC, PLA); 156.94 (C1); 156.29 (C5); 146.64 (C3); 133.82 (d, 2JCP = 11.1, CHorthoPPh3); 132.12 (d, 1JCP = 42.3, Cq, PPh3); 131.09 (CHparaPPh3); 129.42 (d, 3JCP = 10.1, CHmetaPPh3); 123.95 (C2); 122.04 (C4); 79.42 (Cp); 69.82 (CA, PLA); 65.02 (C6); 62.55 (C13); 60.58 (C15); 58.14 (C12); 42.65 (C16); 33.90 (C8); 29.84 (under the signal of the solvent, C10, C11); 25.32 (C9); 17.06 (CB, PLA). 31P NMR [(CD3)2CO, δ/ppm]: 51.13 (s, PPh3). UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1cm−1)]: 295 (29.5), 336 (8.1), 430 (4.8), 481 (Sh). [DMSO, λmax/nm (ε × 103/M−1cm−1)]: 296 (35.5), 361 (Sh), 426 (5.7), 477 (Sh). FTIR [KBr, cm−1]: 3416 (υN-H amine), 3075 (υC-H Cp and aromatic rings); 2995-2878 (υC-H alkanes), 1757 (υC=O PLA and υC=O ester), 1454 (υC=C Cp and aromatic rings), 1273, 1186, 1030 (υ(CF3SO3−)), 1186 (υC-O ester). Mn, calculated by NMR: 6233.7 g/mol.
- Complex 2: 1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.54 (m, 2, H1); 8.21 (m, 2, H4); 7.41 (m, 2, H2); 7.15 (m, 12, P(PhF)3); 5.36 (m, 4, H6); 5.20 (m, 85, HA, PLA); 5.00 (s, 5, η5-C5H5); 4.69 (m, 2, H15); 4.61 (m, 2, H13); 3.24 (m, 2, H12); 2.85 (m, under de signal of the water of solvent, H16); 2.41 (m, 4, H8); 1.70 (m, 4, H9); 1.55 (m, 278, H10+H11+HB, PLA). 13C NMR [(CD3)2CO, δ/ppm]: 173.11 (C7); 170.10 (CC, PLA); 164.76 (dd, 1JCF = 250.5; Cq, P(PhF)3); 157.14 (C1); 156.32 (C5); 147.03 (C3); 136.22 (dd, 3JCP = 12.6; 2JCF = 9.0, CHmetaP(PhF)3); 128.20 (dd, 1JCP = 42.3, Cq, P(PhF)3); 124.35 (C2); 122.42 (C4); 116.64 (dd, 2JCP = 21.1; 3JCF = 11.1 Hz, CHorthoP(PhF)3); 79.66 (Cp); 69.82 (CA, PLA); 65.07 (C6); 62.60 (C13); 60.57 (C15); 58.21 (d, C12); 43.11 (C16); 33.91 (C8); 29.84 (under the signal of the solvent, C10, C11); 25.33 (d, C9); 17.06 (CB, PLA). 31P NMR [(CD3)2CO, δ/ppm]: 50.09 (s, P(PhF)3. UV-Vis [CH2Cl2, λmax/nm (ε × 103/M−1cm−1)]: 295 (30.2); 336 (7.3); 427 (4.9); 479 (Sh). [DMSO, λmax/nm (ε × 103/M−1cm−1)]: 296 (27.0); 346 (Sh); 426 (4.2); 478 (Sh). FTIR [KBr, cm−1]: 3420 (υN-H amine); 3069 (υC-H Cp and aromatic rings); 2997–2876 (υC-H alkanes), 1757 (υC=O PLA and υC=O ester), 1454 (υC=C Cp and aromatic rings), 1284, 1186, 1028 (υ(CF3SO3−)), 1186 (υC-O ester). Mn, calculated by NMR: 5941.6 g/mol.
- Complex 3: 1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.52 (d, 2, 3JHH = 8.0, H1); 8.14 (s, 2, H4); 7.37 (m, 2, H2); 7.02 (m, 6, HorthoP(PhOCH3)3); 6.87 (m, 6, HmetaP(PhOCH3)3); 5.35 (m, 4, H6); 5.22 (m, 86, HA, PLA); 4.93 (s, 5, η5-C5H5); 4.52 (m, 2, H15); 4.36 (m, 2, H13); 3.83 (s, 9, OCH3); 3.26 (m, 2, H12); 2.85 (m, under the signal of the water of solvent, H16) 2.40 (m, 4, H8), 1.55 (m, 278, H9+H10+H11+HB, PLA). 13C NMR [(CD3)2CO, δ/ppm]: 173.20 (C7); 170.10 (CC, PLA); 161.78 (Cq, P(PhOCH3)3); 156.91 (C1); 156.31 (C5); 146.12 (C3); 135.29 (d, 2JCP = 13.1, CHorthoP(PhOCH3)3); 123.87 (C2); 123.52 (d, 1JCP = 47.3, Cq, P(PhOCH3)3); 122.36 (C4); 114.76 (d, 3JCP = 10.1, CHmetaP(PhOCH3)3); 79.20 (Cp); 69.82 (CA, PLA); 65.08 (C6); 62.36 (C13); 60.73 (C15); 58.01 (C12); 55.70 (OCH3); 39.75 (C16); 33.97 (C8); 29.84 (under the signal of the solvent, C10, C11); 25.49 (C9); 17.05 (CB, PLA). 31P NMR [(CD3)2CO, δ/ppm]: 47.02 (s, P(PhOCH3)3). UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1cm−1)]: 246 (53.8); 295 (20.2); 336 (5.9); 435 (3.2); 492 (Sh). [DMSO, λmax/nm (ε × 103/M−1cm−1)]: 294 (18.9); 341 (Sh); 433 (2.9); 492 (Sh). FTIR [KBr, cm−1]: 3410 (υN-H amine); 3073 (υC-H Cp and aromatic rings); 2995–2878 (υC-H alkanes); 1757 (υC=O PLA and υC=O ester), 1456 (υC=C Cp and aromatic rings), 1278, 1186, 1028 (υ(CF3SO3−)), 1186 (υC-O ester). Mn, calculated by NMR: 4319.4 g/mol.
