
Development of Irinotecan Liposome Armed with Dual-Target Anti-Epidermal Growth Factor Receptor and Anti-Fibroblast Activation Protein-Specific Antibody for Pancreatic Cancer Treatment

Abstract
:1. Introduction
2. Materials and Methods
2.1. Production of Dual Target Anti-EGFR/Anti-FAP Specific Antibody
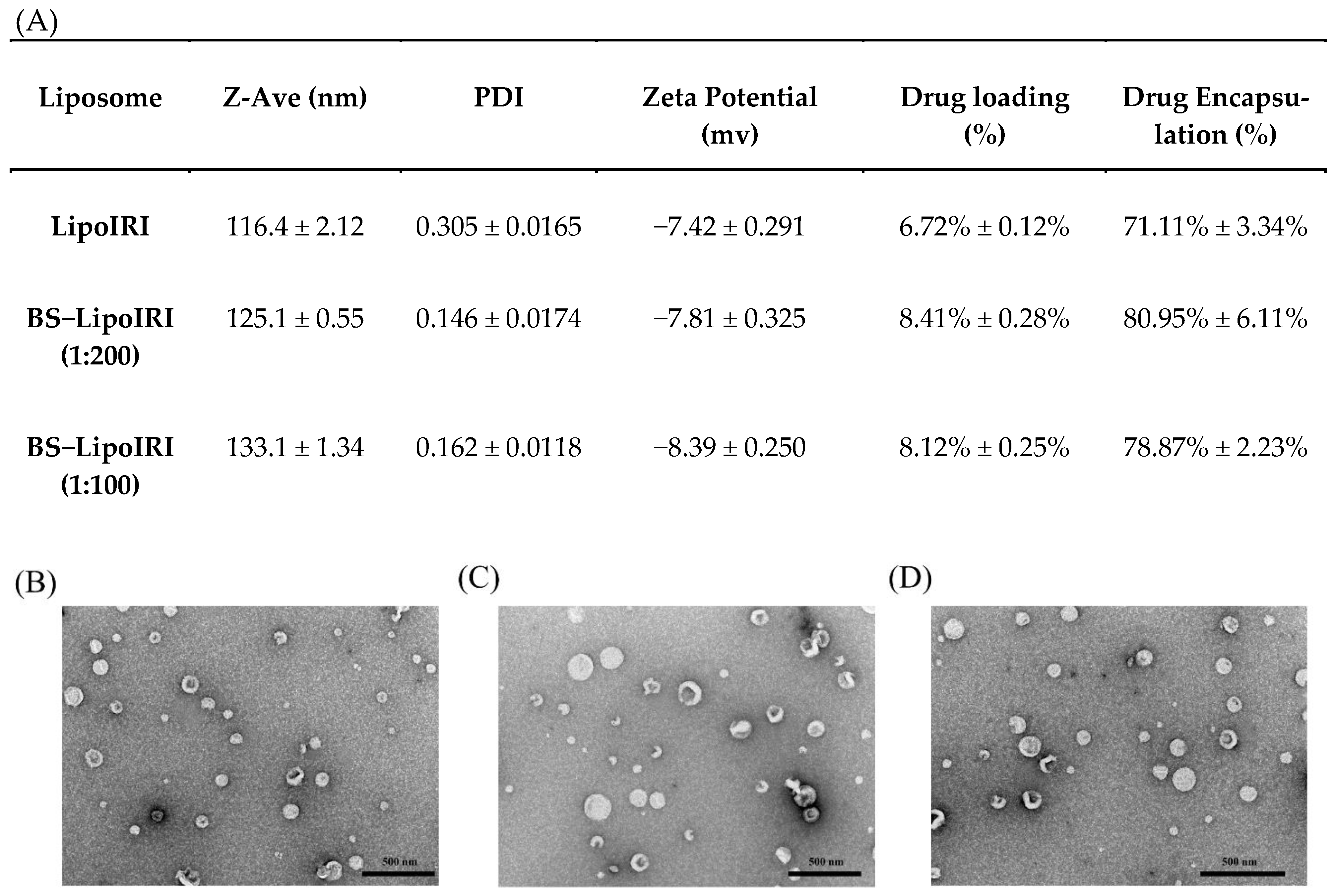
2.2. Characterization of Irinotecan Liposomes
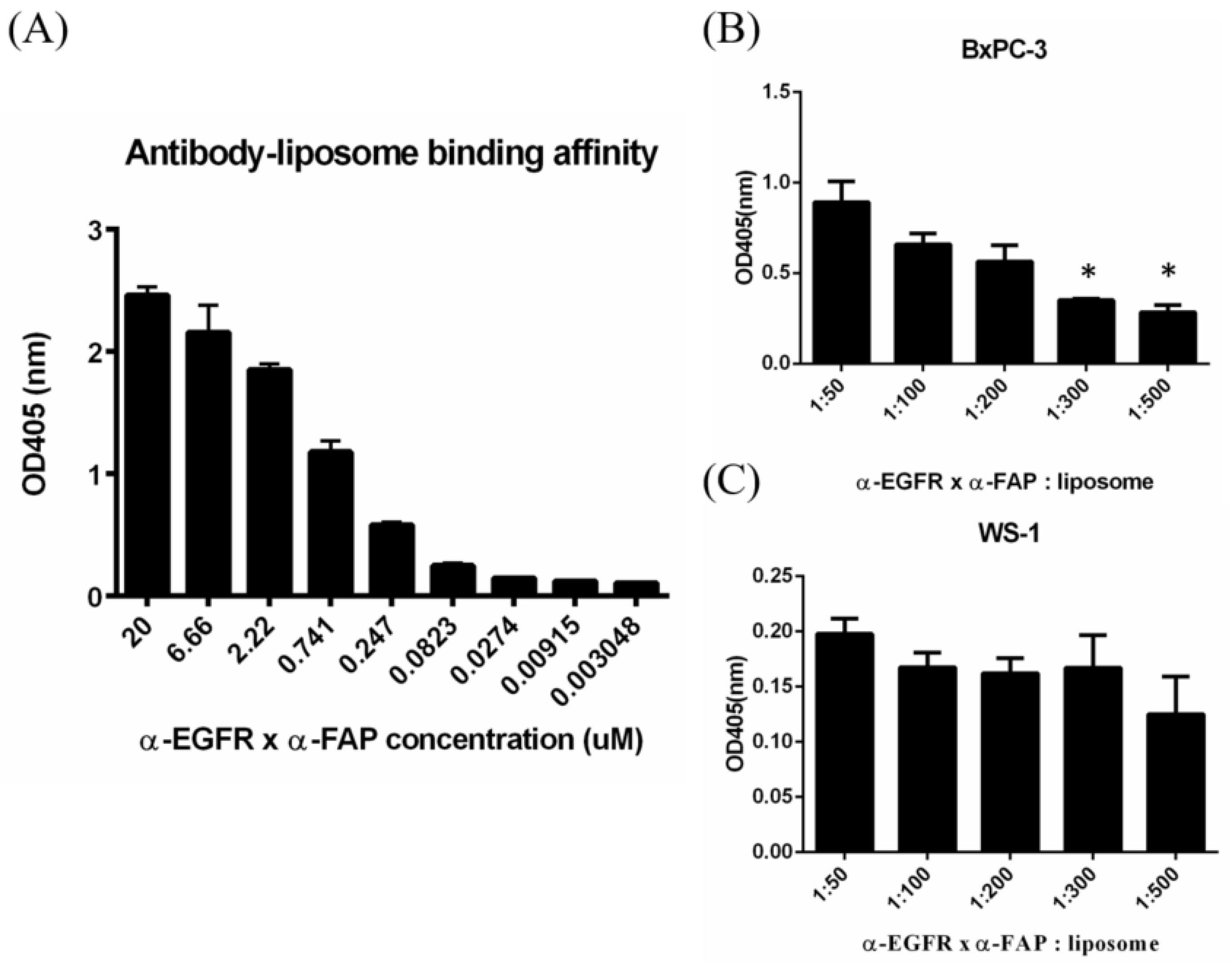
2.3. In Vitro Targeting Ability of Specific Antibody Liposomes
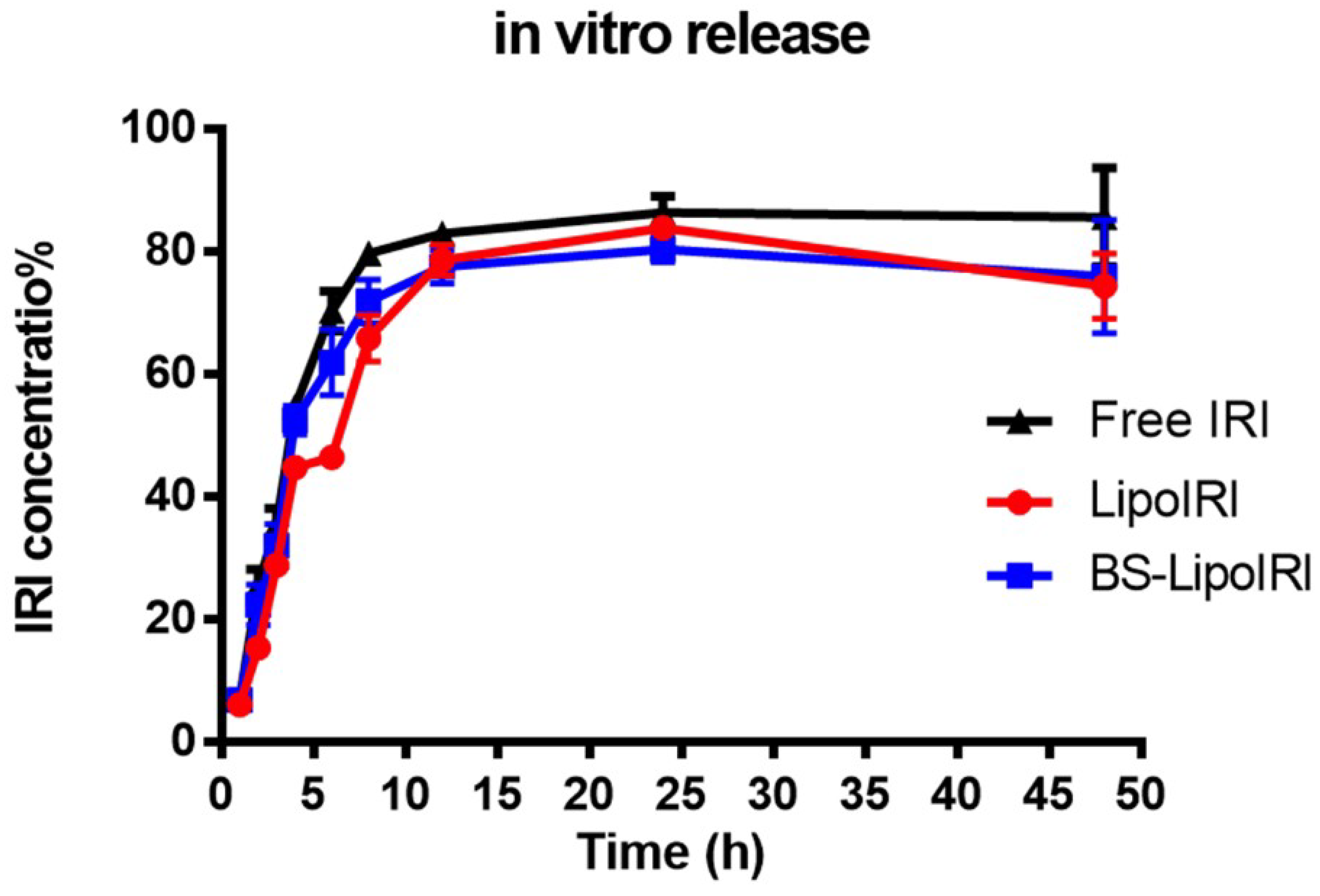
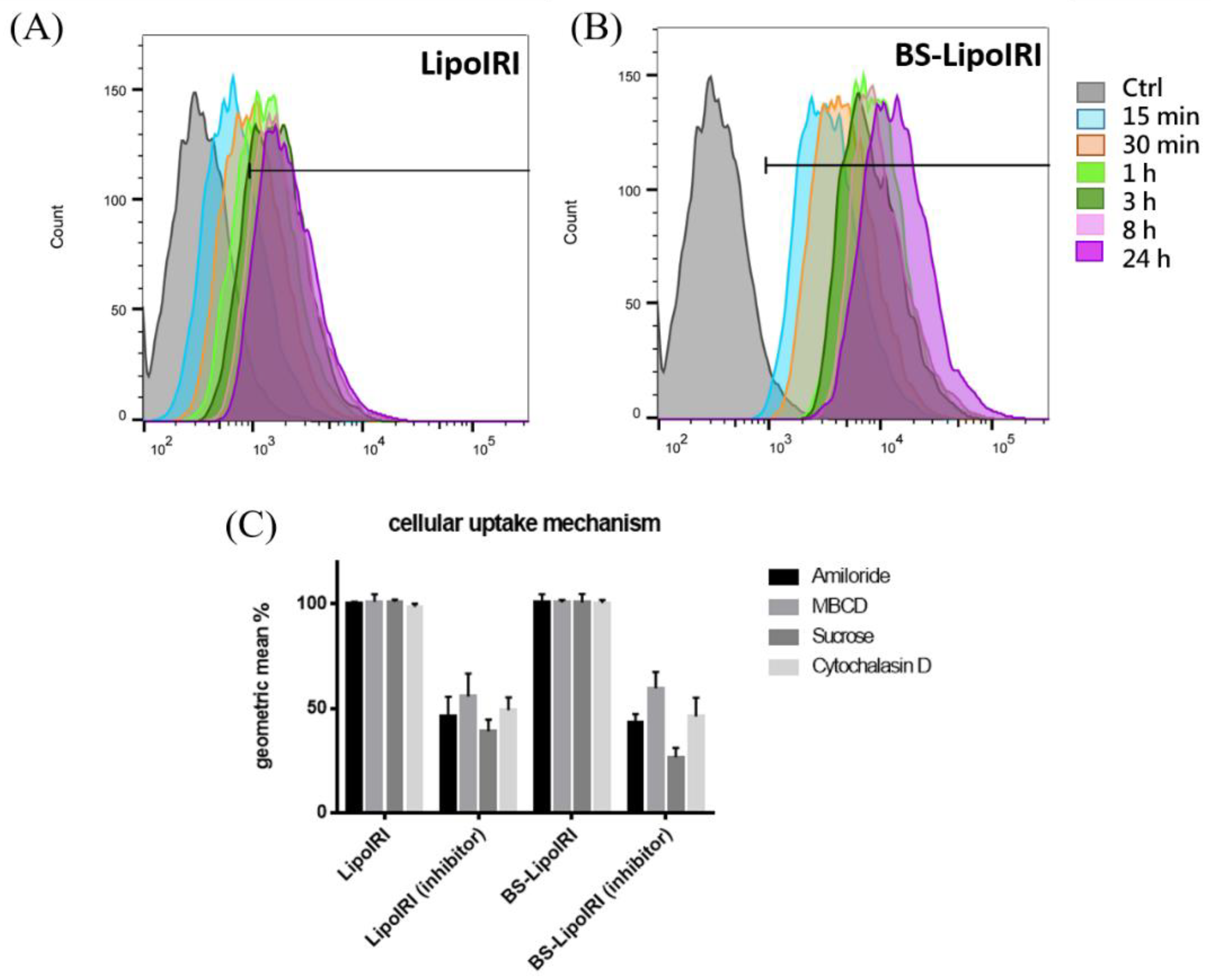
2.4. Drug Release and Cellular Uptake Studies
2.5. In Vitro Pancreatic Tumor Cell Viability
2.6. In Vivo Drug Pharmacokinetic and Distribution Studies
2.7. Administration of BS–LipoIRI against Human Pancreatic Tumor
2.8. Statistical Analysis
3. Results
3.1. Characterization and Stability of LipoIRI
3.2. In Vitro Dual Targeting of BS−LipoIRI
3.3. In Vitro Drug Release Studies
3.4. Cellular Uptake and Mechanism of the BS−LipoIRI
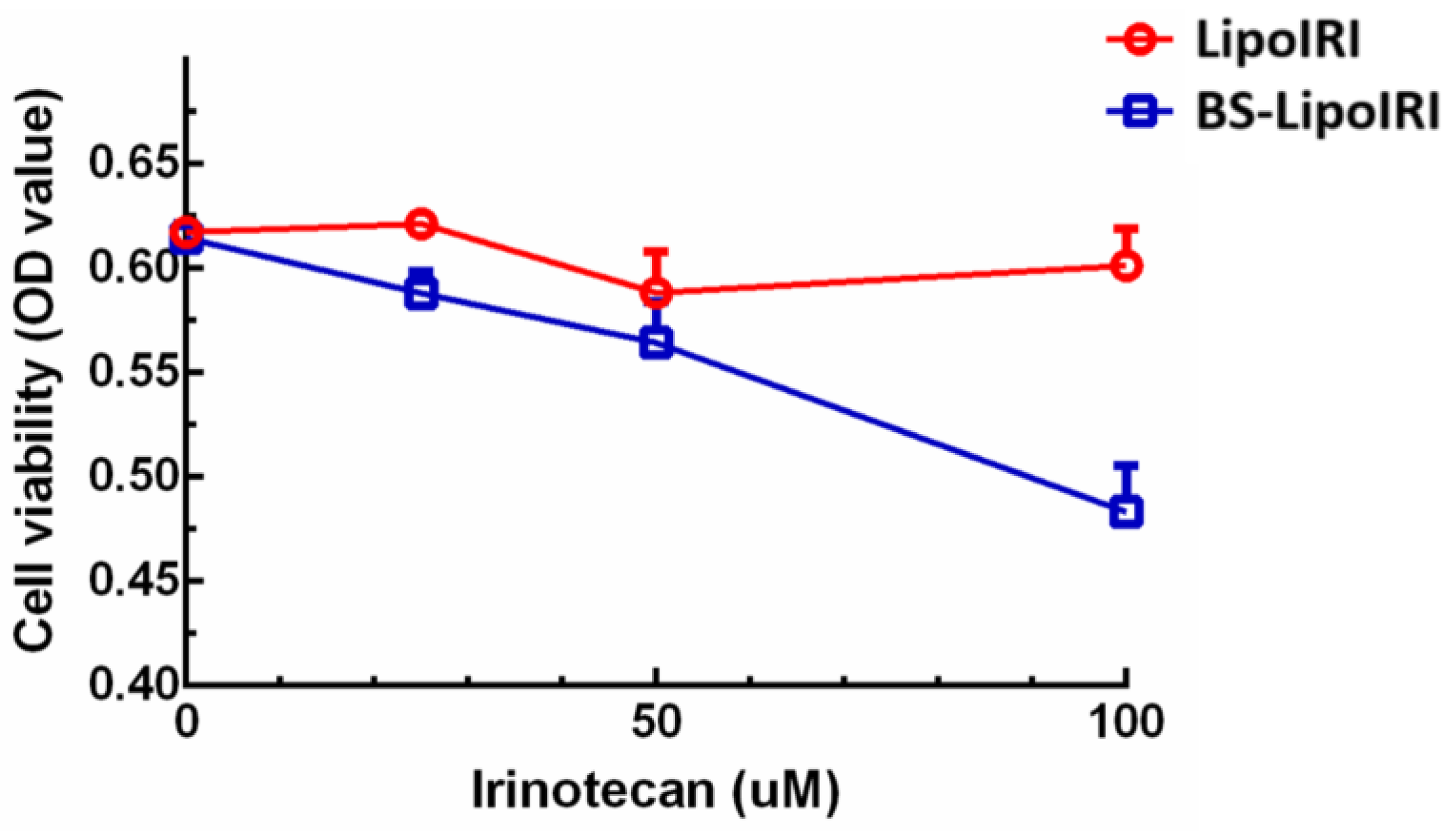
3.5. In Vitro Bxpc3 Cell Viability
