
Liposome-Encapsulated Tobramycin and IDR-1018 Peptide Mediated Biofilm Disruption and Enhanced Antimicrobial Activity against Pseudomonas aeruginosa

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of the Antibacterial Minimum Inhibitory Concentration (MIC)
2.3. Liposomal Nanoparticle Preparation
2.4. Determination of Encapsulation Efficiency (EE%) Using Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometer (UHPLC-MS/MS)
2.5. Particle Size and Zeta Potential Measurement
2.6. In Vitro Cytotoxicity Assessment
2.7. Anti-Biofilm Inhibition Assay
2.8. Statistical Analysis
3. Results and Discussion
3.1. Determination of the Antibacterial Minimum Inhibitory Concentration (MIC)
3.2. Determination of Tobramycin Encapsulation Efficiency% (EE%) Using Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometer (UHPLC-MS/MS)
3.3. Particle Size and Zeta Potential Measurement
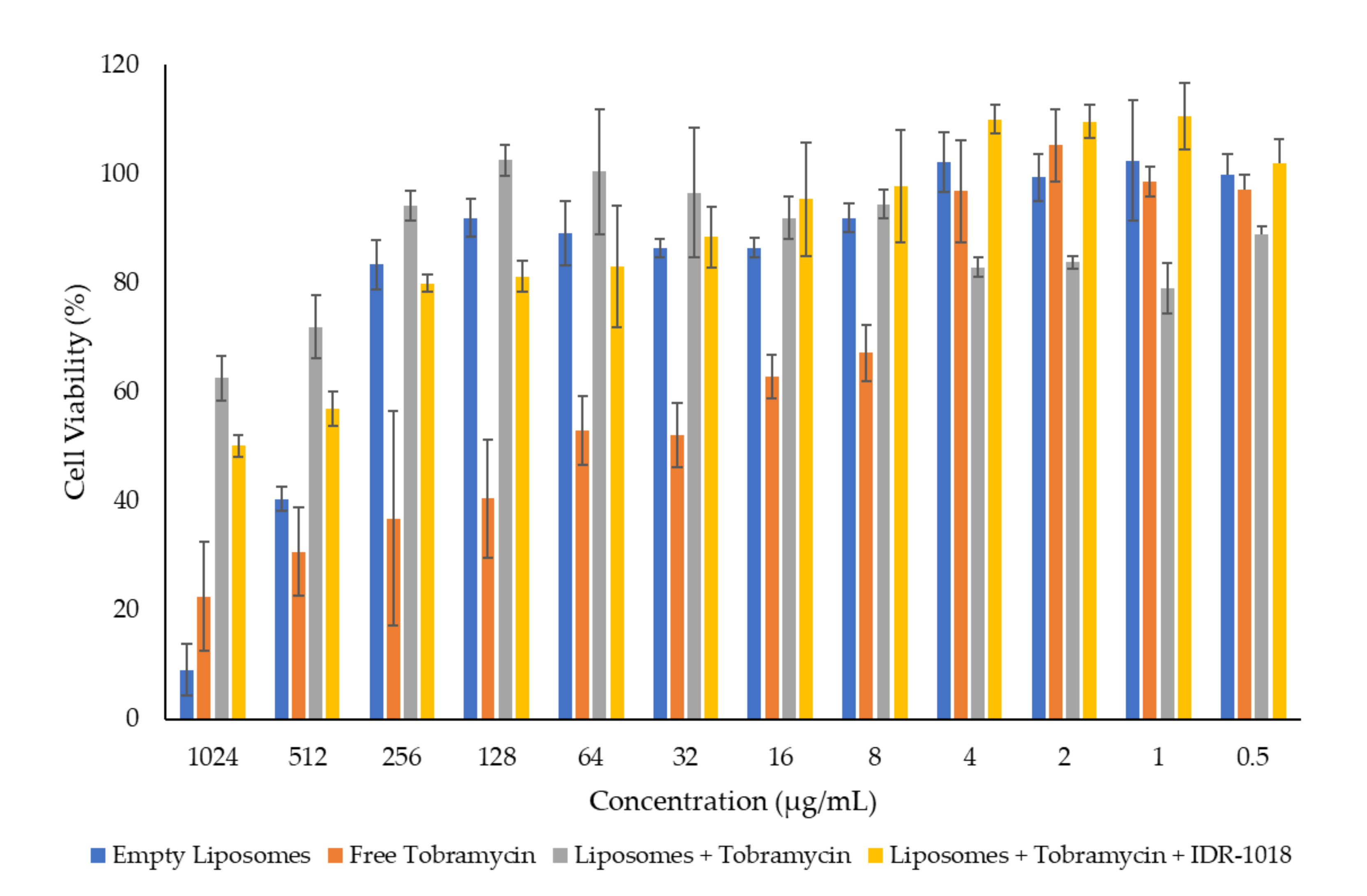
3.4. In Vitro Cytotoxicity Assessment
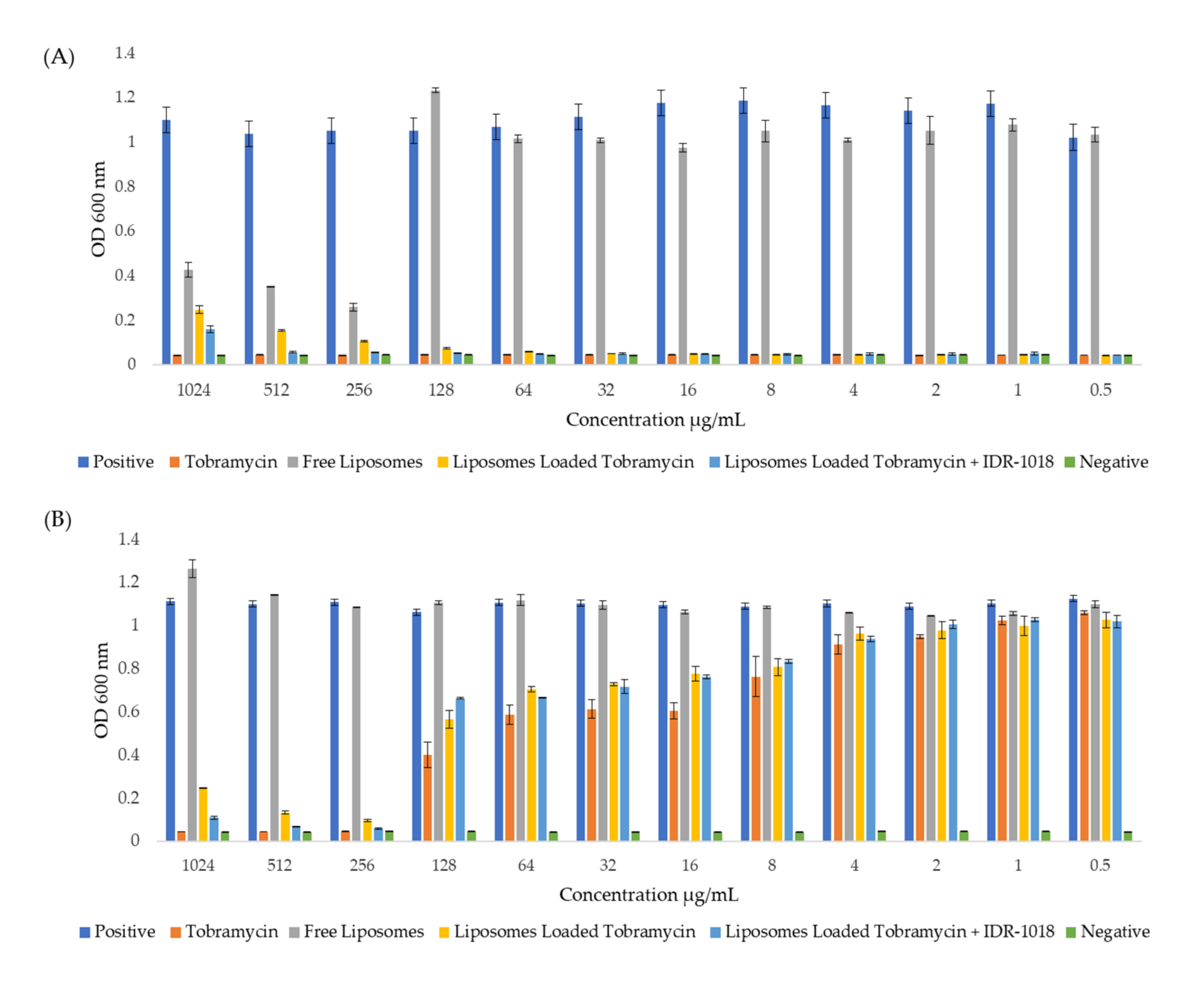
3.5. Effect of Tobramycin-Loaded Liposomes against P. aeruginosa
3.6. Effect of Tobramycin-Loaded Liposomes against Matured P. aeruginosa Biofilms
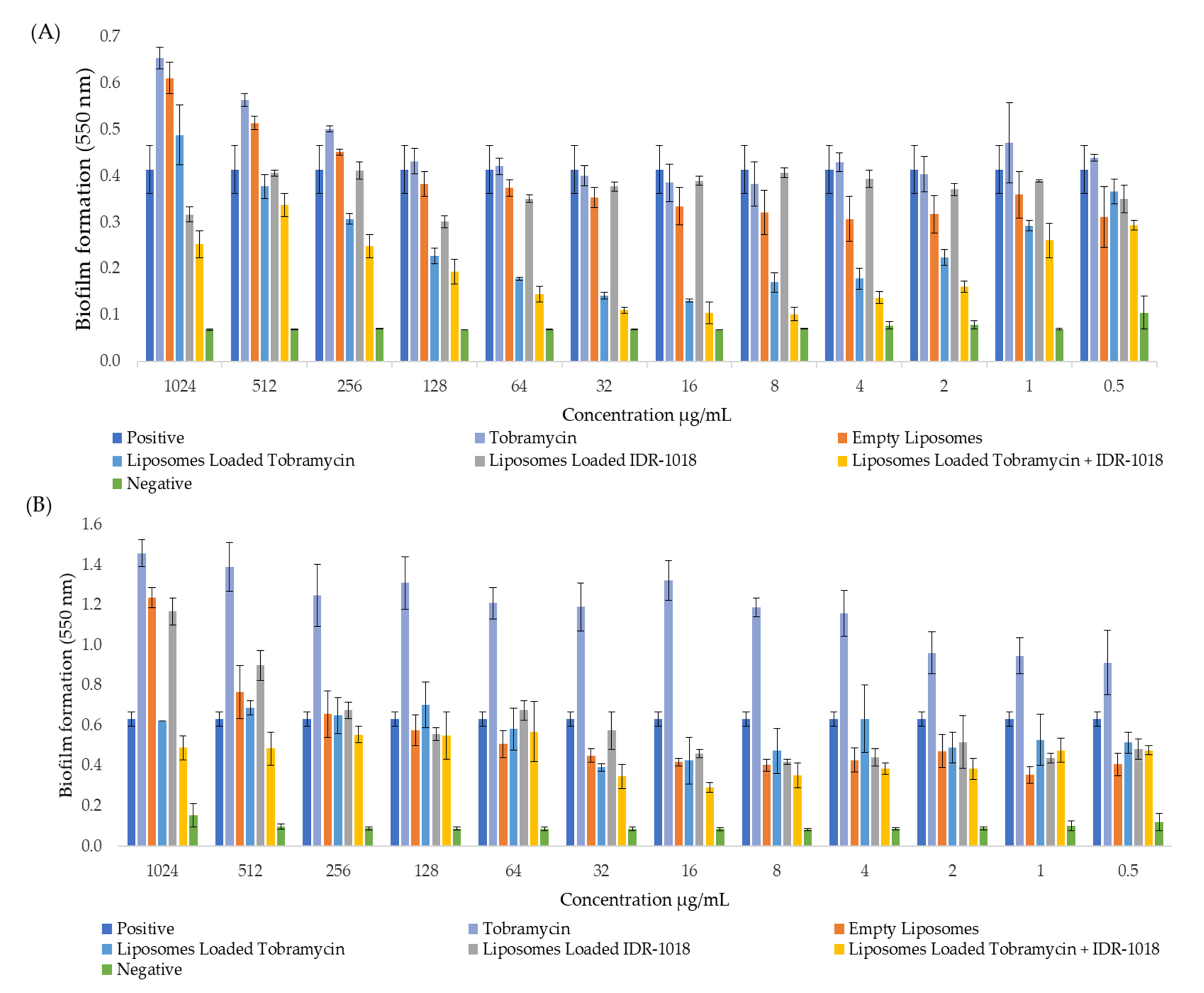
3.7. Effect of Tobramycin-Loaded Liposomes against Biofilm Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almughem, F.A.; Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alharbi, W.S.; Alshahrani, M.Y.; Alshehri, A.A. Cystic Fibrosis: Overview of the Current Development Trends and Innovative Therapeutic Strategies. Pharmaceutics 2020, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Jovic, S.; Shikhagaie, M.; Mörgelin, M.; Kjellström, S.; Erjefalt, J.; Olin, A.I.; Frick, I.M.; Egesten, A. Expression of MIG/CXCL9 in cystic fibrosis and modulation of its activities by elastase of Pseudomonas aeruginosa. J. Innate Immun. 2014, 6, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thabaut, A.; Meyran, M. Activit~ bactericide compar~ e de I’imip/~, n~ me et d’autres antibiotiques sur Pseudomonas aeruginosa. Path. Biol. 1988, 36, 472–476. [Google Scholar]