- Complex 4: 1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.47 (m, 2, H1); 8.11 (m, 2, H4); 7.42 (m, 4, H2+HparaPPh3); 7.33 (m, 7, HmetaPPh3); 7.11 (m, 6, HorthoPPh3); 5.36 (m, 4, H6); 5.20 (m, 64, HA, PLA); 4.77 (s, 2, Hd, Cp); 4.71 (m, 2, H15); 4.67 (m, 2, Hc, Cp); 4.55 (m, 2, H13); 3.39 (m, 2, H12); 3.05 (m, 2, H16); 2.88 (m, 2, H16); 2.40 (m, 4, H8); 1.68 (m, 7, H9 + Ha, Cp); 1.55 (m, 211, H10+H11+HB, PLA). 13C NMR [(CD3)2CO, δ/ppm]: 173.14 (C7); 170.09 (CC, PLA); 156.49 (C1); 156.22 (C5); 146.44 (C3); 133.75 (d, 2JCP = 11.1, CHorthoPPh3); 132.29 (d, 1JCP = 41.3, Cq, PPh3); 131.01 (CHparaPPh3); 129.40 (d, 3JCP = 10.1, CHmetaPPh3); 124.32 (C2); 123.72 (C4); 103.38 (Cb, Cp); 77.00 (Cc + Cd, Cp); 69.81 (CA, PLA); 64.99 (C6); 63.27 (C13); 61.74 (C15); 56.47 (C12); 40.89 (C16); 33.95 (C8); 29.84 (under the signal of the solvent, C10, C11); 25.41 (C9); 17.05 (CB, PLA); 11.69 (Ca, Cp). 31P NMR [(CD3)2CO, δ/ppm]: 51.63 (s, PPh3). UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1cm−1)]: 294 (27.1), 334 (9.0), 435 (4.8), 489 (Sh). [DMSO, λmax/nm (ε × 103/M−1cm−1)]: 296 (12.6), 343 (3.8), 431 (2.1), 493 (Sh). FTIR [KBr, cm−1]: 3307 (υN-H amine), 3069 (υC-H Cp and aromatic rings); 2997–2878 (υC-H alkanes); 2878 (υC-H Cp and aromatic rings); 1753 (υC=O PLA and υC=O ester), 1456 (υC=C Cp and aromatic rings), 1277, 1186, 1028 (υ(CF3SO3-)), 1186 (υC-O ester). Mn, calculated by NMR: 5296.0 g/mol.
2.2. Biological Studies
2.2.1. Cell Lines and Culture Conditions
2.2.2. Compounds Dilution and Storage
2.2.3. MTT Assay
2.2.4. Intracellular Distribution
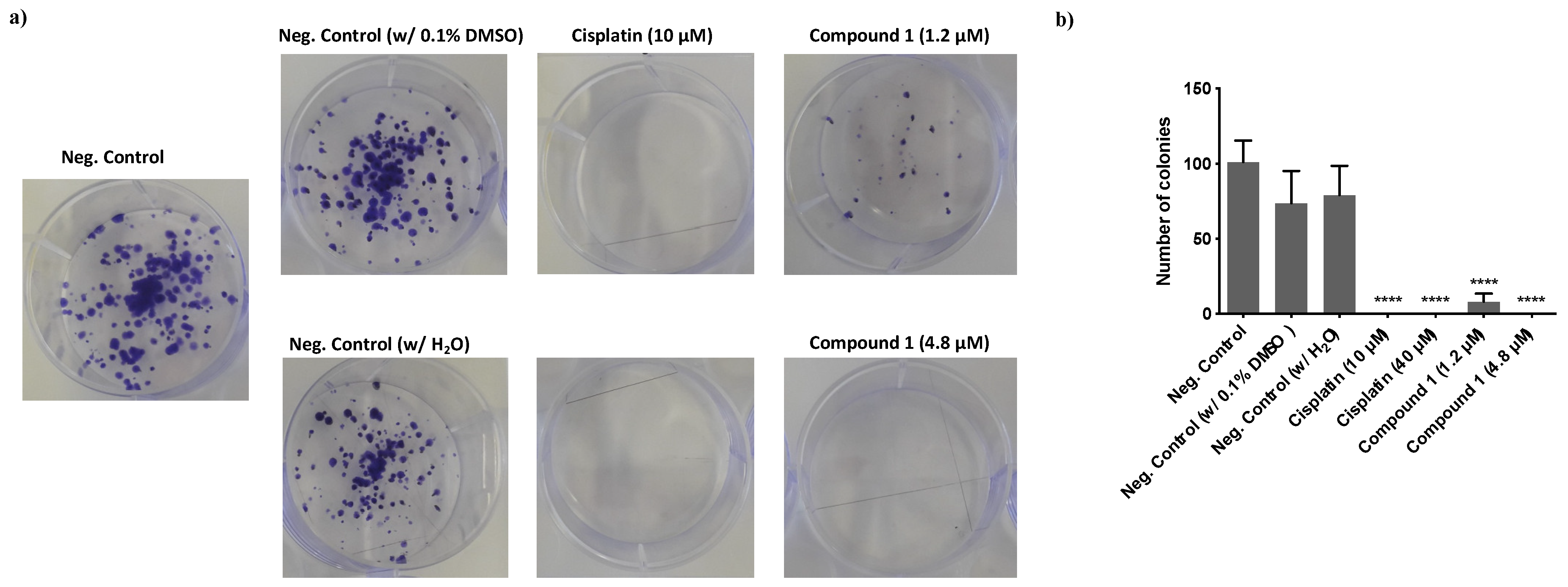
2.2.5. Colony Formation Assay
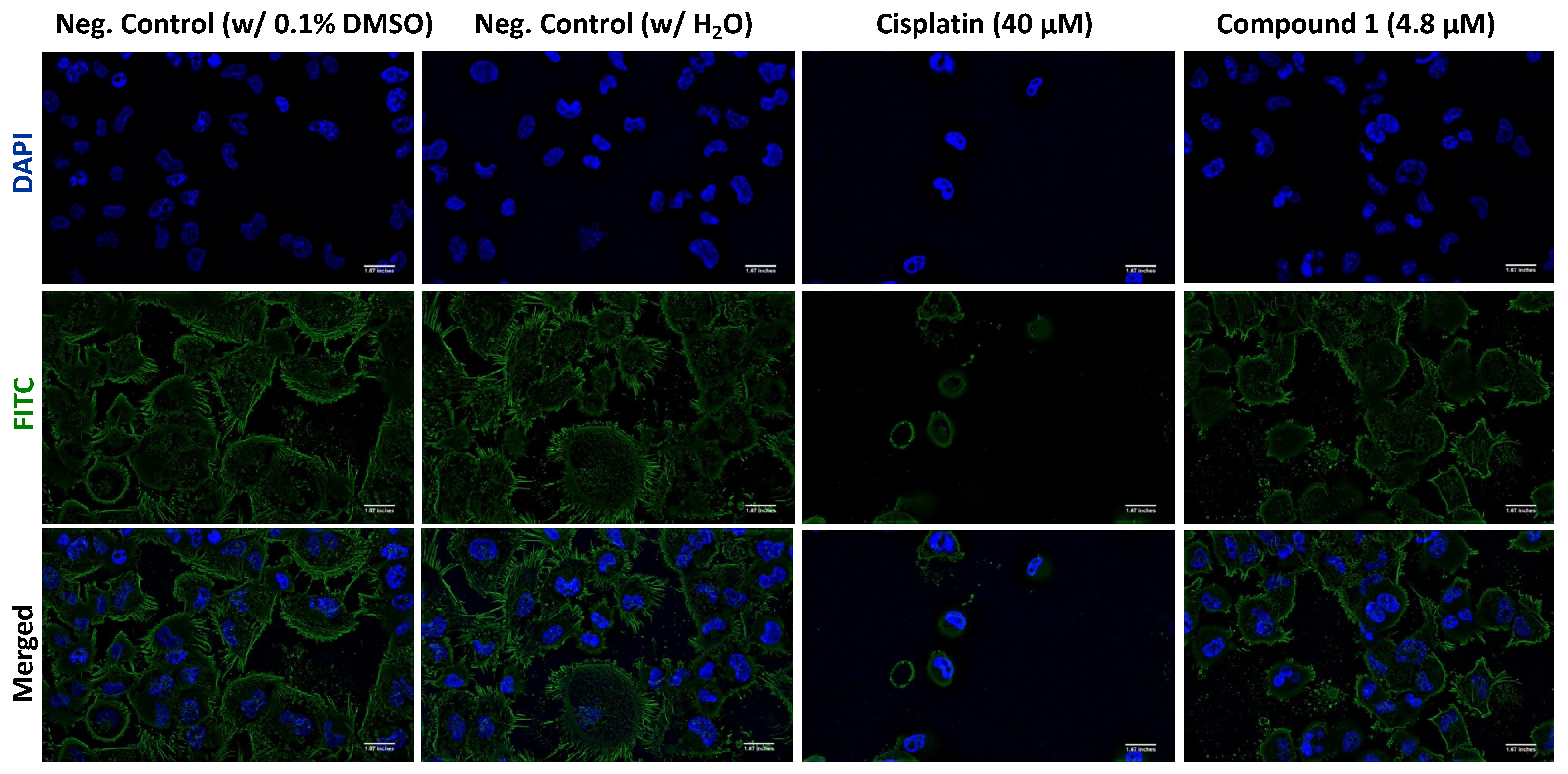
2.2.6. F-Actin Immunofluorescence Assay
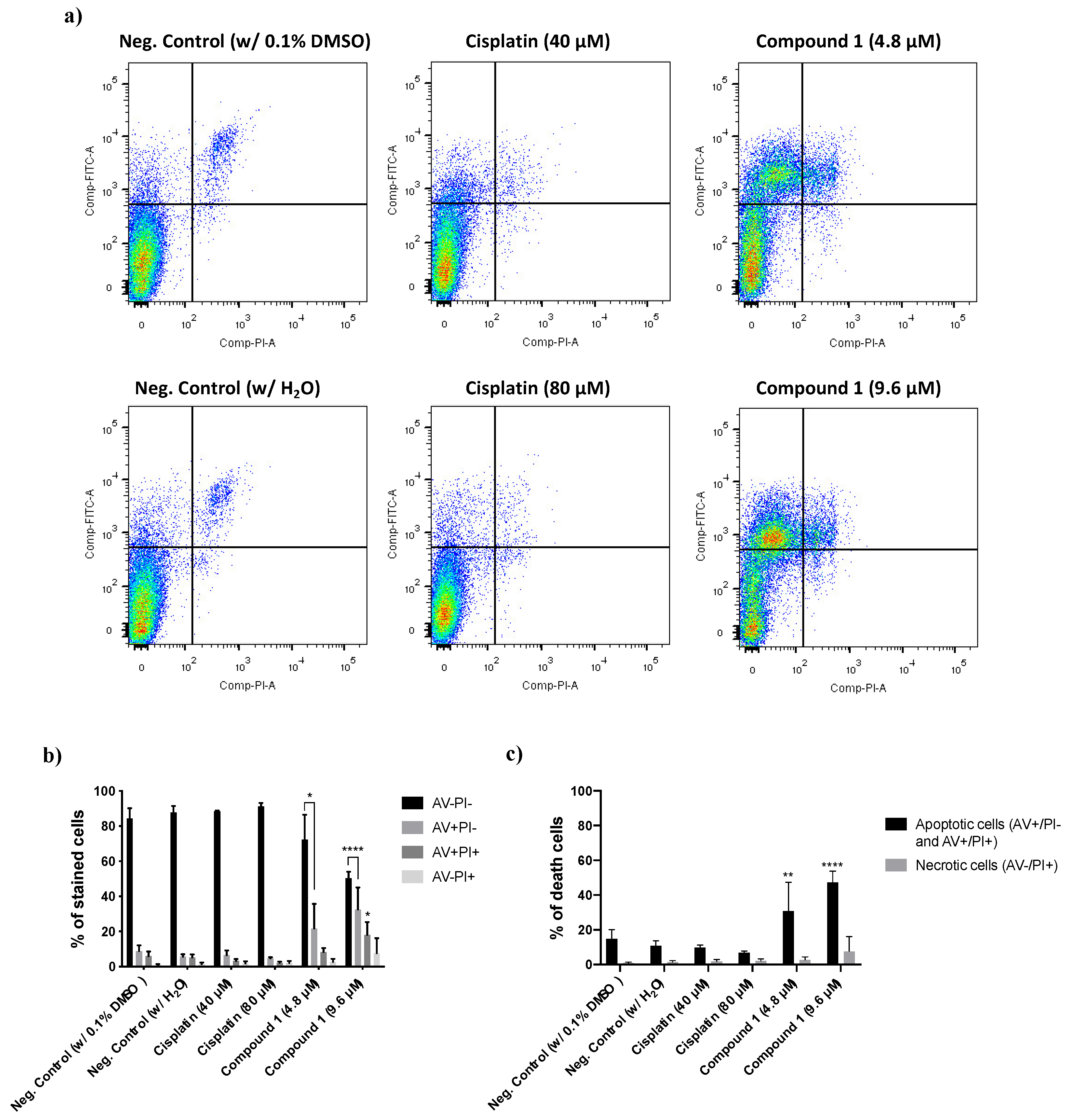
2.2.7. Evaluation of the Cell Death Mechanism by Flow Cytometry
2.2.8. Statistical Analysis for In Vitro Studies
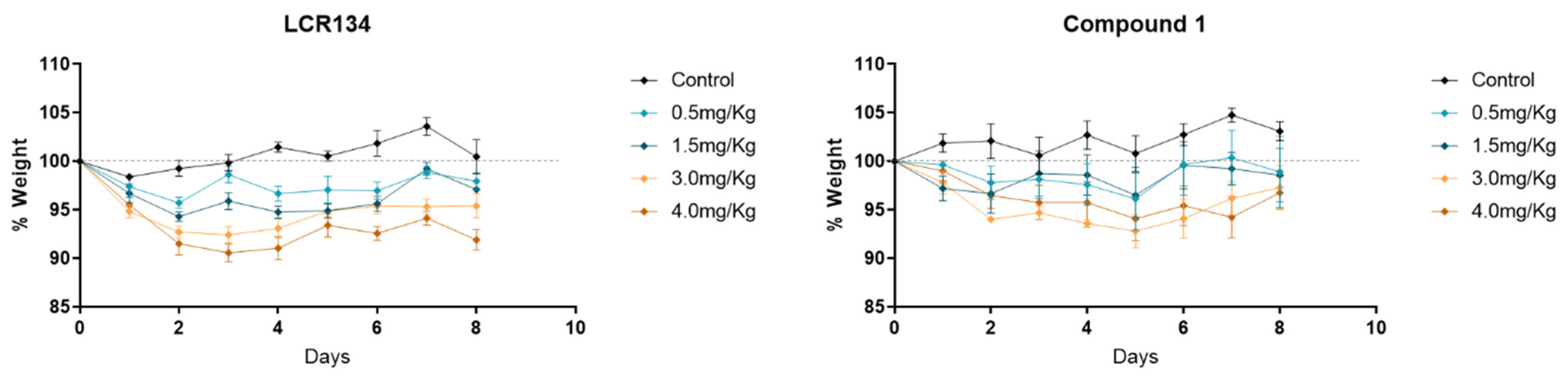
2.3. In Vivo Studies
Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of Ruthenium Compounds
3.2. Biological Evaluation of the Compounds
3.2.1. Analysis of the Anti-Cancer Effect in MDA-MB-231 Breast Cancer Cells