3.6. PK Studies and Biodistribution Assessment of the BS−LipoIRI
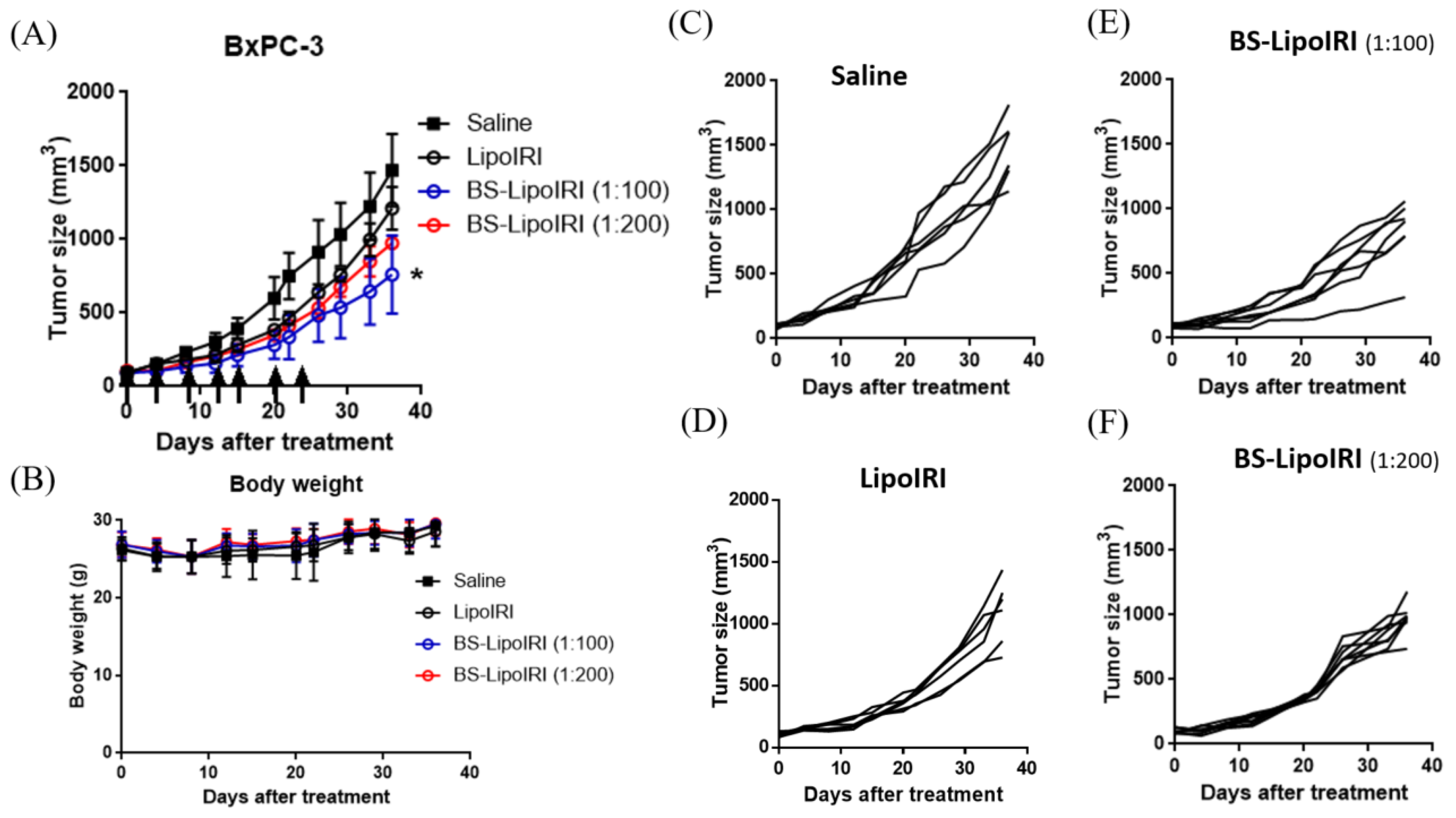
3.7. In Vivo Antitumor Efficacy in Tumor-Bearing Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Timmons, L.J.; Ransom, J.; de Andrade, M.; Petersen, G.M. Pancreatic cancer-associated diabetes mellitus: Prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008, 134, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popescu, I.; Dumitrascu, T. Ppancreatoduodenectomy—past, present and future. Chirurgia 2011, 106, 287–296. [Google Scholar]
- Philip, P.A.; Lutz, M.P. Targeting epidermal growth factor receptor-related signaling pathways in pancreatic cancer. Pancreas 2015, 44, 1046–1052. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase iii trial of the national cancer institute of canada clinical trials group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Moore, M.J.; Au, H.J.; Parulekar, W. Does a statistically significant survival benefit of erlotinib plus gemcitabine for advanced pancreatic cancer translate into clinical significance and value?—Reply. J. Clin. Oncol. 2007, 25, 4508. