
Glioblastoma-Derived Exosomes as Nanopharmaceutics for Improved Glioma Treatment

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
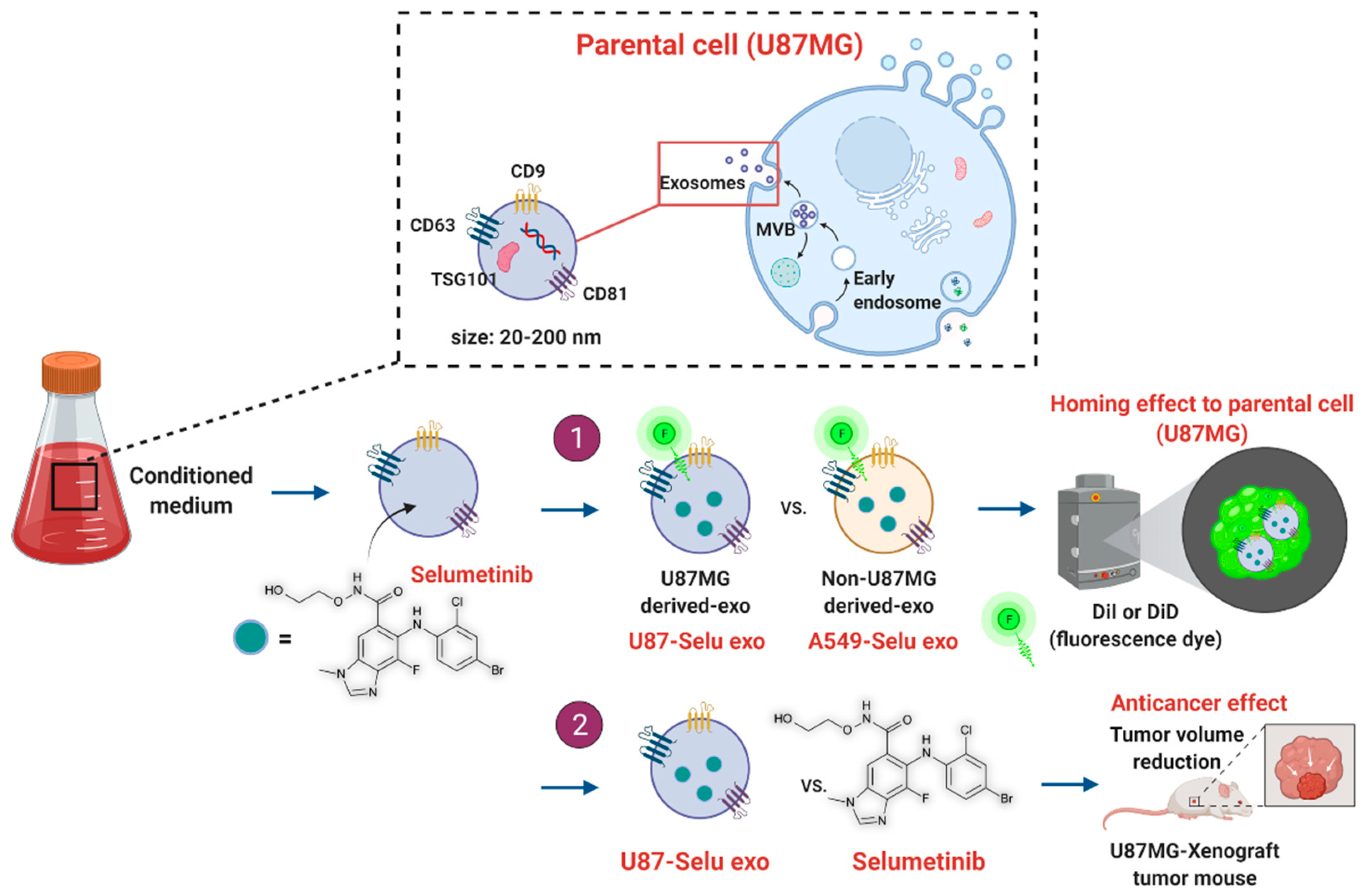
2.1. Experimental Design
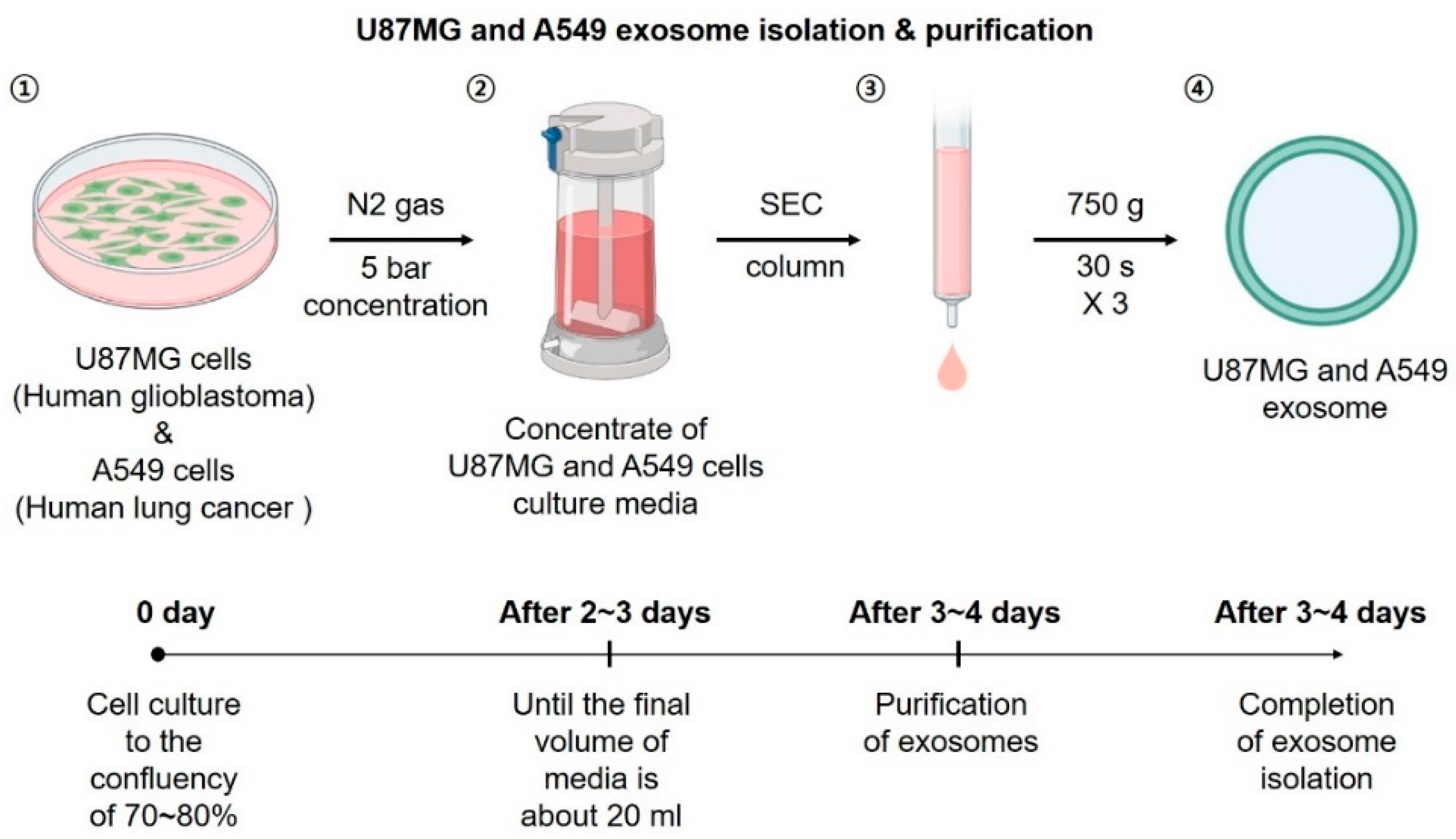
2.2. Cell Culture, Exosome Isolation, Labeling, and Purification
2.3. Fabrication and Purification of Selu-Exo
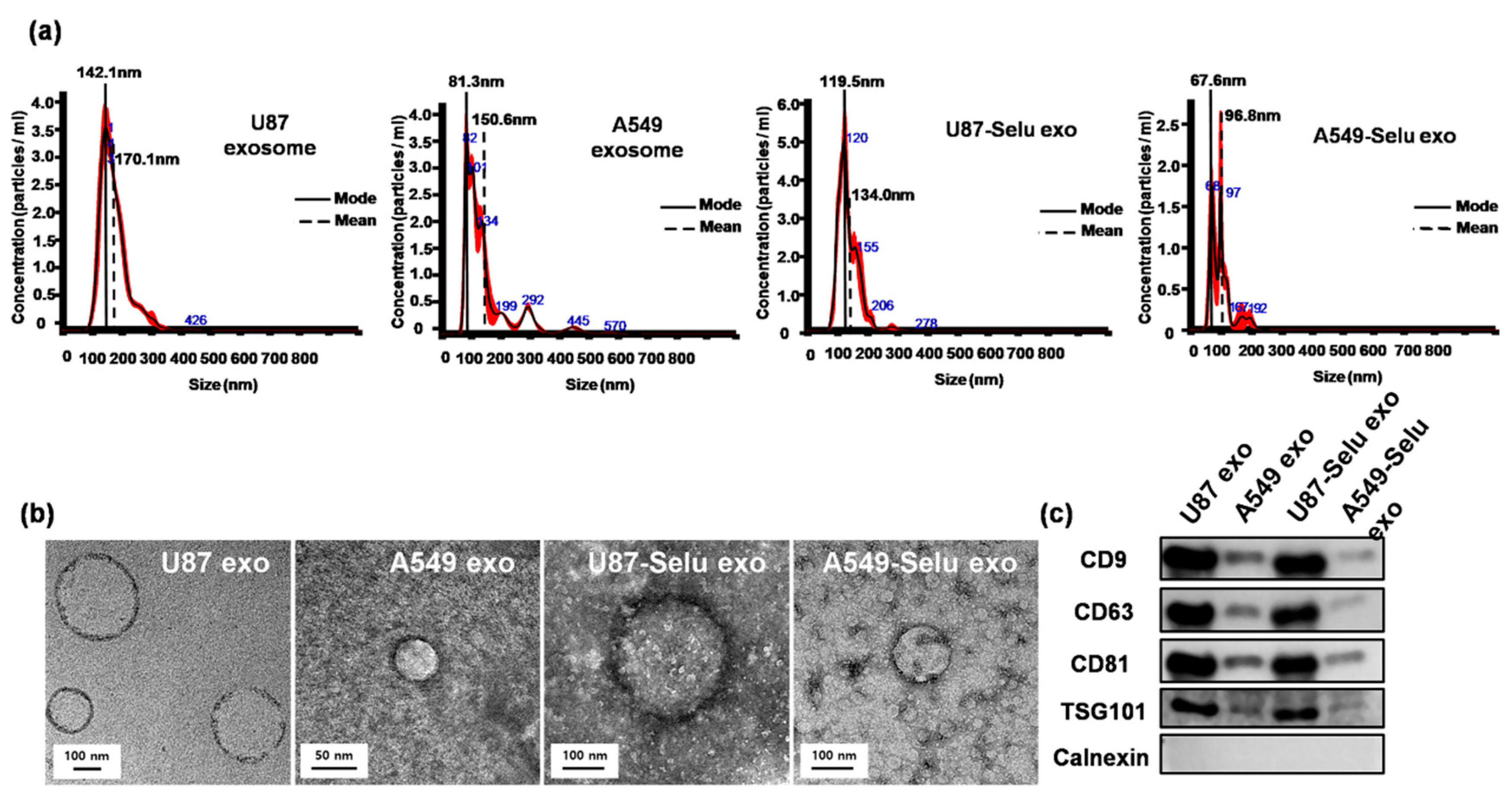
2.4. Characterization of Exosomes
2.4.1. Size Distribution Analysis
2.4.2. Transmission Electron Microscope (TEM)
2.4.3. Western Blot Analysis
2.4.4. Stability of Selumetinib-Loaded Exosomes
2.5. In Vitro Exo and Selu-Exo Internalization to the Parent Cell
2.6. Cell Viability Treated with Exosome, Selumetinib, and U87-Selu Exo
2.7. Animals
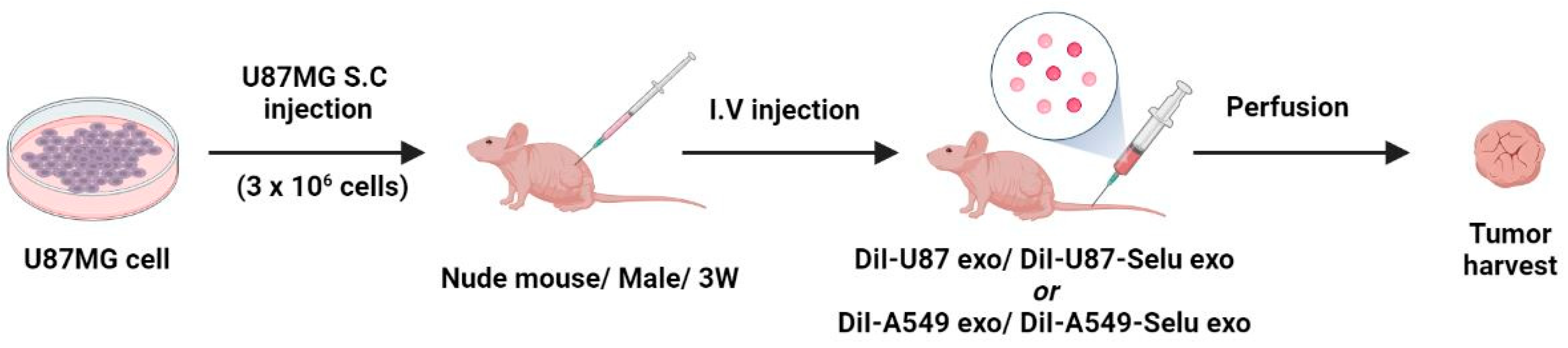
2.8. U87MG Xenograft Model
2.9. In Vivo Exo and Selu-Exo Targeting to the Parental Tumor
2.10. In Vivo Biodistribution of U87-Selu Exo
2.11. In Vivo Anticancer Effect of U87-Selu Exo and A549-Selu Exo
2.12. Histological Analysis for Tumor Tissue
2.13. In Vivo Toxicity of U87-Selu Exo and A549-Selu Exo
2.14. In Vitro Anticancer Effect of U87-Selu Exo
2.15. Flow Cytometry Analysis
2.16. Statistical Analysis
3. Results and Discussion
3.1. Exosome Characterization
