
Regulation of p27 (Kip1) by Ubiquitin E3 Ligase RNF6


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Constructs
2.3. In Vitro Ubiquitination
2.4. Cell Cycle Analysis
2.5. Transient Transfections
2.6. Western Blotting, Immunoprecipitation and Antibodies
2.7. Quantitative Real-Time RT-PCR
2.8. In Vitro Cell Proliferation Assays
2.9. Immunofluorscence Staining
2.10. Cytoplasmic Nuclear Fractionation
2.11. Statistical Analyses
3. Results
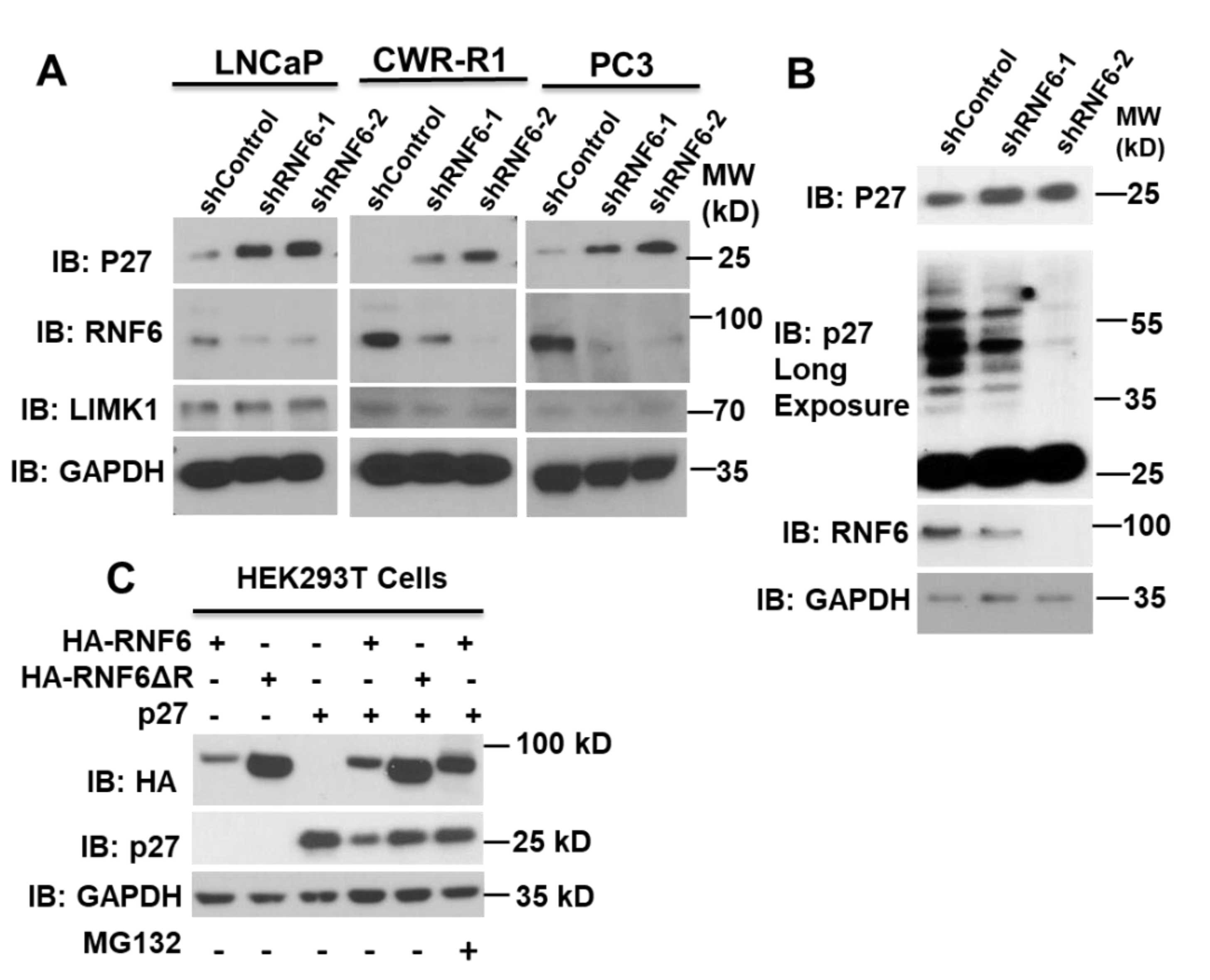
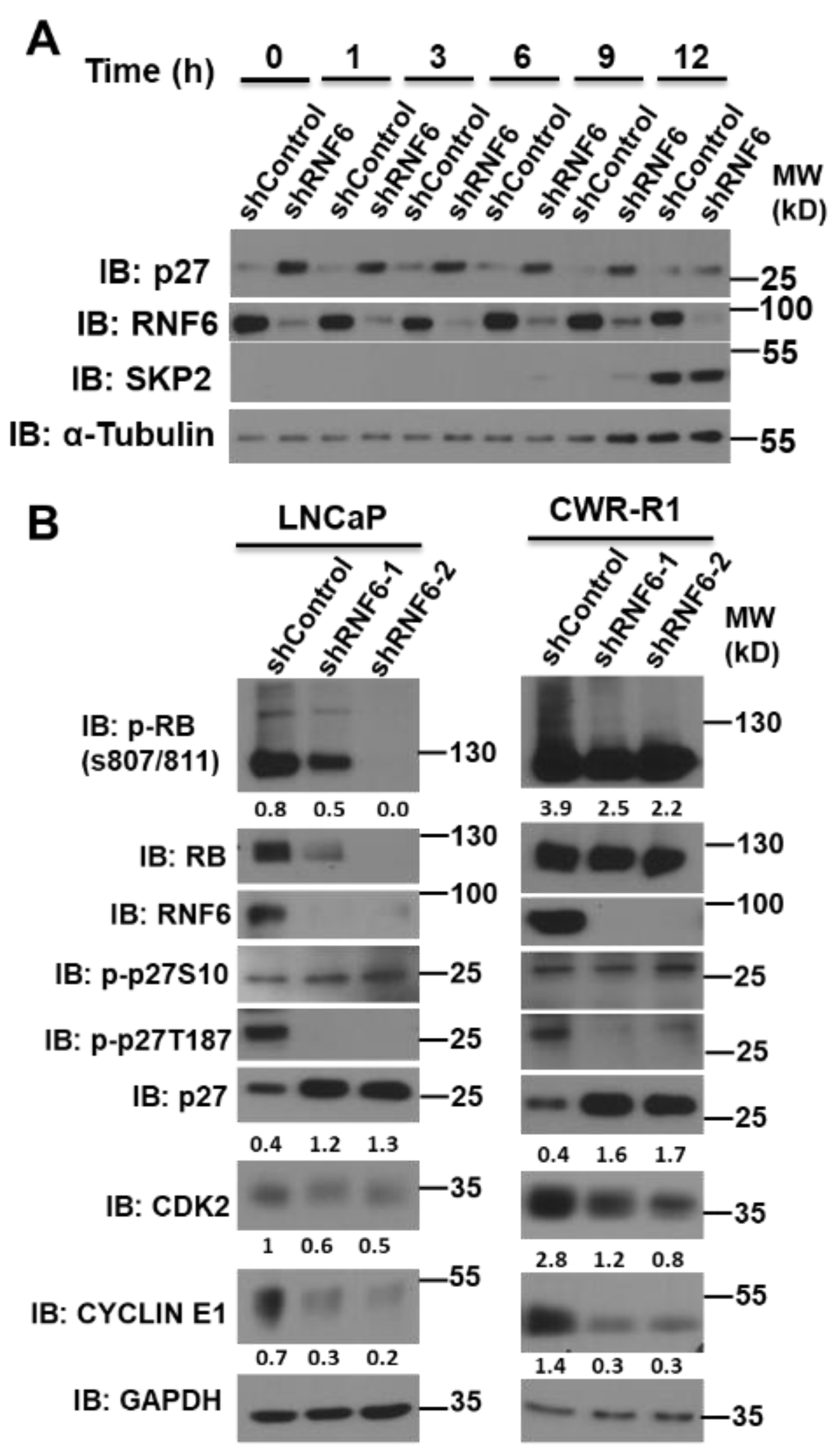
3.1. Knockdown of RNF6 Leads to Increased Stability of p27
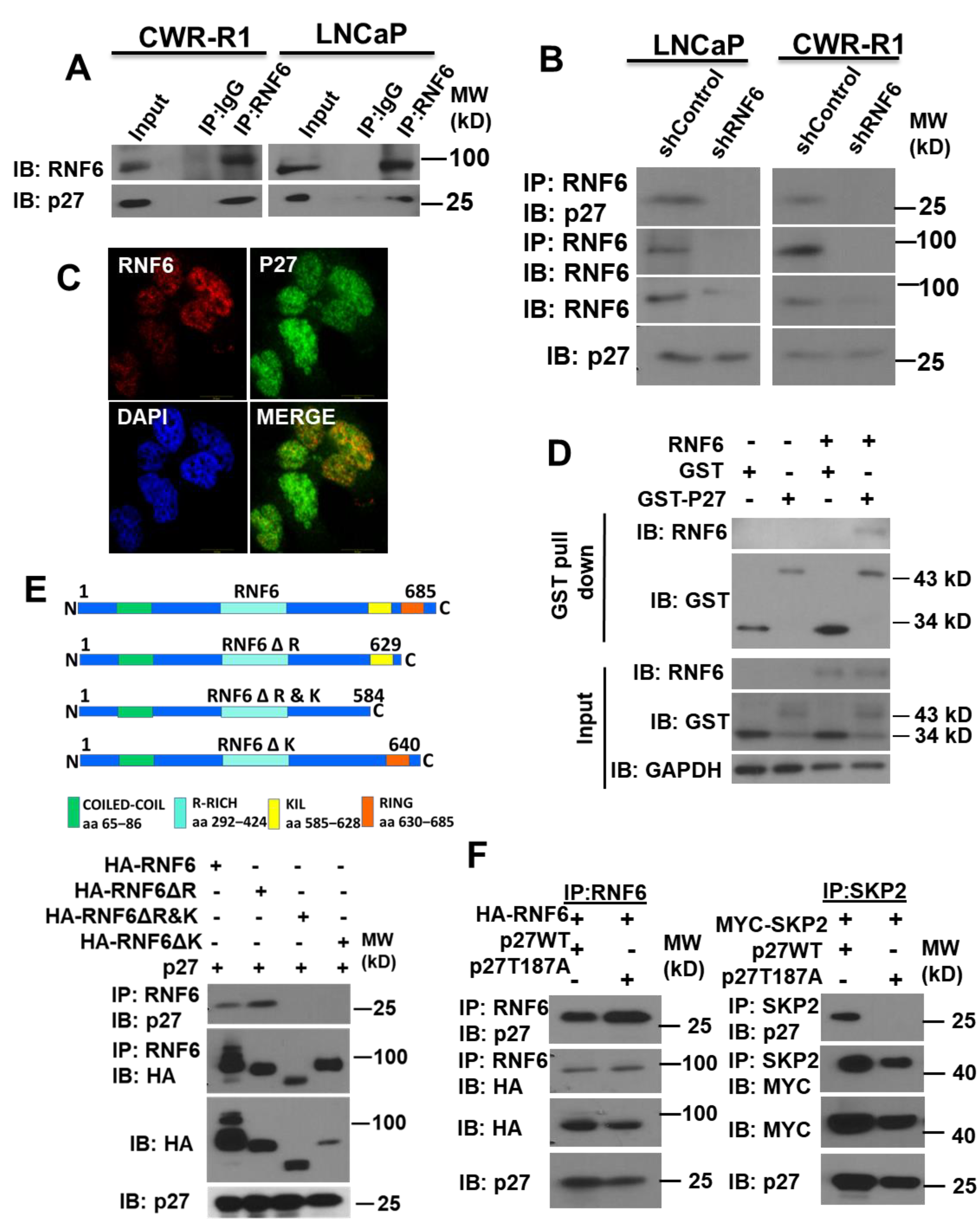
3.2. RNF6 Interacts with P27 via Its KIL Motif in a Phosphorylation-Independent Manner
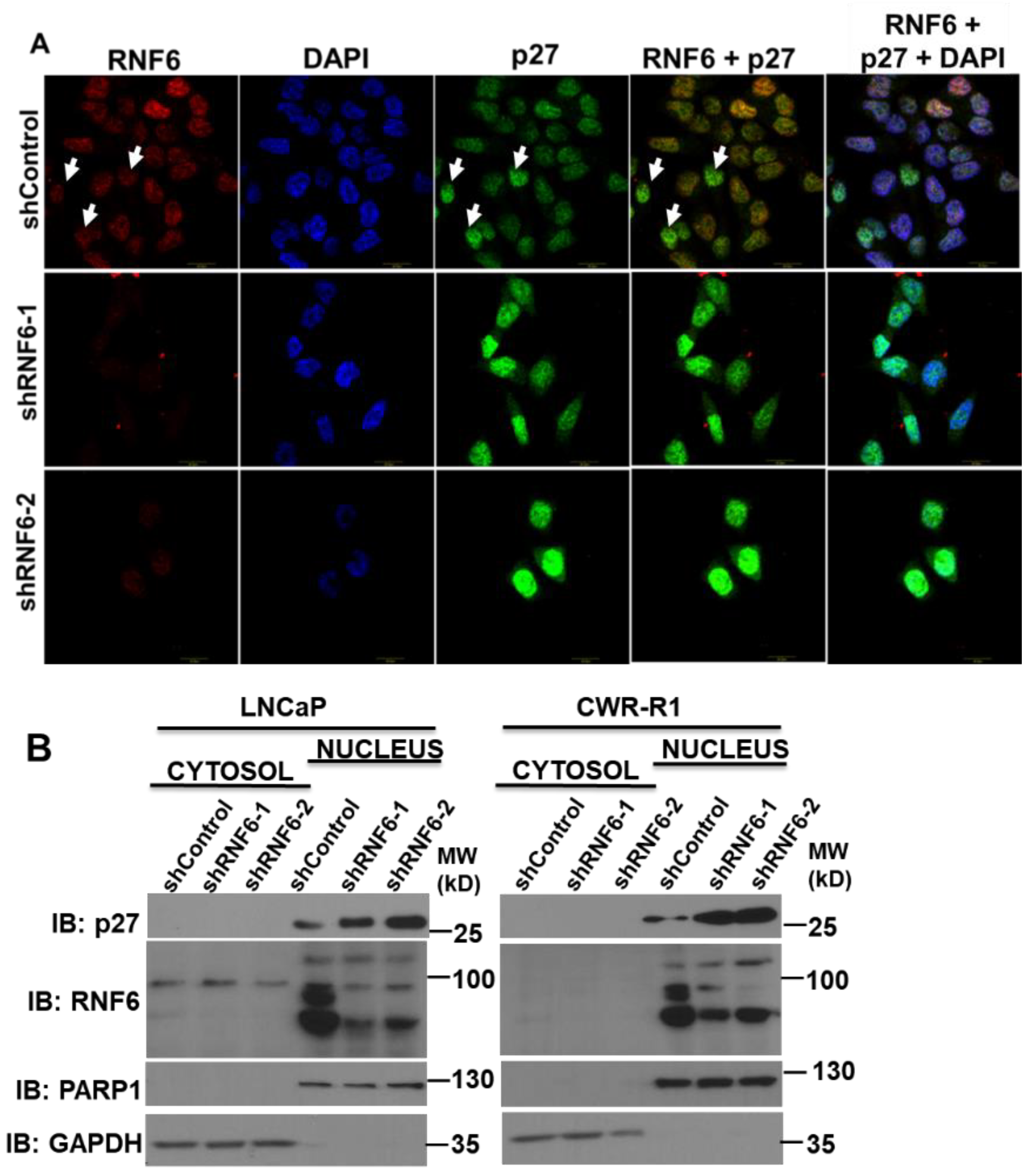
3.3. RNF6 Knockdown Leads to Accumulation of P27 in the Nucleus
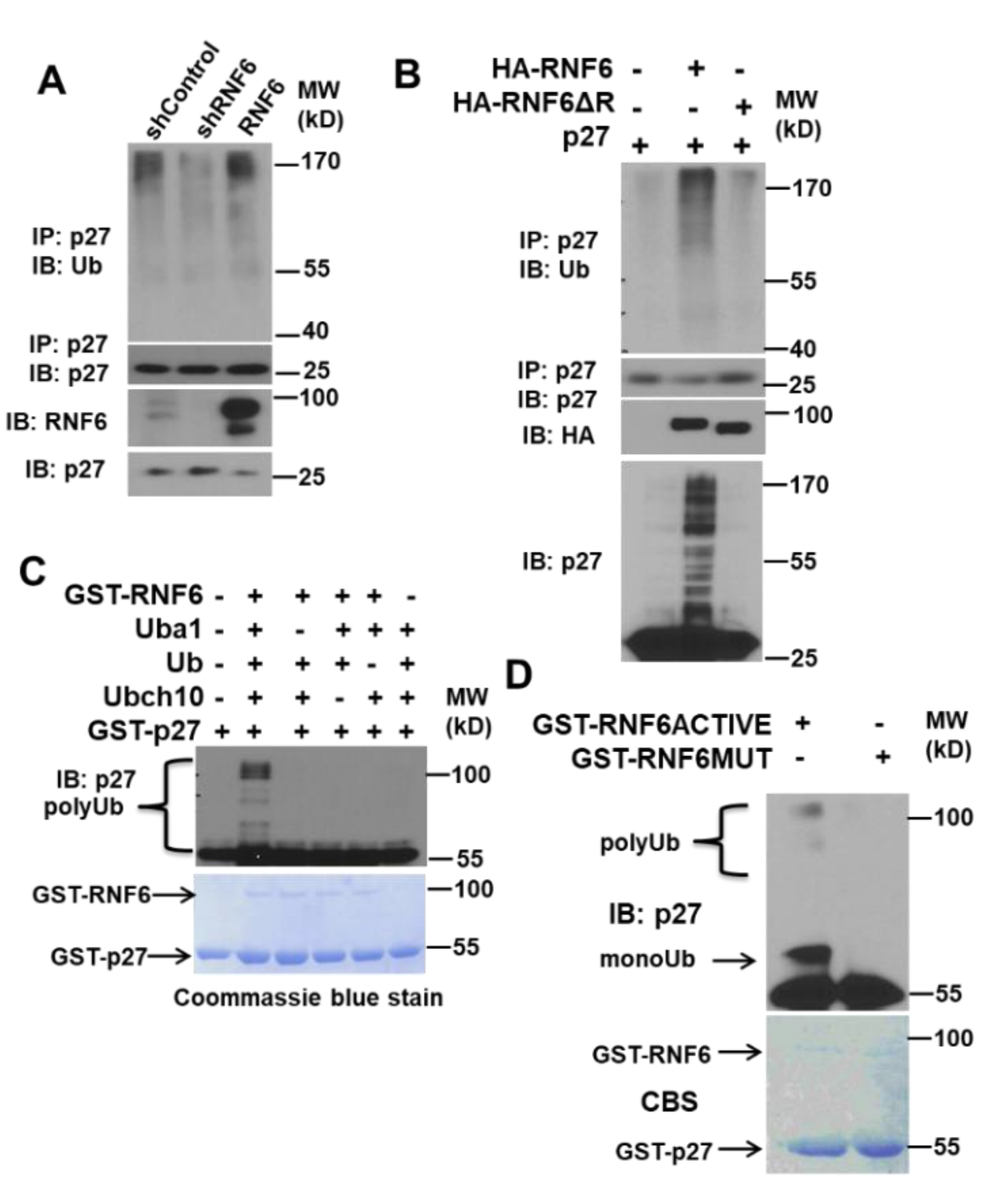