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. Fap promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via stat3-ccl2 signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharm. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Calvet, L.; Santos, A.; Valent, A.; Terrier-Lacombe, M.J.; Opolon, P.; Merlin, J.L.; Aubert, G.; Morizet, J.; Schellens, J.H.; Benard, J.; et al. No topoisomerase i alteration in a neuroblastoma model with in vivo acquired resistance to irinotecan. Br. J. Cancer 2004, 91, 1205–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yueh, M.F.; Bigo, C.; Barbier, O.; Wang, K.; Karin, M.; Nguyen, N.; Tukey, R.H. Intestinal glucuronidation protects against chemotherapy-induced toxicity by irinotecan (cpt-11). Proc. Natl. Acad. Sci. USA 2013, 110, 19143–19148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miya, T.; Fujikawa, R.; Fukushima, J.; Nogami, H.; Koshiishi, Y.; Goya, T. Bradycardia induced by irinotecan: A case report. Jpn. J. Clin. Oncol. 1998, 28, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Bisso, S.; Leroux, J.C. Nanopharmaceuticals: A focus on their clinical translatability. Int. J. Pharm. 2020, 578, 119098. [Google Scholar] [CrossRef]
- Mullard, A. Bispecific antibody pipeline moves beyond oncology. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [Green Version]
- Naito, S.; von Eschenbach, A.C.; Giavazzi, R.; Fidler, I.J. Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 1986, 46, 4109–4115. [Google Scholar]
- Kuhn, D.A.; Vanhecke, D.; Michen, B.; Blank, F.; Gehr, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 2014, 5, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cell. Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Watanabe, A.; Funami, K.; Seya, T.; Matsumoto, M. The clathrin-mediated endocytic pathway participates in dsrna-induced ifn-beta production. J. Immunol. 2008, 181, 5522–5529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barenholz, Y.C. Doxil (r)—The first fda-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Zhang, H. Onivyde for the therapy of multiple solid tumors. Oncotargets Ther. 2016, 9, 3001–3007. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, U.S. Liposomes in drug delivery: Progress and limitations. Int. J. Pharm. 1997, 154, 123–140. [Google Scholar] [CrossRef]