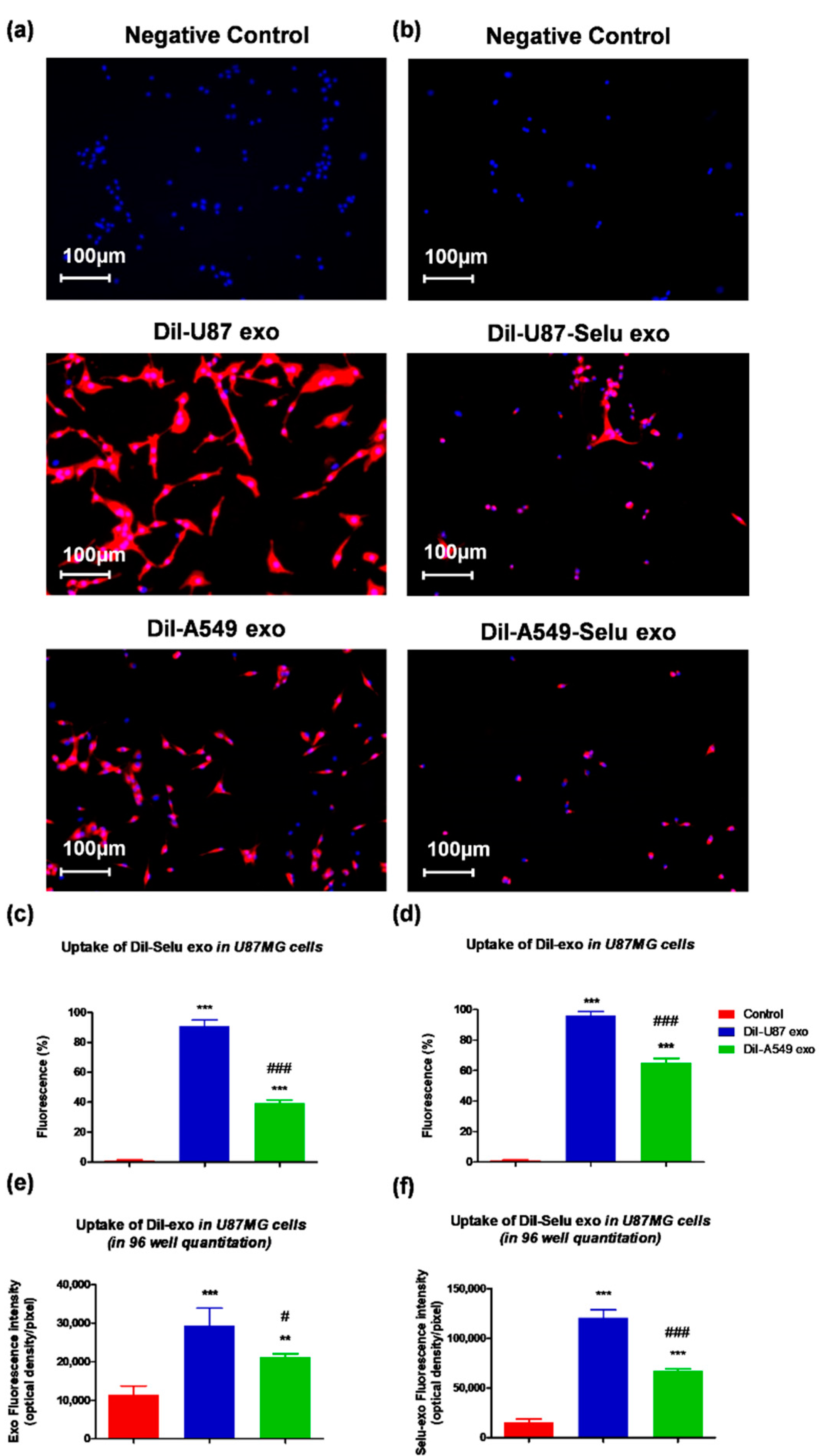
3.2. U87MG Exosome Targeting In Vitro and In Vivo
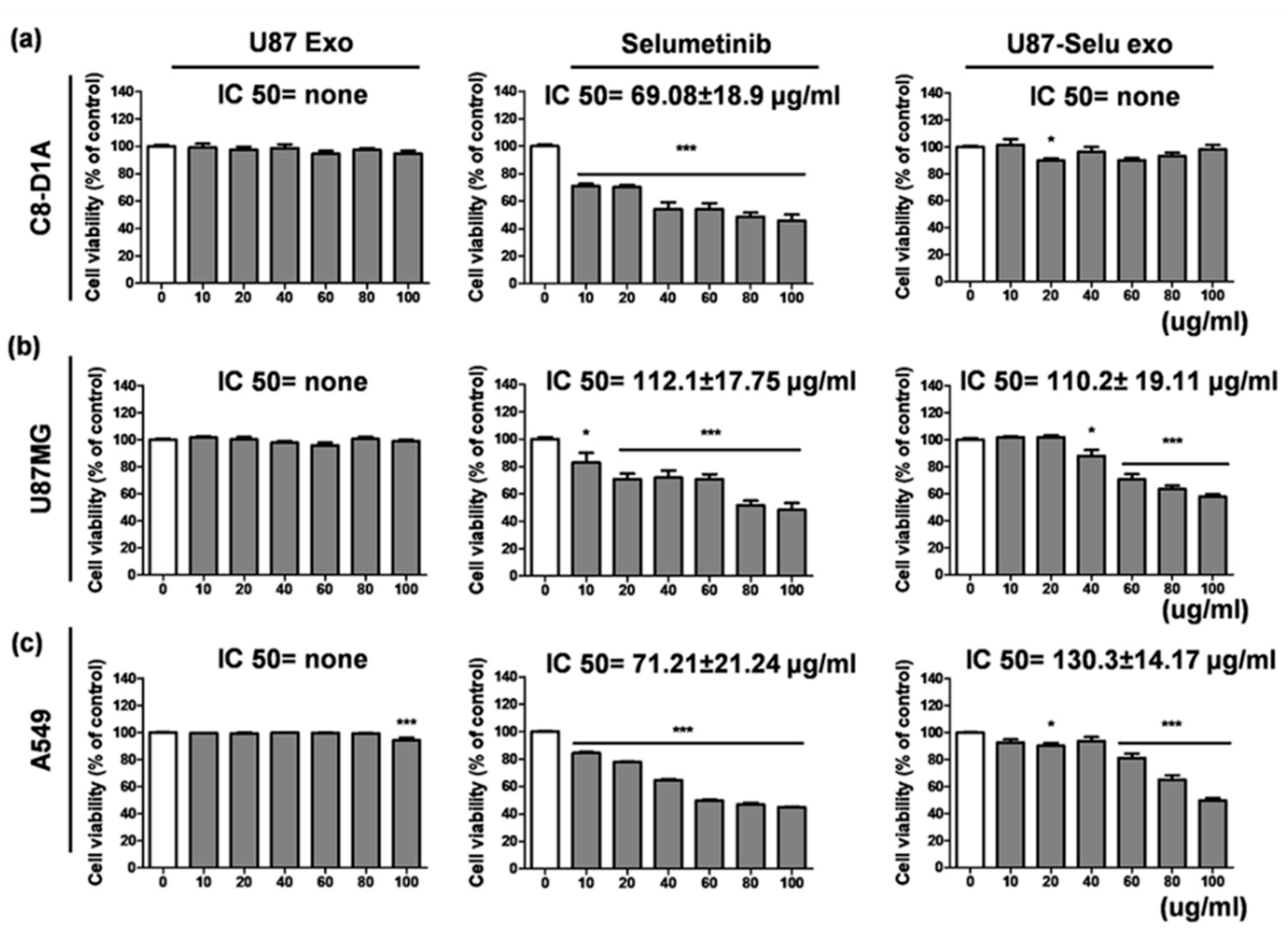
3.3. Cytotoxicity of U87 Exo, Selumetinib and U87-Selu Exo
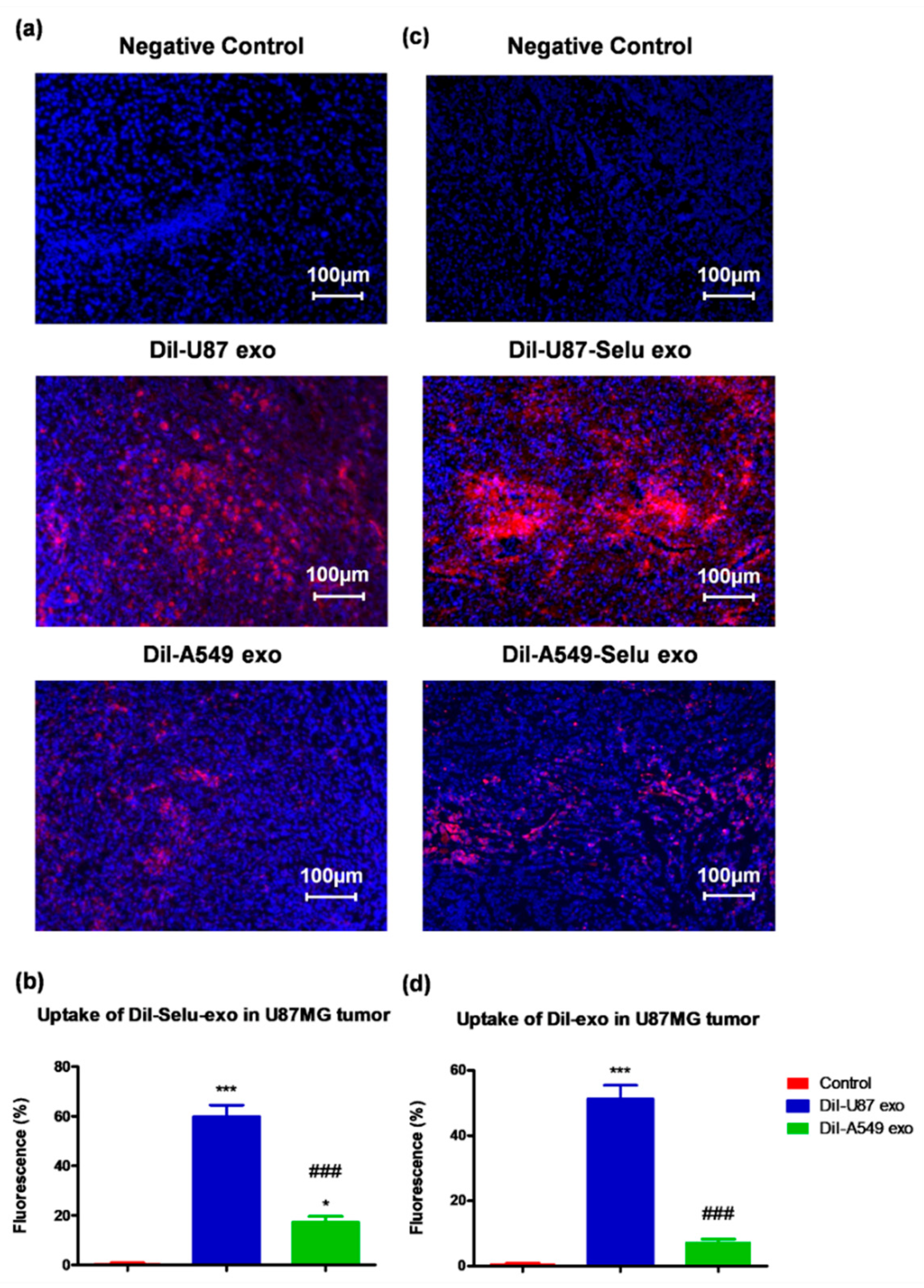
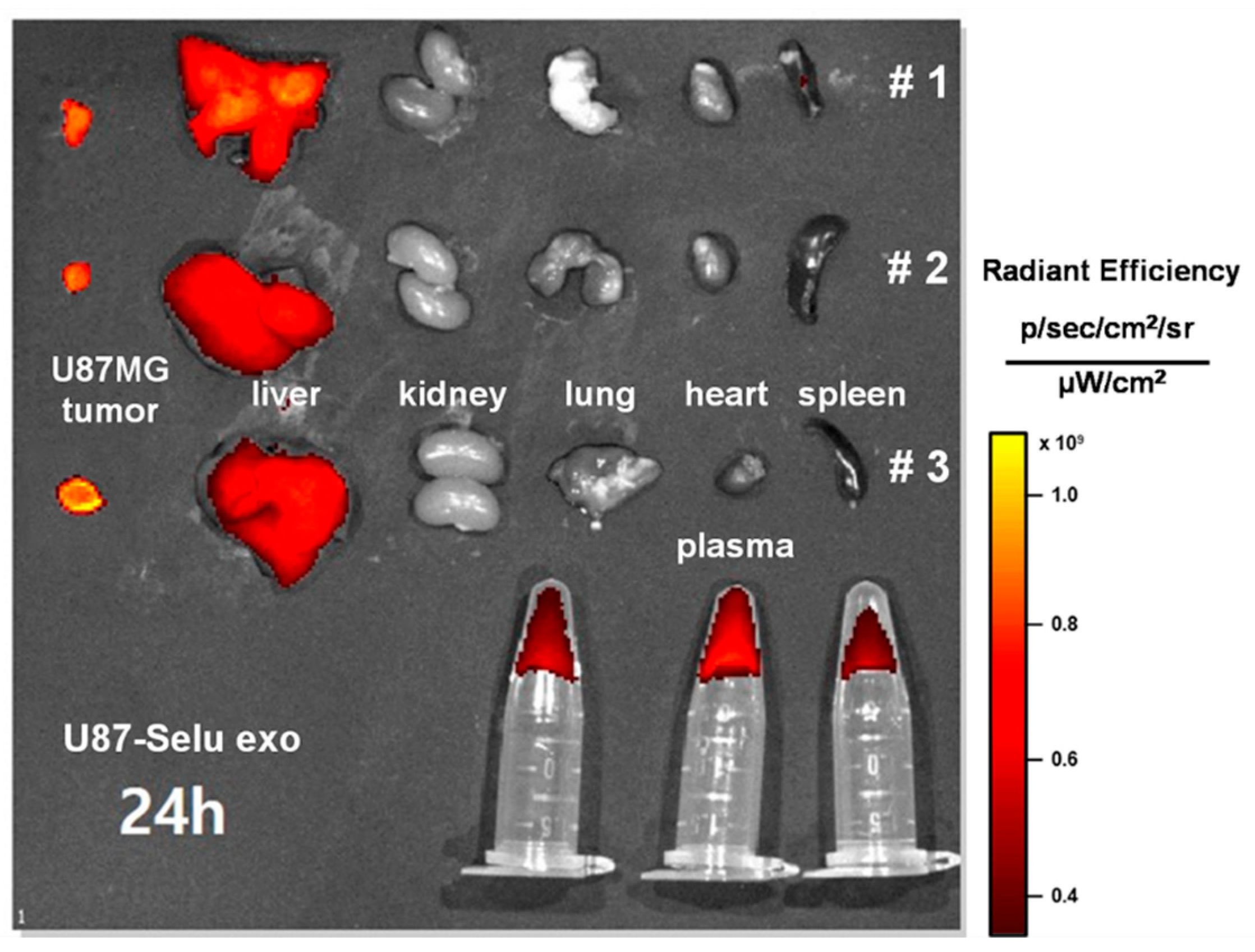
3.4. In Vivo Biodistribution of U87-Selu Exo
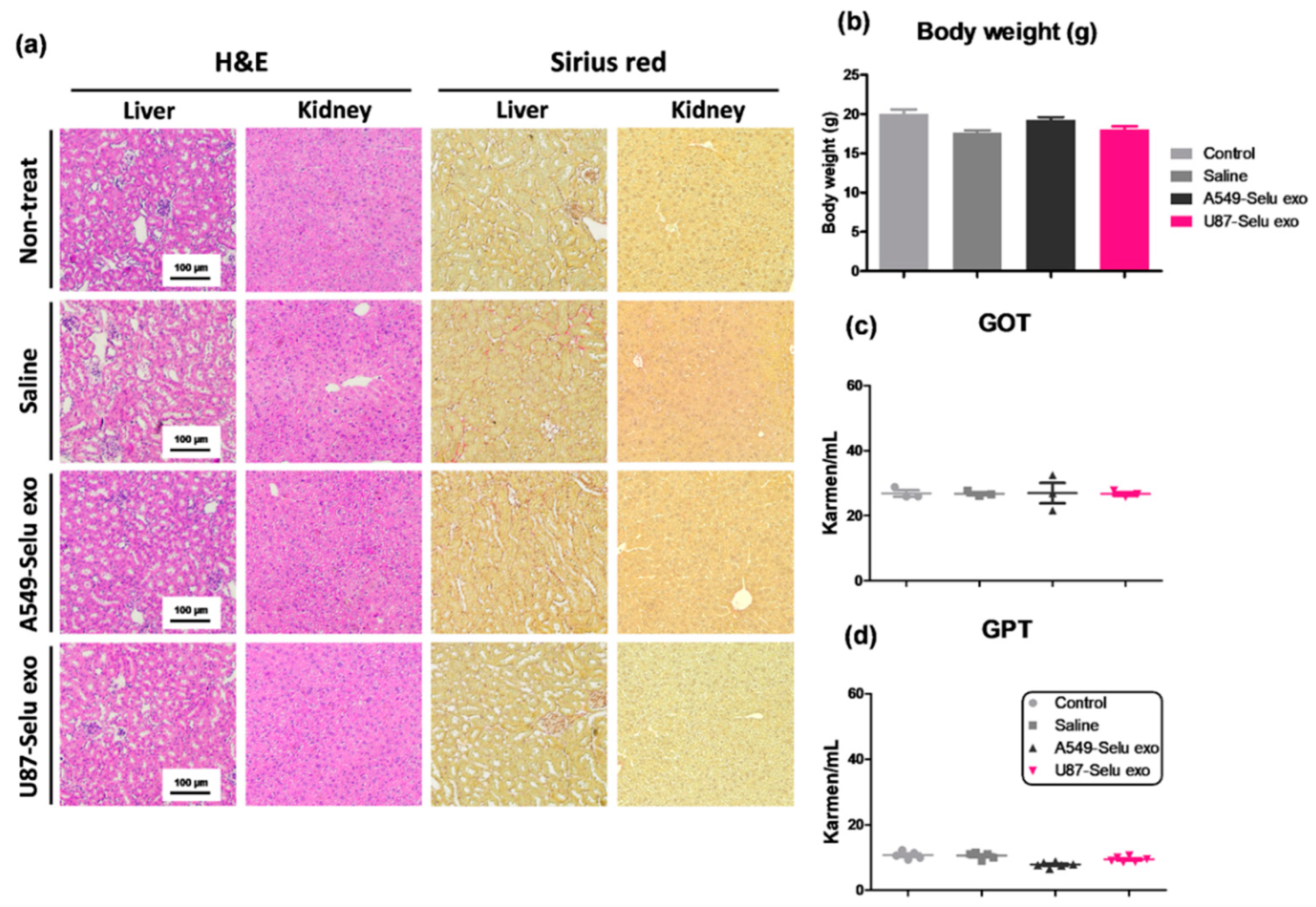
3.5. Liver and Kidney Toxicity of Selu-Exo (U87-Selu Exo and A549-Selu Exo) In Vivo
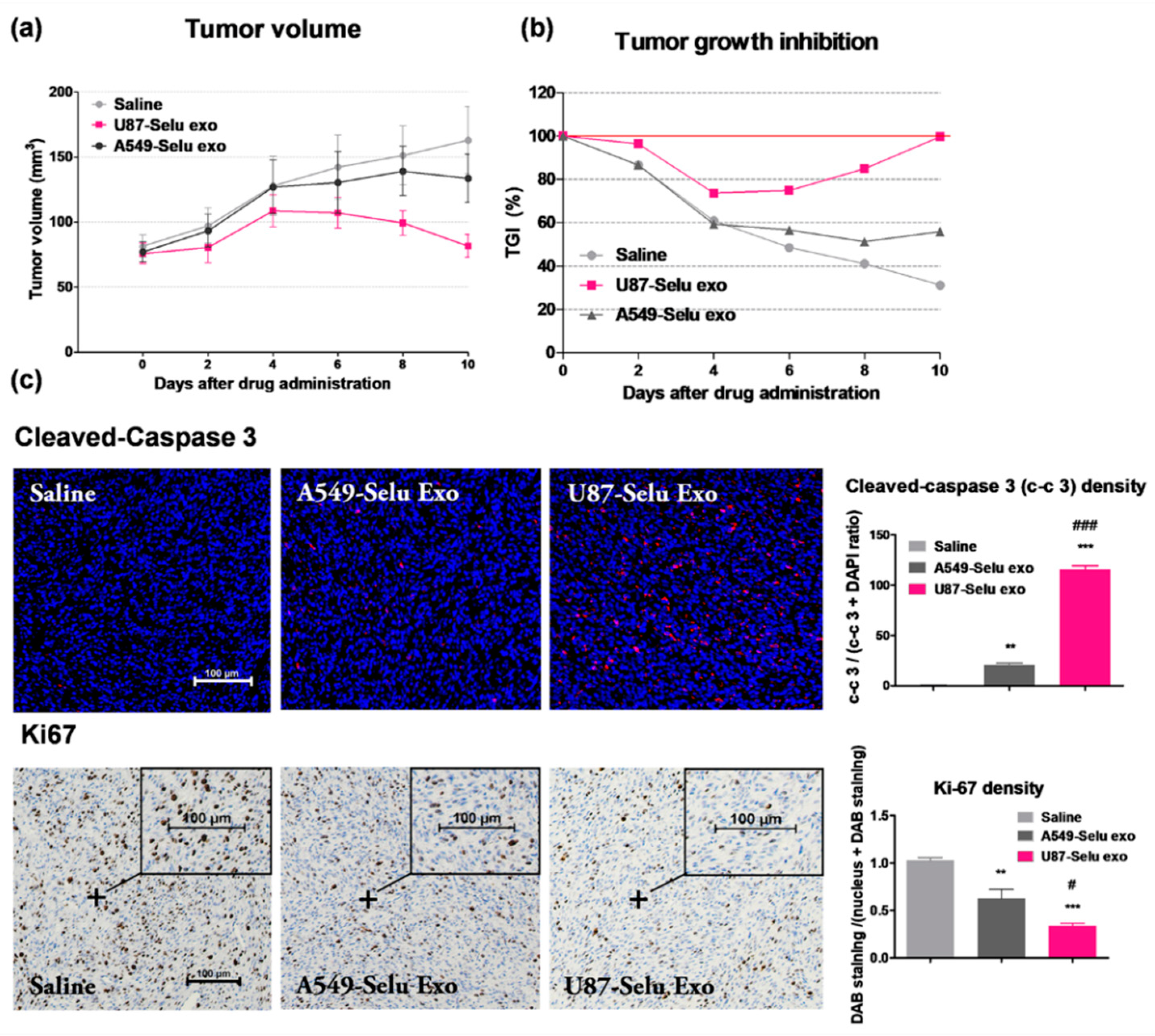
3.6. In Vivo Anticancer Effects of U87-Selu Exo and A549-Selu Exo
3.7. In Vitro Anticancer Effect of U87-Selu Exo
3.7.1. Apoptosis of U87-Selu Exo to U87MG Cells
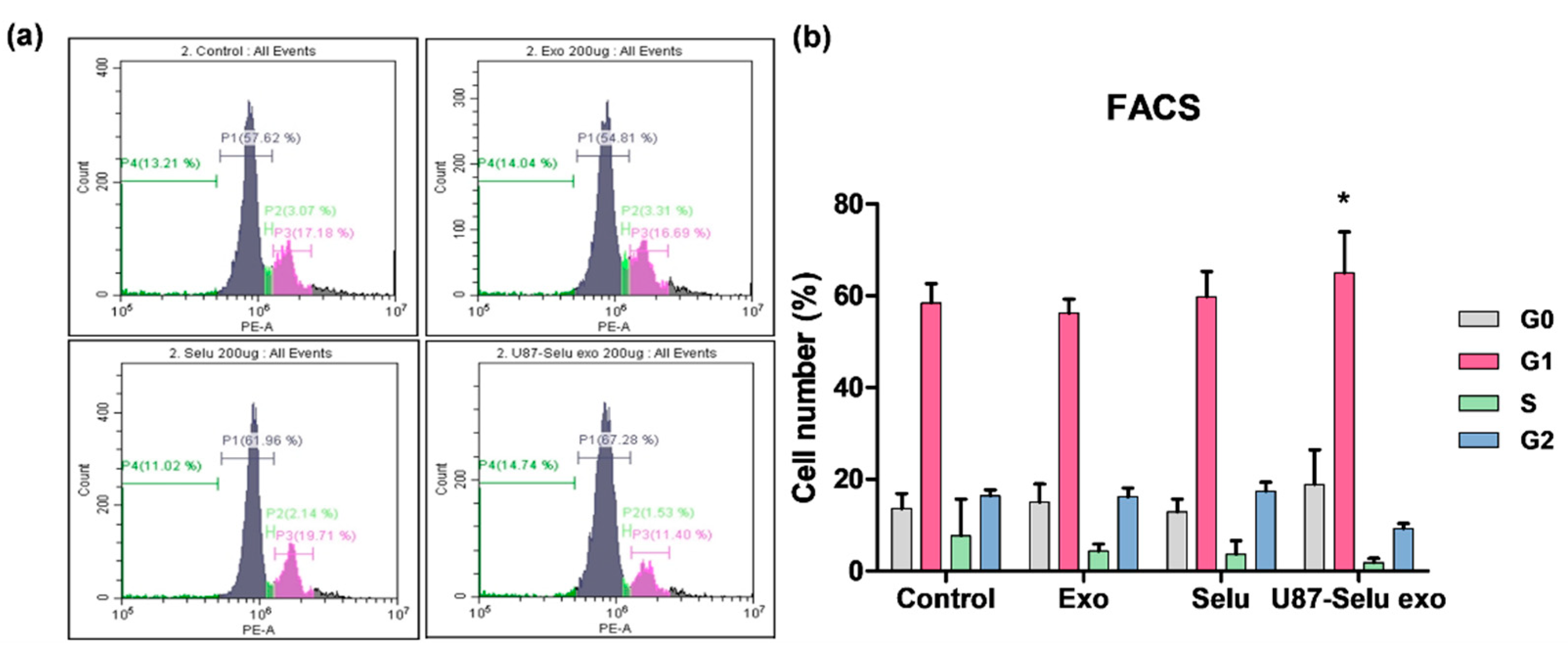
3.7.2. Flow Cytometry
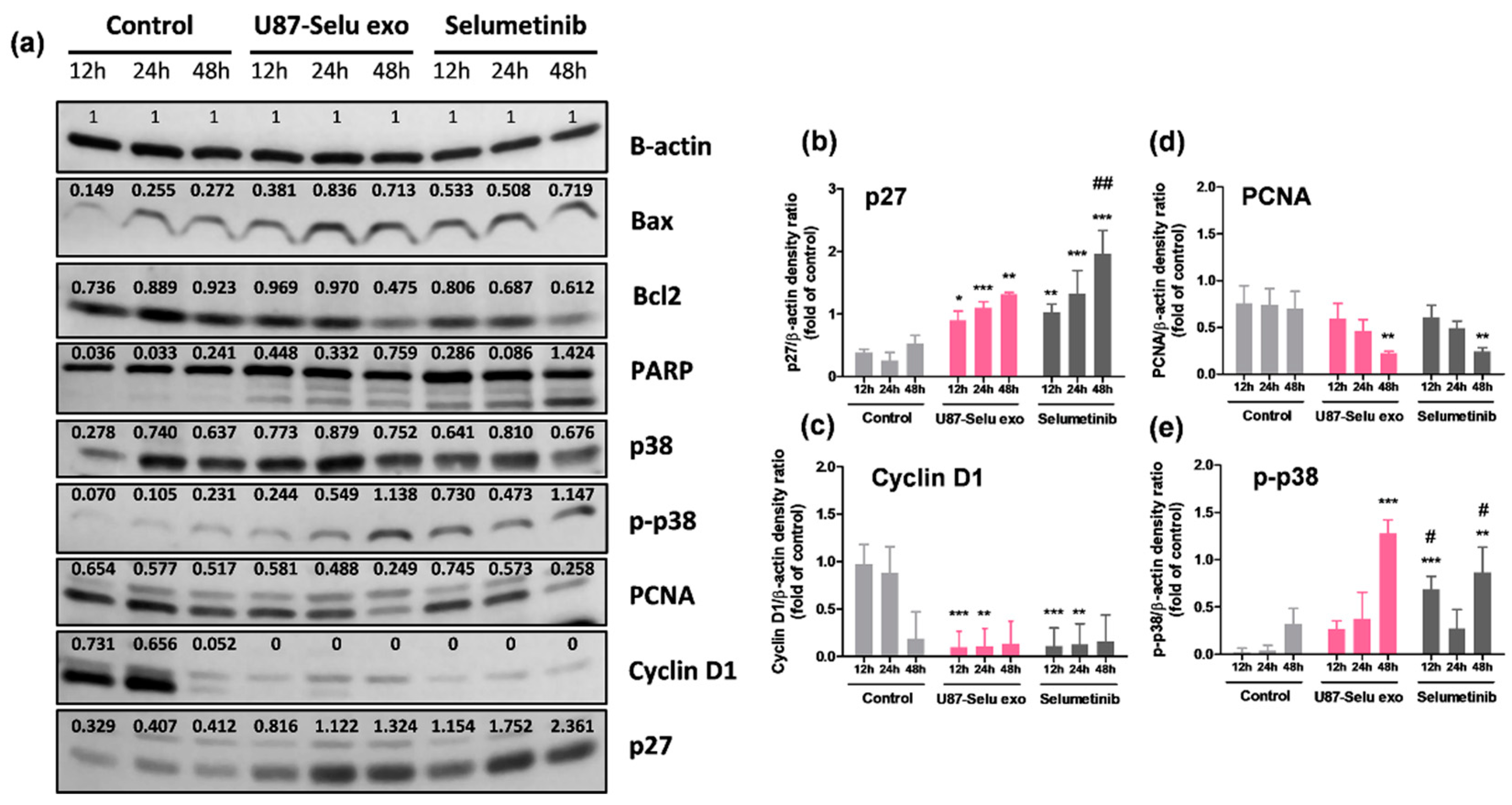
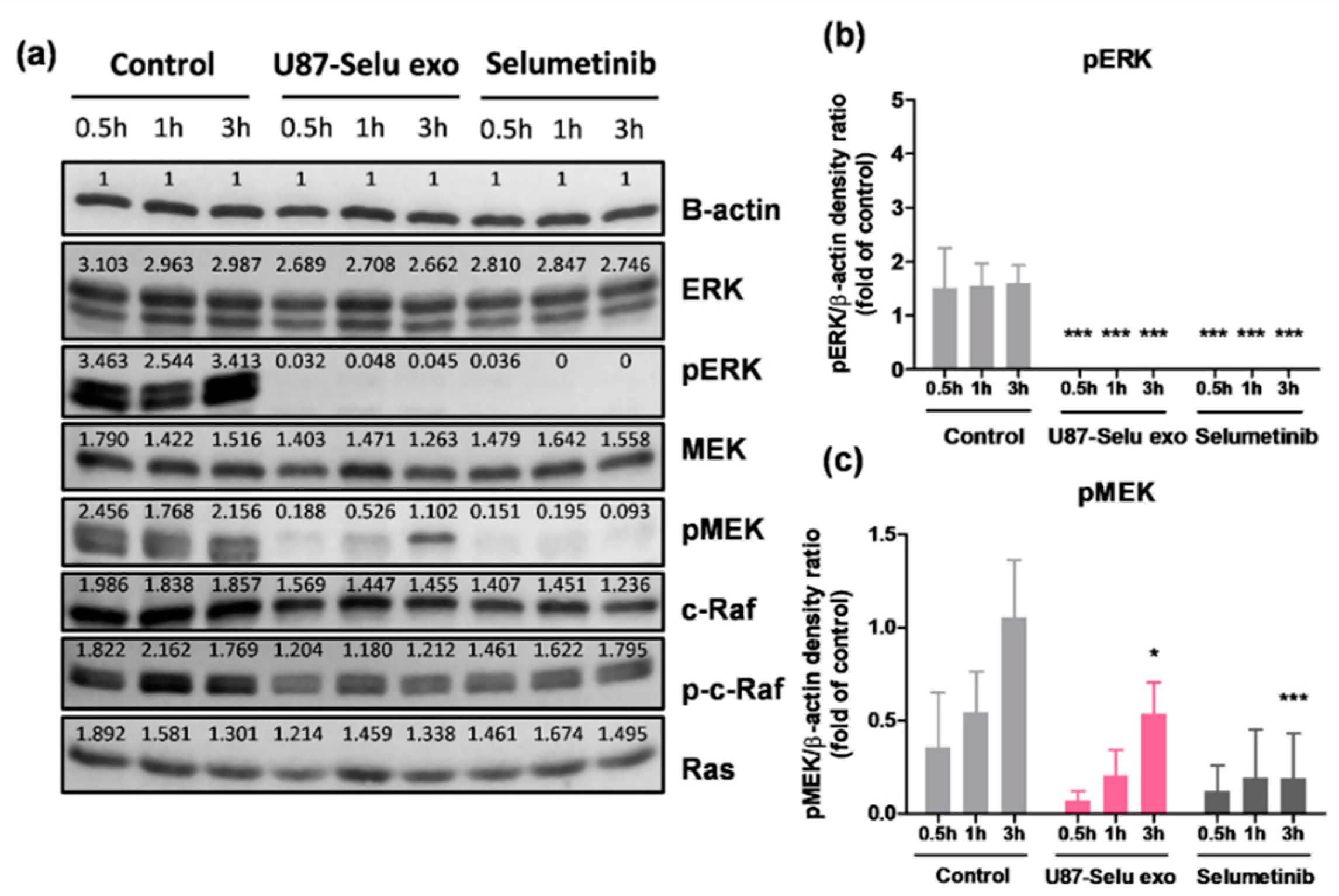
3.8. Anticancer Mechanism of U87-Selu Exo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.R. Q&A: What are exosomes, exactly? BMC Biol. 2016, 14, 46. [Google Scholar]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. A comprehensive review on factors influences biogenesis, functions, therapeutic and clinical implications of exosomes. Int. J. Nanomed. 2021, 16, 1281–1312. [Google Scholar] [CrossRef]