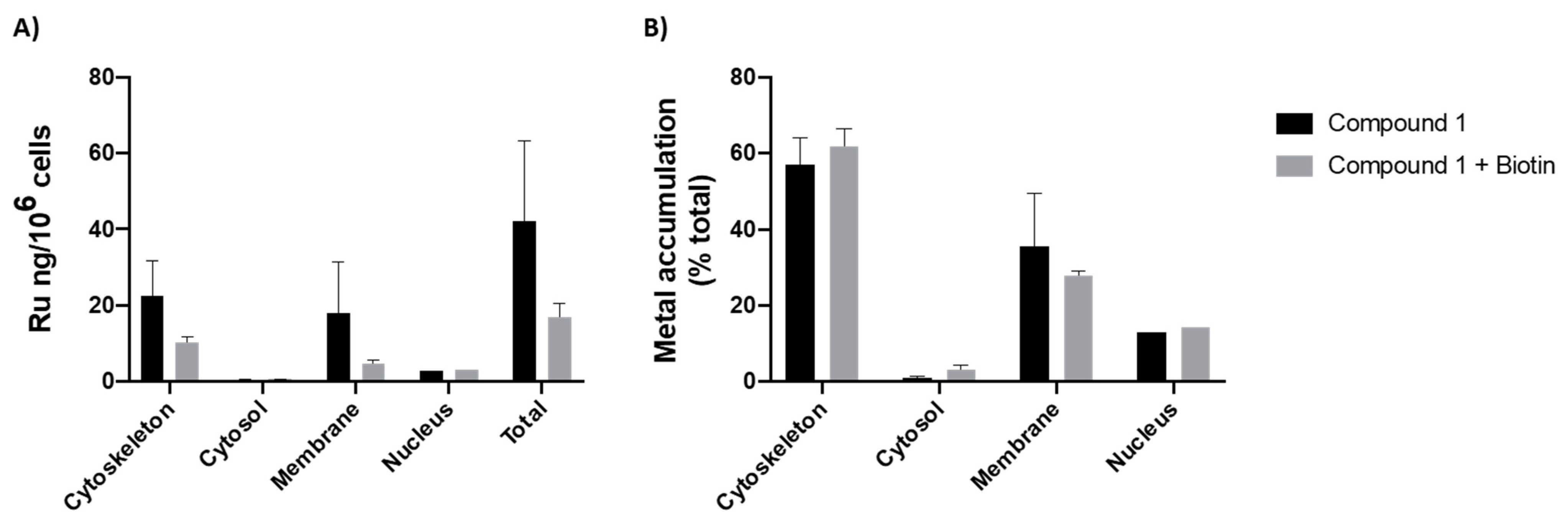
3.2.2. Intracellular Distribution
3.2.3. Determination of the Type of Cell Death Induced by Compound 1
3.2.4. Effect of Compound 1 in the Clonogenic Potential of MDA-MB-231 Breast Cancer Cells
3.2.5. Evaluation of the Effect of Compound 1 on the Actin Cytoskeleton
3.3. In Vivo Studies
3.3.1. Compounds’ Preparation
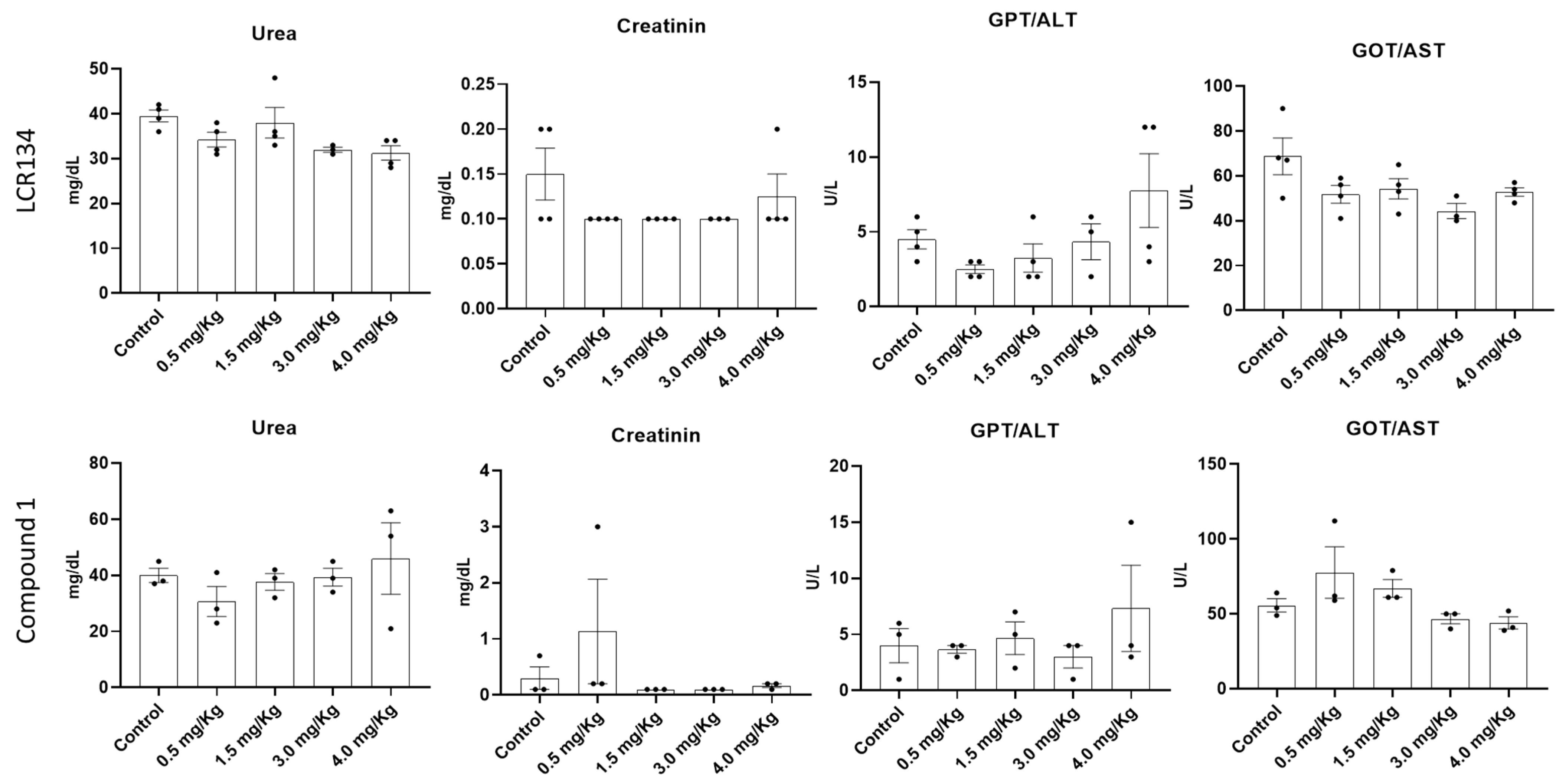
3.3.2. Toxicology Assay
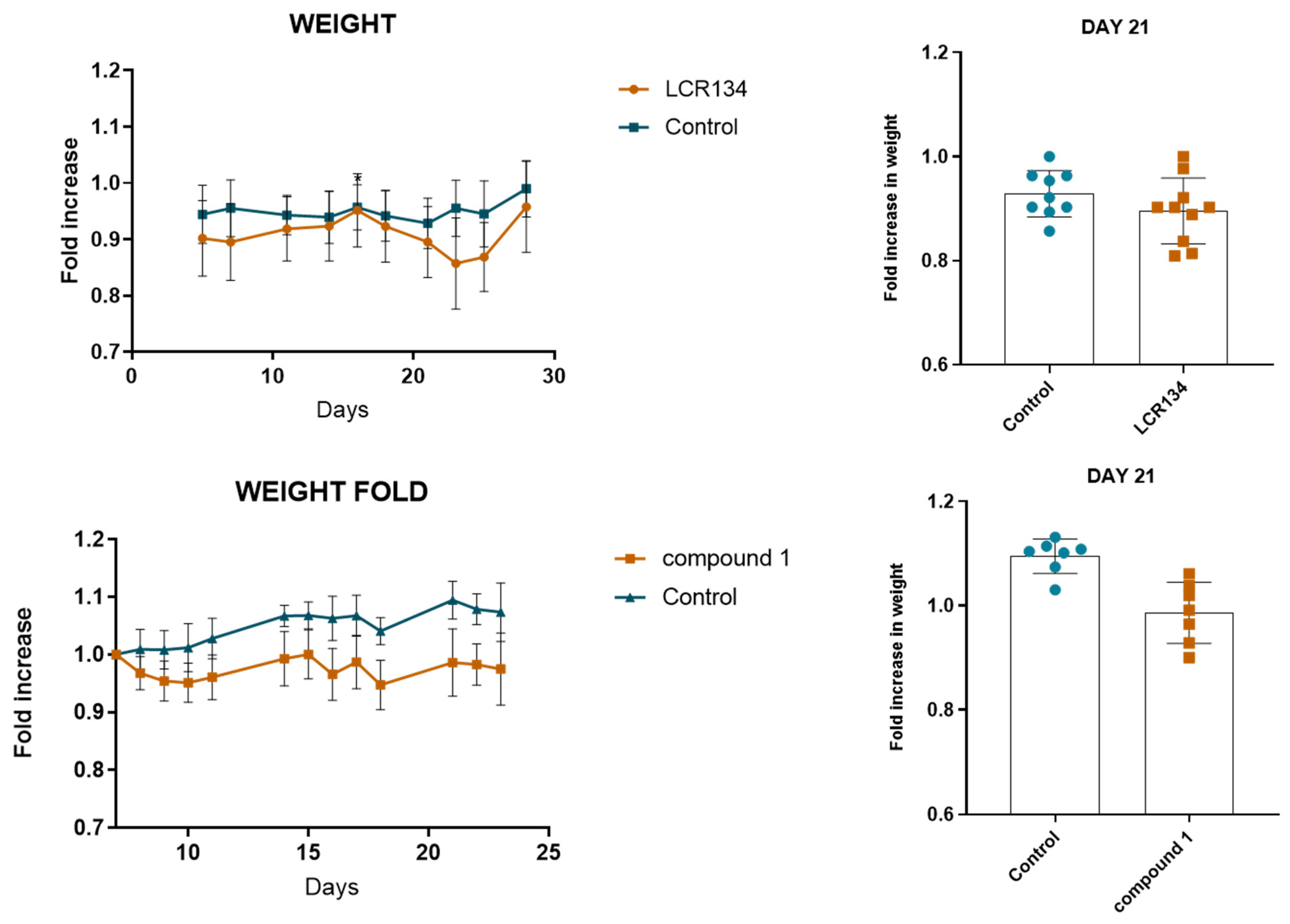
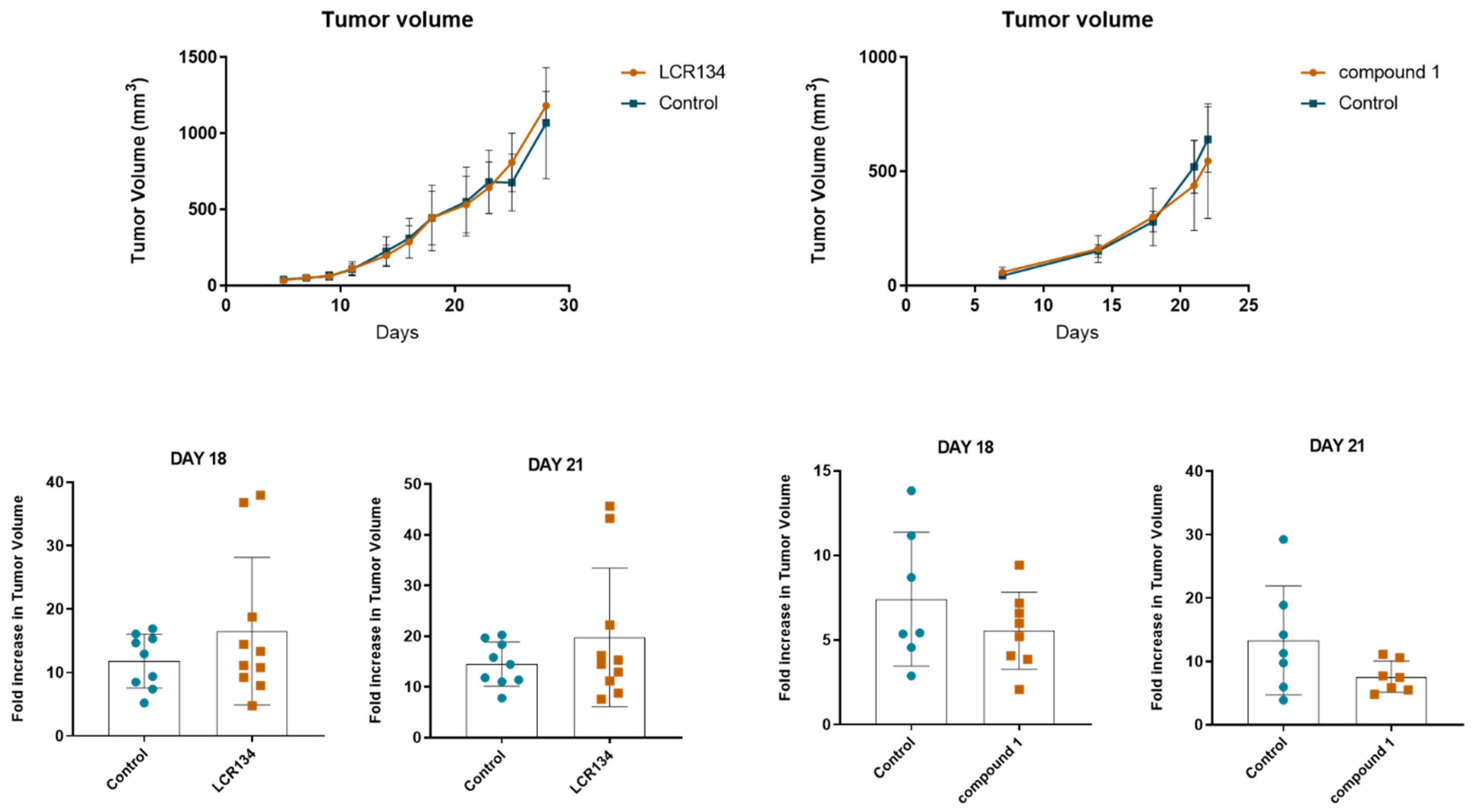
3.3.3. Tumour Growth
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hafeez, M.N.; Celia, C.; Petrikaite, V. Challenges towards targeted drug delivery in cancer nanomedicines. Processes 2021, 9, 1527. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yang, L.; Chen, G.; Xu, F.; Yang, F.; Yu, H.; Li, L.; Dong, X.; Han, J.; Cao, C.; et al. A Review on drug delivery system for tumor therapy. Front. Pharmacol. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Elvira, C.; Gallardo, A.; Roman, J.S.; Cifuentes, A. Covalent polymer-drug conjugates. Molecules 2005, 10, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Jing, X. Biodegradable amphiphilic polymer–drug conjugate micelles. Expert Opin. Drug Deliv. 2009, 6, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Seifu, M.F.; Nath, L.K. Polymer-drug conjugates: Novel carriers for cancer chemotherapy. Polym. Technol. Mater. 2019, 58, 158–171. [Google Scholar] [CrossRef]
- Girase, M.L.; Patil, P.G.; Ige, P.P. Polymer-drug conjugates as nanomedicine: A review. Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 990–1014. [Google Scholar] [CrossRef]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Kedar, U.; Phutane, P.; Shidhaye, S.; Kadam, V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew. Chem.-Int. Ed. 2014, 53, 12320–12364. [Google Scholar] [CrossRef] [PubMed]