- Çağdaş, M.; Sezer, A.D.; Bucak, S. Liposomes as potential drug carrier systems for drug delivery. Appl. Nanotechnol. Drug Deliv. 2014, 1, 1–50. [Google Scholar]
- Passero, F.C., Jr.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert Rev. Anticancer 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, X.; Wang, Z.; Zhang, H.; Chen, P.; Zhou, G.; Sun, C.; Gu, N.; Ji, M. Novel lipophilic sn38 prodrug forming stable liposomes for colorectal carcinoma therapy. Int. J. Nanomed. 2019, 14, 5201–5213. [Google Scholar] [CrossRef] [Green Version]
- Kalra, A.V.; Kim, J.; Klinz, S.G.; Paz, N.; Cain, J.; Drummond, D.C.; Nielsen, U.B.; Fitzgerald, J.B. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014, 74, 7003–7013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhuang, H.; Zhuang, Z.; Lu, Y.; Xia, R.; Gan, L.; Wu, Y. Development of docetaxel liposome surface modified with cd133 aptamers for lung cancer targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1864–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Soroush, F.; Tong, Z.; Kiani, M.F.; Wang, B. Targeted multidrug delivery system to overcome chemoresistance in breast cancer. Int. J. Nanomed. 2017, 12, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, G.H.; Alzghari, S.K.; Chee, W.; Sankari, S.S.; La-Beck, N.M. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J. Control. Release 2016, 232, 255–264. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Dan, N. Drug release through liposome pores. Colloids Surf. B Biointerfaces 2015, 126, 80–86. [Google Scholar] [CrossRef]
- Mathews, P.D.; Mertins, O.; Angelov, B.; Angelova, A. Cubosomal lipid nanoassemblies with ph-sensitive shells created by biopolymer complexes: A synchrotron saxs study. J. Colloid Interface Sci. 2022, 607, 440–450. [Google Scholar] [CrossRef]
- Mokhtar, S.; Khattab, S.N.; Elkhodairy, K.A.; Teleb, M.; Bekhit, A.A.; Elzoghby, A.O.; Sallam, M.A. Methotrexate-lactoferrin targeted exemestane cubosomes for synergistic breast cancer therapy. Front. Chem. 2022, 10, 847573. [Google Scholar] [CrossRef]