- Lowenstein, P.R.; Mandel, R.J.; Xiong, W.D.; Kroeger, K.; Castro, M.G. Immune responses to adenovirus and adeno-associated vectors used for gene therapy of brain diseases: The role of immunological synapses in understanding the cell biology of neuroimmune interactions. Curr. Gene Ther. 2007, 7, 347–360. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.S.; Liu, F.; Huang, L. Implications of pharmacokinetic behavior of lipoplex for its inflammatory toxicity. Adv. Drug Deliv. Rev. 2005, 57, 689–698. [Google Scholar] [CrossRef]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef]
- Arranja, A.G.; Pathak, V.; Lammers, T.; Shi, Y. Tumor-targeted nanomedicines for cancer theranostics. Pharm. Res. 2017, 115, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kim, E.H.; Kwak, G.; Chi, S.G.; Kim, S.H.; Yang, Y. Exosomes: Cell-derived nanoplatforms for the delivery of cancer therapeutics. Int. J. Mol. Sci. 2020, 22, 14. [Google Scholar] [CrossRef]
- Jia, G.; Han, Y.; An, Y.; Ding, Y.; He, C.; Wang, X.; Tang, Q. NRP–1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Yim, N.; Ryu, S.W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef]
- Lamichhane, T.N.; Raiker, R.S.; Jay, S.M. Exogenous DNA Loading into Extracellular Vesicles via Electroporation is Size-Dependent and Enables Limited Gene Delivery. Mol. Pharm. 2015, 12, 3650–3657. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Gao, J.; Ai, H.; Chen, X. Applications and potential toxicity of magnetic iron oxide nanoparticles. Small 2013, 9, 1533–1545. [Google Scholar] [CrossRef]
- Sun, W.; Luo, J.D.; Jiang, H.; Duan, D.D. Tumor exosomes: A double-edged sword in cancer therapy. Acta Pharm. Sin. 2018, 39, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, H.; Sahebkar, A.; Jaafari, M.R.; Goodarzi, M.; Mirzaei, H.R. Diagnostic and Therapeutic Potential of Exosomes in Cancer: The Beginning of a New Tale? J. Cell. Physiol. 2017, 232, 3251–3260. [Google Scholar] [CrossRef]
- Qiao, L.; Hu, S.; Huang, K.; Su, T.; Li, Z.; Vandergriff, A.; Cores, J.; Dinh, P.U.; Allen, T.; Shen, D.; et al. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474–3487. [Google Scholar] [CrossRef]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Toda, Y.; Takata, K.; Nakagawa, Y.; Kawakami, H.; Fujioka, S.; Kobayashi, K.; Hattori, Y.; Kitamura, Y.; Akaji, K.; Ashihara, E. Effective internalization of U251-MG-secreted exosomes into cancer cells and characterization of their lipid components. Biochem. Biophys. Res. Commun. 2015, 456, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Giannatale, A.D.