- Blunden, B.M.; Rawal, A.; Lu, H.; Stenzel, M.H. Superior chemotherapeutic benefits from the ruthenium-based anti-metastatic drug NAMI-A through conjugation to polymeric micelles. Macromolecules 2014, 47, 1646–1655. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Carie, A.; Costich, T.; Burke, B.; Skaff, H.; Panicucci, R.; Sill, K.J. A Versatile Polymer Micelle Drug Delivery System for Encapsulation and InVivo Stabilization of Hydrophobic Anticancer Drugs. J. Drug Deliv. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hussein, Y.H.A.; Youssry, M. Polymeric micelles of biodegradable diblock copolymers: Enhanced encapsulation of hydrophobic drugs. Materials 2018, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Majumder, N.; Das, N.G.; Das, S.K. Polymeric micelles for anticancer drug delivery. Ther. Deliv. 2020, 11, 613–635. [Google Scholar] [CrossRef]
- Cabral, H.; Kataoka, K. Progress of drug-loaded polymeric micelles into clinical studies. J. Control. Release 2014, 190, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Sadhukha, T.; Prabha, S. Encapsulation in nanoparticles improves anti-cancer efficacy of carboplatin. AAPS Pharmscitech 2014, 15, 1029–1038. [Google Scholar] [CrossRef]
- McNamara, K.; Tofail, S.A.M. Nanoparticles in biomedical applications. Adv. Phys. X 2017, 2, 54–88. [Google Scholar] [CrossRef]
- Gavas, S.; Quazi, S.; Karpiński, T.M. Nanoparticles for Cancer Therapy: Current Progress and Challenges. Nanoscale Res. Lett. 2021, 16, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Gao, L.; Chen, K.; Zhang, W.; Zhang, Q.; Li, Q.; Hu, K. Nanoparticles: A new approach to upgrade cancer diagnosis and treatment. Nanoscale Res. Lett. 2021, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Côrte-Real, L.; Karas, B.; Brás, A.R.; Pilon, A.; Avecilla, F.; Marques, F.; Preto, A.; Buckley, B.T.; Cooper, K.R.; Doherty, C.; et al. Ruthenium–Cyclopentadienyl Bipyridine–Biotin Based Compounds: Synthesis and Biological Effect. Inorg. Chem. 2019, 58, 9135–9149. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.W.; Kwon, S.H.; Choi, J.H.; Lee, A. A promising biocompatible platform: Lipid-based and bio-inspired smart drug delivery systems for cancer therapy. Int. J. Mol. Sci. 2018, 19, 3859. [Google Scholar] [CrossRef] [Green Version]
- Qian, Q.; Zhu, L.; Zhu, X.; Sun, M.; Yan, D. Drug-polymer hybrid macromolecular engineering: Degradable PEG integrated by platinum (IV) for cancer therapy. Matter 2019, 1, 1618–1630. [Google Scholar] [CrossRef] [Green Version]