- Zhang, L.; Parente, J.; Harris, S.M.; Woods, D.E.; Hancock, R.E.; Falla, T.J. Antimicrobial peptide therapeutics for cystic fibrosis. Antimicrob. Agents Chemother. 2005, 49, 2921–2927. [Google Scholar] [CrossRef] [Green Version]
- Jefferson, K.K. What drives bacteria to produce a biofilm? FEMS Microbiol. Lett. 2004, 236, 163–173. [Google Scholar] [CrossRef]
- Maukonen, J.; Mättö, J.; Wirtanen, G.; Raaska, L.; Mattila-Sandholm, T.; Saarela, M. Methodologies for the characterization of microbes in industrial environments: A review. J. Ind. Microbiol. Biotechnol. 2003, 30, 327–356. [Google Scholar] [CrossRef]
- Di Somma, A.; Moretta, A.; Canè, C.; Cirillo, A.; Duilio, A. Antimicrobial and Antibiofilm Peptides. Biomolecules 2020, 10, 652. [Google Scholar] [CrossRef] [Green Version]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Hsiao, F.S.; Ho, Y.H.; Chen, C.S. The proteome targets of intracellular targeting antimicrobial peptides. Proteomics 2016, 16, 1225–1237. [Google Scholar] [CrossRef]
- Yasir, M.; Willcox, M.D.P.; Dutta, D. Action of Antimicrobial Peptides against Bacterial Biofilms. Materials 2018, 11, 2468. [Google Scholar] [CrossRef] [Green Version]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J. Bacterial Extracellular Polysaccharides in Biofilm Formation and Function. Microbiol. Spectr. 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 1401. [Google Scholar] [CrossRef]
- Wieczorek, M.; Jenssen, H.; Kindrachuk, J.; Scott, W.R.; Elliott, M.; Hilpert, K.; Cheng, J.T.; Hancock, R.E.; Straus, S.K. Structural studies of a peptide with immune modulating and direct antimicrobial activity. Chem. Biol. 2010, 17, 970–980. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Deslouches, B.; Montelaro, R.C.; Di, Y.P. Prevention of ESKAPE pathogen biofilm formation by antimicrobial peptides WLBU2 and LL37. Int. J. Antimicrob. Agents 2018, 52, 667–672. [Google Scholar] [CrossRef]
- Mansour, S.C.; de la Fuente-Núñez, C.; Hancock, R.E. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef]
- Liu, J.; Chen, C.; Wei, T.; Gayet, O.; Loncle, C.; Borge, L.; Dusetti, N.; Ma, X.; Marson, D.; Laurini, E. Dendrimeric nanosystem consistently circumvents heterogeneous drug response and resistance in pancreatic cancer. Exploration 2021, 1, 21–34. [Google Scholar] [CrossRef]