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bălașa, A.; Șerban, G.; Chinezu, R.; Hurghiș, C.; Tămaș, F.; Manu, D. The involvement of exosomes in glioblastoma development, diagnosis, prognosis, and treatment. Brain Sci. 2020, 10, 553. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoinmmunology 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.; Rao, A.; Braganhol, E.; Whiteside, T. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neurooncol. Adv. 2020, 2, vdaa056. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [Green Version]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Wu, A.; Kong, L.Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE 2011, 6, e16195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Q.; Lv, X.W.; Xu, Y.L.; Cai, R.P.; Dai, R.X.; Yang, X.H.; Zhao, W.K.; Kong, B.H. Exosomal LINC00174 derived from vascular endothelial cells attenuates myocardial I/R injury via p53-mediated autophagy and apoptosis. Mol. Nucleic Acids 2021, 23, 1304–1322. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiang, B.; Wang, X.; Xiang, C. Exosomes derived from human menstrual blood-derived stem cells alleviate fulminant hepatic failure. Stem Cell Res. Ther. 2017, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Zhu, Y.L.; Hu, F.H.; Wang, Y.Y.; Huang, N.P.; Xiao, Z.D. Dynamics of exosome internalization and trafficking. J. Cell. Physiol. 2013, 228, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR–124 Promotes Neurogenesis after Ischemia. Mol. Nucleic Acids 2017, 7, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Alptekin, A.; Liu, Y.; Arbab, A.S. Generation of Novel Diagnostic and Therapeutic Exosomes to Detect and Deplete Protumorigenic M2 Macrophages. Adv. Ther. 2020, 3, 1900209. [Google Scholar] [CrossRef]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Naryzhny, S.; Volnitskiy, A.; Kopylov, A.; Zorina, E.; Kamyshinsky, R.; Bairamukov, V.; Garaeva, L.; Shlikht, A.; Shtam, T. Proteome of Glioblastoma-Derived Exosomes as a Source of Biomarkers. Biomedicines 2020, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; Lim, J.W.E.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteom. 2006, 6, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Day, T.; Kopp, N.; van Bodegom, D.; Davids, M.S.; Ryan, J.; Bird, L.; Kommajosyula, N.; Weigert, O.; Yoda, A.; et al. BCL2 suppresses PARP1 function and nonapoptotic cell death. Cancer Res. 2012, 72, 4193–4203. [Google Scholar] [CrossRef] [Green Version]