- Karim, K.J.A.; Utama, R.H.; Lua, H.; Stenzel, M.H. Enhanced drug toxicity by conjugation of platinum drugs to polymers with guanidine containing zwitterionic functional groups that mimic cell-penetrating peptides. Polym. Chem. 2014, 5, 6600–6610. [Google Scholar] [CrossRef]
- Aderibigbe, B.A.; Mugogodi, A.; Nwamadi, M.; Ray, S.S.; Steenkamp, V.; Balogun, M.O.; Matshe, W.M.R. Polyamidoamine-drug conjugates containing metal-based anticancer compounds. J. Inorg. Organomet. Polym. Mater. 2020, 30, 1503–1518. [Google Scholar] [CrossRef]
- Uivarosi, V.; Olar, R.; Badea, M. Nanoformulation as a tool for improve the pharmacological profile of platinum and ruthenium anticancer drugs. In Descriptive Inorganic Chemistry Researches of Metal Compounds; IntechOpen: London, UK, 2017; Chapter 1; pp. 1–26. [Google Scholar]
- Alven, S.; Nqoro, X.; Buyana, B.; Aderibigbe, B.A. Polymer-drug conjugate, a potential therapeutic to combat breast and lung cancer. Pharmaceutics 2020, 12, 406. [Google Scholar] [CrossRef] [PubMed]
- Blunden, B.M.; Lu, H.; Stenzel, M.H. Enhanced delivery of the RAPTA-C macromolecular chemotherapeutic by conjugation to degradable polymeric micelles. Biomacromolecules 2013, 14, 4177–4188. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Chen, F.; Noy, J.M.; Lu, H.; Stenzel, M.H. Enhanced Antimetastatic Activity of the Ruthenium Anticancer Drug RAPTA-C Delivered in Fructose-Coated Micelles. Macromol. Biosci. 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Li, S.; Häupler, B.; Liu, J.; Jin, S.; Steffen, W.; Schubert, U.S.; Butt, H.J.; Liang, X.J.; Wu, S. An Amphiphilic Ruthenium Polymetallodrug for Combined Photodynamic Therapy and Photochemotherapy In Vivo. Adv. Mater. 2017, 29, 1603702. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Huang, H.; Zhao, Z.; Gunatillake, P.A.; Hao, X. Brush-shaped RAFT polymer micelles as nanocarriers for a ruthenium (II) complex photodynamic anticancer drug. Eur. Polym. J. 2019, 113, 267–275. [Google Scholar] [CrossRef]