- Adjei, I.M.; Sharma, B.; Labhasetwar, V. Nanoparticles: Cellular uptake and cytotoxicity. Adv. Exp. Med. Biol. 2014, 811, 73–91. [Google Scholar]
- Xing, J.; Liu, D.; Zhou, G.; Li, Y.; Wang, P.; Hu, K.; Gu, N.; Ji, M. Liposomally formulated phospholipid-conjugated novel near-infrared fluorescence probe for particle size effect on cellular uptake and biodistribution in vivo. Colloids Surf. B Biointerfaces 2018, 161, 588–596. [Google Scholar] [CrossRef]
- Patel, S.; Kim, J.; Herrera, M.; Mukherjee, A.; Kabanov, A.V.; Sahay, G. Brief update on endocytosis of nanomedicines. Adv. Drug Deliv. Rev. 2019, 144, 90–111. [Google Scholar] [CrossRef] [PubMed]
- Abu Lila, A.S.; Ishida, T. Liposomal delivery systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Z-Ave (nm) | PDI | Zeta Potential (mv) | Drug Loading (%) | Drug Encapsulation (%) |
|---|---|---|---|---|---|
| 2 Weeks | 114.6 ± 0.16 | 0.257 ± 0.0255 | −7.21 ± 0.181 | 8.23% | 78.11% |
| 4 Weeks | 116.8 ± 0.31 | 0.246 ± 0.1284 | −8.11 ± 0.523 | 8.10% | 77.93% |