- Hashemi-Niasari, F.; Rabbani-Chadegani, A.; Razmi, M.; Fallah, S. Synergy of theophylline reduces necrotic effect of berberine, induces cell cycle arrest and PARP, HMGB1, Bcl-2 family mediated apoptosis in MDA-MB-231 breast cancer cells. Biomed. Pharmacother. 2018, 106, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Murad, H.; Hawat, M.; Ekhtiar, A.; AlJapawe, A.; Abbas, A.; Darwish, H.; Sbenati, O.; Ghannam, A. Induction of G1-phase cell cycle arrest and apoptosis pathway in MDA-MB-231 human breast cancer cells by sulfated polysaccharide extracted from Laurencia papillosa. Cancer Cell Int. 2016, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fofaria, N.M.; Kim, S.H.; Srivastava, S.K. Piperine causes G1 phase cell cycle arrest and apoptosis in melanoma cells through checkpoint kinase-1 activation. PLoS ONE 2014, 9, e94298. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Xie, X.; Pitner, M.K.; Kondo, K.; Dadbin, A.; Lee, J.; Saso, H.; Smith, P.D.; Dalby, K.N.; Ueno, N.T. MEK Inhibitor Selumetinib (AZD6244; ARRY-142886) Prevents Lung Metastasis in a Triple-Negative Breast Cancer Xenograft Model. Mol. Cancer Ther. 2015, 14, 2773–2781. [Google Scholar] [CrossRef] [Green Version]