- Shahriar, S.M.S.; Mondal, J.; Hasan, M.N.; Revuri, V.; Lee, D.Y.; Lee, Y.K. Electrospinning Nanofibers for Therapeutics Delivery. Nanomaterials 2019, 9, 532. [Google Scholar] [CrossRef] [Green Version]
- Rade, P.P.; Giram, P.S.; Shitole, A.A.; Sharma, N.; Garnaik, B. Physicochemical and in Vitro Antibacterial Evaluation of Metronidazole Loaded Eudragit S-100 Nanofibrous Mats for the Intestinal Drug Delivery. Adv. Fiber Mater. 2022, 4, 76–88. [Google Scholar] [CrossRef]
- Ferreira, M.; Ogren, M.; Dias, J.N.R.; Silva, M.; Gil, S.; Tavares, L.; Aires-da-Silva, F.; Gaspar, M.M.; Aguiar, S.I. Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance. Molecules 2021, 26, 2047. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Ishida, T. Liposomal Delivery Systems: Design Optimization and Current Applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A. Liposomes as delivery systems for antibiotics. Int. J. Pharm. 2010, 387, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Banerjee, A.; Önyüksel, H. Improvement of drug safety by the use of lipid-based nanocarriers. J. Control. Release 2012, 163, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Desgranges, S.; Ruddle, C.C.; Burke, L.P.; McFadden, T.M.; O’Brien, J.E.; Fitzgerald-Hughes, D.; Humphreys, H.; Smyth, T.P.; Devocelle, M. β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. RSC Adv. 2012, 2, 2480–2492. [Google Scholar] [CrossRef]
- Li, W.; O’Brien-Simpson, N.M.; Holden, J.A.; Otvos, L.; Reynolds, E.C.; Separovic, F.; Hossain, M.A.; Wade, J.D. Covalent conjugation of cationic antimicrobial peptides with a β-lactam antibiotic core. Pept. Sci. 2018, 110, e24059. [Google Scholar] [CrossRef]
- Vanić, Ž.; Rukavina, Z.; Manner, S.; Fallarero, A.; Uzelac, L.; Kralj, M.; Amidžić Klarić, D.; Bogdanov, A.; Raffai, T.; Virok, D.P.; et al. Azithromycin-liposomes as a novel approach for localized therapy of cervicovaginal bacterial infections. Int. J. Nanomed. 2019, 14, 5957–5976. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y. Liposome as a delivery system for the treatment of biofilm-mediated infections. J. Appl. Microbiol. 2021, 131, 2626–2639. [Google Scholar] [CrossRef]