| 6 Weeks | 115.7 ± 0.18 | 0.592 ± 0.1119 | −7.33 ± 0.115 | 7.43% | 71.27% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-J.; Liang, T.-L.; Chang, Y.-Y.; Liu, D.-Z.; Fan, J.-Y.; Roffler, S.R.; Lin, S.-Y. Development of Irinotecan Liposome Armed with Dual-Target Anti-Epidermal Growth Factor Receptor and Anti-Fibroblast Activation Protein-Specific Antibody for Pancreatic Cancer Treatment. Pharmaceutics 2022, 14, 1202. https://doi.org/10.3390/pharmaceutics14061202
Lin H-J, Liang T-L, Chang Y-Y, Liu D-Z, Fan J-Y, Roffler SR, Lin S-Y. Development of Irinotecan Liposome Armed with Dual-Target Anti-Epidermal Growth Factor Receptor and Anti-Fibroblast Activation Protein-Specific Antibody for Pancreatic Cancer Treatment. Pharmaceutics. 2022; 14(6):1202. https://doi.org/10.3390/pharmaceutics14061202
Chicago/Turabian StyleLin, Hung-Jun, Tien-Li Liang, Yao-Yuan Chang, Der-Zen Liu, Jia-Yu Fan, Steve R. Roffler, and Shyr-Yi Lin. 2022. "Development of Irinotecan Liposome Armed with Dual-Target Anti-Epidermal Growth Factor Receptor and Anti-Fibroblast Activation Protein-Specific Antibody for Pancreatic Cancer Treatment" Pharmaceutics 14, no. 6: 1202. https://doi.org/10.3390/pharmaceutics14061202