- Holt, S.V.; Logié, A.; Odedra, R.; Heier, A.; Heaton, S.P.; Alferez, D.; Davies, B.R.; Wilkinson, R.W.; Smith, P.D. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br. J. Cancer. 2012, 106, 858–866. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression of Factor | U87-Selu Exo (Fold) | Selumetinib (Fold) |
|---|---|---|
| p27 | 4.2 ± 0.3 | 5.1 ± 1.2 |
| PCNA | 0.6 ± 0.1 | 0.7 ± 0.1 |
| Cyclin D1 | 0.1 ± 0.2 | 0.1 ± 0.2 |
| p-p38 | 10.8 ± 6.5 | 7.8 ± 4.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Bae, K.; Baek, A.-R.; Kwon, E.-B.; Kim, Y.-H.; Nam, S.-W.; Lee, G.H.; Chang, Y. Glioblastoma-Derived Exosomes as Nanopharmaceutics for Improved Glioma Treatment. Pharmaceutics 2022, 14, 1002. https://doi.org/10.3390/pharmaceutics14051002
Lee H, Bae K, Baek A-R, Kwon E-B, Kim Y-H, Nam S-W, Lee GH, Chang Y. Glioblastoma-Derived Exosomes as Nanopharmaceutics for Improved Glioma Treatment. Pharmaceutics. 2022; 14(5):1002. https://doi.org/10.3390/pharmaceutics14051002
Chicago/Turabian StyleLee, Hyeji, Kanghye Bae, Ah-Rum Baek, Eun-Bin Kwon, Yeoun-Hee Kim, Sung-Wook Nam, Gang Ho Lee, and Yongmin Chang. 2022. "Glioblastoma-Derived Exosomes as Nanopharmaceutics for Improved Glioma Treatment" Pharmaceutics 14, no. 5: 1002. https://doi.org/10.3390/pharmaceutics14051002