- Nguyen, S.; Hiorth, M.; Rykke, M.; Smistad, G. The potential of liposomes as dental drug delivery systems. Eur. J. Pharm. Biopharm. 2011, 77, 75–83. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A.; Gubernator, J.; Gula, G.; Bocer, T.; Doroszkiewicz, W. The interaction between Pseudomonas aeruginosa cells and cationic PC:Chol:DOTAP liposomal vesicles versus outer-membrane structure and envelope properties of bacterial cell. Int. J. Pharm. 2009, 367, 211–219. [Google Scholar] [CrossRef]
- Wong, J.P.; Yang, H.; Blasetti, K.L.; Schnell, G.; Conley, J.; Schofield, L.N. Liposome delivery of ciprofloxacin against intracellular Francisella tularensis infection. J. Control. Release 2003, 92, 265–273. [Google Scholar] [CrossRef]
- Okusanya, O.O.; Bhavnani, S.M.; Hammel, J.; Minic, P.; Dupont, L.J.; Forrest, A.; Mulder, G.J.; Mackinson, C.; Ambrose, P.G.; Gupta, R. Pharmacokinetic and pharmacodynamic evaluation of liposomal amikacin for inhalation in cystic fibrosis patients with chronic pseudomonal infection. Antimicrob. Agents Chemother. 2009, 53, 3847–3854. [Google Scholar] [CrossRef] [Green Version]
- Weers, J.; Metzheiser, B.; Taylor, G.; Warren, S.; Meers, P.; Perkins, W.R. A gamma scintigraphy study to investigate lung deposition and clearance of inhaled amikacin-loaded liposomes in healthy male volunteers. J. Aerosol Med. Pulm. Drug Deliv. 2009, 22, 131–138. [Google Scholar] [CrossRef]
- Hill, M.; Cunningham, R.N.; Hathout, R.M.; Johnston, C.; Hardy, J.G.; Migaud, M.E. Formulation of Antimicrobial Tobramycin Loaded PLGA Nanoparticles via Complexation with AOT. J. Funct. Biomater. 2019, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Halwani, M.; Hebert, S.; Suntres, Z.E.; Lafrenie, R.M.; Azghani, A.O.; Omri, A. Bismuth-thiol incorporation enhances biological activities of liposomal tobramycin against bacterial biofilm and quorum sensing molecules production by Pseudomonas aeruginosa. Int. J. Pharm. 2009, 373, 141–146. [Google Scholar] [CrossRef]