3.4. RNF6 Polyubiquitinates P27 In Vivo and In Vitro
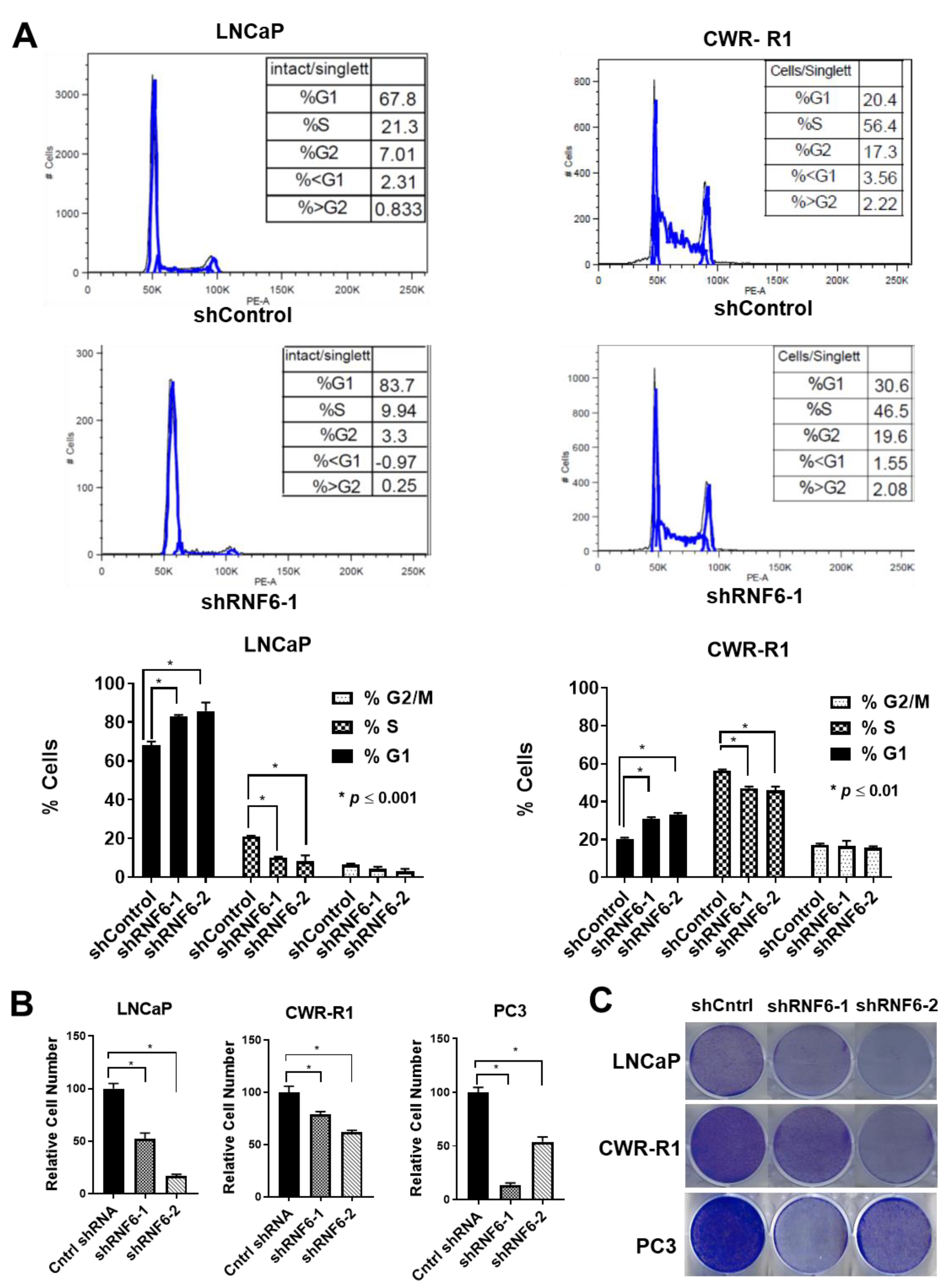
3.5. RNF6 Knockdown Leads to P27 Accumulation in Early G1 and to Reduced Rb Phosphorylation
3.6. Knockdown of RNF6 Leads to G1/S Block and to Reduced Cell Proliferation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiao, B.; Taniguchi-Ishigaki, N.; Güngör, C.; Peters, M.A.; Chen, Y.-W.; Riethdorf, S.; Drung, A.; Ahronian, L.G.; Shin, J.; Pagnis, R.; et al. Functional activity of RLIM/Rnf12 is regulated by phosphorylation-dependent nucleocytoplasmic shuttling. Mol. Biol. Cell 2013, 24, 3085–3096. [Google Scholar] [CrossRef]
- Macdonald, D.H.; Lahiri, D.; Sampath, A.; Chase, A.; Sohal, J.; Cross, N.C. Cloning and characterization of RNF6, a novel RING finger gene mapping to 13q12. Genomics 1999, 58, 94–97. [Google Scholar] [CrossRef]