- Soliman, N.; McKenzie, L.K.; Karges, J.; Bertrand, E.; Tharaud, M.; Jakubaszek, M.; Guérineau, V.; Goud, B.; Hollenstein, M.; Gasser, G.; et al. Ruthenium-initiated polymerization of lactide: A route to remarkable cellular uptake for photodynamic therapy of cancer. Chem. Sci. 2020, 11, 2657–2663. [Google Scholar] [CrossRef] [Green Version]
- Karges, J.; Li, J.; Zeng, L.; Chao, H.; Gasser, G. Polymeric encapsulation of a ruthenium polypyridine complex for tumor targeted one-and two-photon photodynamic therapy. ACS Appl. Mater. Interfaces 2020, 12, 54433–54444. [Google Scholar] [CrossRef]
- Moreira, T.; Francisco, R.; Comsa, E.; Duban-Deweer, S.; Labas, V.; Teixeira-Gomes, A.-P.; Combes-Soia, L.; Marques, F.; Matos, A.; Favrelle, A.; et al. Polymer “ruthenium-cyclopentadienyl” conjugates-New emerging anti-cancer drugs. Eur. J. Med. Chem. 2019, 168, 373–384. [Google Scholar] [CrossRef]
- Valente, A.; Helena, M.; Marques, F.; Miao, Y.; Rousseau, C.; Zinck, P. First polymer “ruthenium-cyclopentadienyl” complex as potential anticancer agent. J. Inorg. Biochem. 2013, 127, 79–81. [Google Scholar] [CrossRef]
- Moreno, V.; Font-Bardia, M.; Calvet, T.; Lorenzo, J.; Avilés, F.X.; Garcia, M.H.; Morais, T.S.; Valente, A.; Robalo, M.P. DNA interaction and cytotoxicity studies of new ruthenium (II) cyclopentadienyl derivative complexes containing heteroaromatic ligands. J. Inorg. Biochem. 2011, 105, 241–249. [Google Scholar] [CrossRef]
- Isabel, A.; Jakusch, T.; Morais, T.S.; Marques, F.; de Almeida, R.F.M.; Mendes, F.; Enyedy, É.A.; Santos, I.; Costa, J.; Kiss, T.; et al. [RuII (η5-C5H5)(bipy)(PPh3)]+, a promising large spectrum antitumor agent: Cytotoxic activity and interaction with human serum albumin. J. Inorg. Biochem. 2012, 117, 261–269. [Google Scholar]
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Côrte-Real, L.; Karas, B.; Gírio, P.; Moreno, A.; Avecilla, F.; Marques, F.; Buckley, B.T.; Cooper, K.R.; Doherty, C.; Falson, P.; et al. Unprecedented inhibition of P-gp activity by a novel ruthenium-cyclopentadienyl compound bearing a bipyridine-biotin ligand. Eur. J. Med. Chem. 2019, 163, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Karas, B.F.; Hotz, J.M.; Gural, B.M.; Terez, K.R.; DiBona, V.L.; Côrte-Real, L.; Valente, A.; Buckley, B.T.; Cooper, K.R. Anti-cancer activity and in vitro to in vivo mechanistic recapitulation of novel ruthenium-based metallodrugs in the zebrafish model. Toxicol. Sci. 2021, 182, 29–43. [Google Scholar] [CrossRef]