- Ye, T.; Sun, S.; Sugianto, T.D.; Tang, P.; Parumasivam, T.; Chang, Y.K.; Astudillo, A.; Wang, S.; Chan, H.K. Novel combination proliposomes containing tobramycin and clarithromycin effective against Pseudomonas aeruginosa biofilms. Int. J. Pharm. 2018, 552, 130–138. [Google Scholar] [CrossRef]
- Wayne, P. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fourth Informational Supplement, M100–S24; Clinical Laboratory Standards Institute (CLSI): Malvern, PA, USA, 2011; Volume 31, pp. 100–121. [Google Scholar]
- Alkahtani, M.; Alfahd, A.; Alsofyani, N.; Almuqhim, A.A.; Qassem, H.; Alshehri, A.A.; Almughem, F.A.; Hemmer, P. Photostable and Small YVO(4):Yb, Er Upconversion Nanoparticles in Water. Nanomaterials 2021, 11, 1535. [Google Scholar] [CrossRef]
- Messiaen, A.S.; Nelis, H.; Coenye, T. Investigating the role of matrix components in protection of Burkholderia cepacia complex biofilms against tobramycin. J. Cyst. Fibros. 2014, 13, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Lozano, C.; López, M.; Rojo-Bezares, B.; Sáenz, Y. Antimicrobial Susceptibility Testing in Pseudomonas aeruginosa Biofilms: One Step Closer to a Standardized Method. Antibiotics 2020, 9, 880. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.T.A. Goodman Gilmans the Pharmacological Basis of Therapeutics, 13th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Kapoor, P.; Murphy, P. Combination antibiotics against Pseudomonas aeruginosa, representing common and rare cystic fibrosis strains from different Irish clinics. Heliyon 2018, 4, e00562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolter, D.J.; Lister, P.D. Mechanisms of β-lactam resistance among Pseudomonas aeruginosa. Curr. Pharm. Des. 2013, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.A.; Blaser, M.; Santanam, P.; Hächler, H.; Kayser, F.; Hare, R.; Miller, G. Appearance of amikacin and tobramycin resistance due to 4’-aminoglycoside nucleotidyltransferase [ANT (4’)-II] in gram-negative pathogens. Antimicrob. Agents Chemother. 1990, 34, 2381–2386. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.B.; Betageri, G.V.; Singh, M. Factors affecting microencapsulation of drugs in liposomes. J. Microencapsul. 1995, 12, 229–246. [Google Scholar] [CrossRef]