- Tursun, B.; Schlüter, A.; Peters, M.A.; Viehweger, B.; Ostendorff, H.P.; Soosairajah, J.; Drung, A.; Bossenz, M.; Johnsen, S.A.; Schweizer, M.; et al. The ubiquitin ligase Rnf6 regulates local LIM kinase 1 levels in axonal growth cones. Genes Dev. 2005, 19, 2307–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Chen, H.; et al. Regulation of Androgen Receptor Transcriptional Activity and Specificity by RNF6-Induced Ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Uematsu, A.; Yamanaka, S.; Imamura, M.; Nakajima, T.; Doi, K.; Yasuoka, S.; Takahashi, C.; Takeda, H.; Sawasaki, T. Establishment of a Wheat Cell-Free Synthesized Protein Array Containing 250 Human and Mouse E3 Ubiquitin Ligases to Identify Novel Interaction between E3 Ligases and Substrate Proteins. PLoS ONE 2016, 11, e0156718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, P.; Vidal, F.; Martin, L.; Lopez-Fernandez, L.; Rual, J.-F.; Rosen, B.S.; Cuzin, F.; Rassoulzadegan, M. Gene Control in Germinal Differentiation: Rnf6, a Transcription Regulatory Protein in the Mouse Sertoli Cell. Mol. Cell. Biol. 2002, 22, 3488–3496. [Google Scholar] [CrossRef] [Green Version]
- Susaki, E.; Nakayama, K.I. Functional similarities and uniqueness of p27 and p57: Insight from a knock-in mouse model. Cell Cycle 2009, 8, 2497–2501. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Hengst, L.; Reed, S.I. Inhibitors of the Cip/Kip Family. Curr. Top. Microbiol. Immunol. 1998, 227, 25–41. [Google Scholar] [CrossRef]
- Polyak, K.; Kato, J.Y.; Solomon, M.J.; Sherr, C.J.; Massague, J.; Roberts, J.M.; Koff, A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994, 8, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Nigg, E. Targets of cyclin-dependent protein kinases. Curr. Opin. Cell Biol. 1993, 5, 187–193. [Google Scholar] [CrossRef]