- Bruce, M.I.; Windsor, N.J. Cyclopentadienyl-ruthenium and-osmium chemistry. IV. Convenient high-yield synthesis of some cyclopentadienyl ruthenium or osmium tertiary phosphine halide complexes. Aust. J. Chem. 1977, 30, 1601–1604. [Google Scholar] [CrossRef]
- Teixeira, R.G.; Brás, A.R.; Côrte-Real, L.; Tatikonda, R.; Sanches, A.; Robalo, M.P.; Avecilla, F.; Moreira, T.; Garcia, M.H.; Haukka, M.; et al. Novel ruthenium methylcyclopentadienyl complex bearing a bipyridine perfluorinated ligand shows strong activity towards colorectal cancer cells. Eur. J. Med. Chem. 2018, 143, 503–514. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer molecular weight analysis by 1H NMR spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Côrte-Real, L.; Matos, A.P.; Alho, I.; Morais, T.S.; Tomaz, A.I.; Garcia, M.H.; Santos, I.; Bicho, M.P.; Marques, F. Cellular uptake mechanisms of an antitumor ruthenium compound: The endosomal/lysosomal system as a target for anticancer metal-based drugs. Microsc. Microanal. 2013, 19, 1122–1130. [Google Scholar] [CrossRef]
- Meier, M.A.R.; Lohmeijer, B.G.G.; Schubert, U.S. Characterization of defined metal-containing supramolecular block copolymers. Macromol. Rapid Commun. 2003, 24, 852–857. [Google Scholar] [CrossRef]
- Côrte-Real, L.; Teixeira, R.G.; Gírio, P.; Comsa, E.; Moreno, A.; Nasr, R.; Baubichon-Cortay, H.; Avecilla, F.; Marques, F.; Robalo, M.P.; et al. Methyl-cyclopentadienyl ruthenium compounds with 2, 2′-bipyridine derivatives display strong anticancer activity and multidrug resistance potential. Inorg. Chem. 2018, 57, 4629–4639. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.X.; Han, J.; Uhm, S.; Jang, Y.J.; Kang, C.; Kim, J.H.; Kim, J.S. Recent development of biotin conjugation in biological imaging, sensing, and target delivery. Chem. Commun. 2015, 51, 10403–10418. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield (%) a | DPb | Mn,NMRc (g/mol) | |