- Forier, K.; Messiaen, A.S.; Raemdonck, K.; Nelis, H.; De Smedt, S.; Demeester, J.; Coenye, T.; Braeckmans, K. Probing the size limit for nanomedicine penetration into Burkholderia multivorans and Pseudomonas aeruginosa biofilms. J. Control. Release 2014, 195, 21–28. [Google Scholar] [CrossRef]
- Kim, H.-J.; Michael Gias, E.L.; Jones, M.N. The adsorption of cationic liposomes to Staphylococcus aureus biofilms. Colloids Surf. A: Physicochem. Eng. Asp. 1999, 149, 561–570. [Google Scholar] [CrossRef]
- Forier, K.; Raemdonck, K.; De Smedt, S.C.; Demeester, J.; Coenye, T.; Braeckmans, K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J. Control. Release 2014, 190, 607–623. [Google Scholar] [CrossRef] [Green Version]
- Flemming, H.C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Yu, W.; Hallinen, K.M.; Wood, K.B. Interplay between Antibiotic Efficacy and Drug-Induced Lysis Underlies Enhanced Biofilm Formation at Subinhibitory Drug Concentrations. Antimicrob. Agents Chemother. 2018, 62, e01603-17. [Google Scholar] [CrossRef] [Green Version]
- Tahrioui, A.; Duchesne, R.; Bouffartigues, E.; Rodrigues, S.; Maillot, O.; Tortuel, D.; Hardouin, J.; Taupin, L.; Groleau, M.C.; Dufour, A.; et al. Extracellular DNA release, quorum sensing, and PrrF1/F2 small RNAs are key players in Pseudomonas aeruginosa tobramycin-enhanced biofilm formation. NPJ Biofilms Microbiomes 2019, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Fricks-Lima, J.; Hendrickson, C.M.; Allgaier, M.; Zhuo, H.; Wiener-Kronish, J.P.; Lynch, S.V.; Yang, K. Differences in biofilm formation and antimicrobial resistance of Pseudomonas aeruginosa isolated from airways of mechanically ventilated patients and cystic fibrosis patients. Int. J. Antimicrob. Agents 2011, 37, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, W.; Zhu, Y.; Gong, Q.; Yu, W.; Lu, X. Antibiotics at subinhibitory concentrations improve the quorum sensing behavior of Chromobacterium violaceum. FEMS Microbiol. Lett. 2013, 341, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Laureti, L.; Matic, I.; Gutierrez, A. Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics. Antibiotics 2013, 2, 100. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.B. Antibiotic-induced biofilm formation. Int. J. Artif. Organs 2011, 34, 737–751. [Google Scholar] [CrossRef]
- Tseng, B.S.; Zhang, W.; Harrison, J.J.; Quach, T.P.; Song, J.L.; Penterman, J.; Singh, P.K.; Chopp, D.L.; Packman, A.I.; Parsek, M.R. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 2013, 15, 2865–2878. [Google Scholar] [CrossRef] [Green Version]
- Soto, S.M.; Smithson, A.; Martinez, J.A.; Horcajada, J.P.; Mensa, J.; Vila, J. Biofilm formation in uropathogenic Escherichia coli strains: Relationship with prostatitis, urovirulence factors and antimicrobial resistance. J. Urol. 2007, 177, 365–368. [Google Scholar] [CrossRef]
- Fàbrega, A.; Soto, S.M.; Ballesté-Delpierre, C.; Fernández-Orth, D.; Jiménez de Anta, M.T.; Vila, J. Impact of quinolone-resistance acquisition on biofilm production and fitness in Salmonella enterica. J. Antimicrob. Chemother. 2014, 69, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; López, Y.; Muñoz, E.; Rolo, D.; Ardanuy, C.; Martí, S.; Xercavins, M.; Horcajada, J.P.; Bosch, J.; Soto, S.M. Relationship Between Biofilm Formation and Antimicrobial Resistance in Gram-Negative Bacteria. Microb. Drug Resist. 2019, 25, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Thomas, N.; Thierry, B.; Vreugde, S.; Prestidge, C.A.; Wormald, P.J. Distribution and Inhibition of Liposomes on Staphylococcus aureus and Pseudomonas aeruginosa Biofilm. PLoS ONE 2015, 10, e0131806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibaraki, H.; Kanazawa, T.; Chien, W.-Y.; Nakaminami, H.; Aoki, M.; Ozawa, K.; Kaneko, H.; Takashima, Y.; Noguchi, N.; Seta, Y. The effects of surface properties of liposomes on their activity against Pseudomonas aeruginosa PAO-1 biofilm. J. Drug Deliv. Sci. Technol. 2020, 57, 101754. [Google Scholar] [CrossRef]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: A powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [Green Version]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J.P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Antibiotics | MIC (μg/mL) | |||
|---|---|---|---|---|
| PA01 | MDR 7067 | |||
| 24 h | 48 h | 24 h | 48 h | |