|---|---|---|---|
| Bipy-PLA-OH (PLA1) | 95 | 58 | 4392.2 |
| Bipy-PLA-OH (PLA2) | 88 | 66 | 4968.2 |
| Bipy-PLA-OH (PLA3) | 86 | 62 | 4680.2 |
| DP a | |
|---|---|
| L1_PLA1 | 68 |
| L1_PLA2 | 60 |
| L1_PLA3 | 58 |
| DP a | Mn (1H-NMR) b | |
|---|---|---|
| Complex 1 | 70 | 6233.9 g/mol |
| Complex 2 | 64 | 5941.6 g/mol |
| Complex 3 | 42 | 4319.4 g/mol |
| Complex 4 | 56 | 5296.0 g/mol |
| Compound | MDA-MB-231 (μM) |
|---|---|
| [Ru(η5-Cp)(P(C6H5)3)(bipy-biotin)]+ (LCR134) | 3.2 ± 1.1 [23] |
| [Ru(η5-Cp)(P(C6H5)3)(bipy-PLA-biotin)]+ (1) | 2.3 ± 0.1 |
| [Ru(η5-Cp)(P(C6H4F)3)(bipy-biotin)]+ (LCR205) | 7.1 ± 0.6 [23] |
| [Ru(η5-Cp)(P(C6H4F)3)(bipy-PLA-biotin)]+ (2) | 14.6 ± 0.4 |
| [Ru(η5-Cp)(P(C6H4OCH3)3)(bipy-biotin)]+ (LCR234) | 4.0 ± 0.2 [23] |
| [Ru(η5-Cp)(P(C6H4OCH3)3)(bipy-PLA-biotin)]+ (3) | 6.5 ± 0.3 |
| [Ru(η5-MeCp)(P(C6H5)3)(bipy-PLA-biotin)]+ (4) | 3.4 ± 0.1 |
| CDDP | 40 ± 3.7 |
| bipy-PLA-biotin (L1) | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Côrte-Real, L.; Brás, A.R.; Pilon, A.; Mendes, N.; Ribeiro, A.S.; Martins, T.D.; Farinha, J.P.S.; Oliveira, M.C.; Gärtner, F.; Garcia, M.H.; et al. Biotinylated Polymer-Ruthenium Conjugates: In Vitro and In Vivo Studies in a Triple-Negative Breast Cancer Model. Pharmaceutics 2022, 14, 1388. https://doi.org/10.3390/pharmaceutics14071388
Côrte-Real L, Brás AR, Pilon A, Mendes N, Ribeiro AS, Martins TD, Farinha JPS, Oliveira MC, Gärtner F, Garcia MH, et al. Biotinylated Polymer-Ruthenium Conjugates: In Vitro and In Vivo Studies in a Triple-Negative Breast Cancer Model. Pharmaceutics. 2022; 14(7):1388. https://doi.org/10.3390/pharmaceutics14071388
Chicago/Turabian StyleCôrte-Real, Leonor, Ana Rita Brás, Adhan Pilon, Nuno Mendes, Ana Sofia Ribeiro, Tiago D. Martins, José Paulo S. Farinha, M. Conceição Oliveira, Fátima Gärtner, M. Helena Garcia, and et al. 2022. "Biotinylated Polymer-Ruthenium Conjugates: In Vitro and In Vivo Studies in a Triple-Negative Breast Cancer Model" Pharmaceutics 14, no. 7: 1388. https://doi.org/10.3390/pharmaceutics14071388