| Ceftazidime | 2 | >512 | 128 | >1024 |
| Imipenem | 8 | 32 | 512 | >1024 |
| Tobramycin | <0.5 | 8 | 256 | >1024 |
| The Concentration of Loaded Tobramycin (µg/mL) | Average Particle Size (nm) | Poly Dispersity Index | Zeta Potential (mV) |
|---|---|---|---|
| 0 (empty liposomes) | 124 ± 0.1 | 0.26 ± 0.13 | 65.4 ± 2.0 |
| 0.5 | 187 ± 0.2 | 0.39 ± 0.05 | 67.7 ± 0.6 |
| 1 | 124 ± 0.4 | 0.21 ± 0.02 | 55.3 ± 1.4 |
| 2 | 140 ± 0.7 | 0.27 ± 0.07 | 57.9 ± 0.4 |
| 4 | 127 ± 0.2 | 0.40 ± 0.01 | 62.1 ± 0.9 |
| 8 | 145 ± 0.8 | 0.31 ± 0.04 | 58.7 ± 3.2 |
| Formulations | MIC (μg/mL) | |||
|---|---|---|---|---|
| PA01 | MDR 7067 | |||
| 24 h | 48 h | 24 h | 48 h | |
| Tobramycin | ≤0.5 | 32 | 256 | >1024 |
| Liposomes + Tobramycin | ≤0.5 | 32 | 256 | >1024 |
| Liposomes + Tobramycin + IDR-1018 | ≤0.5 | 32 | 256 | >1024 |
| Empty Liposomes | >1024 | >1024 | >1024 | >1024 |
| Liposomes + IDR-1018 | >1024 | >1024 | >1024 | >1024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzahrani, N.M.; Booq, R.Y.; Aldossary, A.M.; Bakr, A.A.; Almughem, F.A.; Alfahad, A.J.; Alsharif, W.K.; Jarallah, S.J.; Alharbi, W.S.; Alsudir, S.A.; et al. Liposome-Encapsulated Tobramycin and IDR-1018 Peptide Mediated Biofilm Disruption and Enhanced Antimicrobial Activity against Pseudomonas aeruginosa. Pharmaceutics 2022, 14, 960. https://doi.org/10.3390/pharmaceutics14050960
Alzahrani NM, Booq RY, Aldossary AM, Bakr AA, Almughem FA, Alfahad AJ, Alsharif WK, Jarallah SJ, Alharbi WS, Alsudir SA, et al. Liposome-Encapsulated Tobramycin and IDR-1018 Peptide Mediated Biofilm Disruption and Enhanced Antimicrobial Activity against Pseudomonas aeruginosa. Pharmaceutics. 2022; 14(5):960. https://doi.org/10.3390/pharmaceutics14050960
Chicago/Turabian StyleAlzahrani, Nouf M., Rayan Y. Booq, Ahmad M. Aldossary, Abrar A. Bakr, Fahad A. Almughem, Ahmed J. Alfahad, Wijdan K. Alsharif, Somayah J. Jarallah, Waleed S. Alharbi, Samar A. Alsudir, and et al. 2022. "Liposome-Encapsulated Tobramycin and IDR-1018 Peptide Mediated Biofilm Disruption and Enhanced Antimicrobial Activity against Pseudomonas aeruginosa" Pharmaceutics 14, no. 5: 960. https://doi.org/10.3390/pharmaceutics14050960