- James, M.K.; Ray, A.; Leznova, D.; Blain, S.W. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol. Cell Biol. 2008, 28, 498–510. [Google Scholar] [CrossRef] [Green Version]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilenšek, Z.; Keidel, E.-M.; Waddell, M.B.; Jäkel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-Inhibitory Activity and Stability of p27Kip1 Are Directly Regulated by Oncogenic Tyrosine Kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Kiyokawa, H.; Kineman, R.D.; OManova-Todorova, K.; Soares, V.C.; Hoffman, E.S.; Ono, M.; Khanam, D.; Hayday, A.C.; AFrohman, L.; Koff, A. Enhanced Growth of Mice Lacking the Cyclin-Dependent Kinase Inhibitor Function of p27Kip1. Cell 1996, 85, 721–732. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996, 85, 707. [Google Scholar] [CrossRef] [Green Version]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Slingerland, J.; Pagano, M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 2000, 183, 10. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell. Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef]
- Malek, N.P.; Sundberg, H.; McGrew, S.; Nakayama, K.; Kyriakidis, T.R.; Roberts, J.M. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 2001, 413, 323–327. [Google Scholar] [CrossRef]
- Bondar, T.; Kalinina, A.; Khair, L.; Kopanja, D.; Nag, A.; Bagchi, S.; Raychaudhuri, P. Cul4A and DDB1 associate with Skp2 to target p27Kip1 for proteolysis involving the COP9 signalosome. Mol. Cell. Biol. 2006, 26, 2531. [Google Scholar] [CrossRef] [Green Version]
- Gstaiger, M.; Jordan, R.; Lim, M.; Catzavelos, C.; Mestan, J.; Slingerland, J.; Krek, W. Skp2 is oncogenic and overexpressed in human cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 5043–5048. [Google Scholar] [CrossRef] [Green Version]
- Imaki, H.; Nakayama, K.; Delehouzee, S.; Handa, H.; Kitagawa, M.; Kamura, T.; INakayama, K. Cell cycle-dependent regulation of the Skp2 promoter by GA-binding protein. Cancer Res. 2003, 63, 4607–4613. [Google Scholar]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Matsumoto, M.; Nakamichi, I.; Kitagawa, K.; Shirane, M.; Tsunematsu, R.; Tsukiyama, T.; Ishida, N.; et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000, 19, 2069. [Google Scholar] [CrossRef] [Green Version]
- Gregory, C.W.; Johnson, R.T., Jr.; Mohler, J.L.; French, F.S.; Wilson, E.M. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001, 61, 2892–2898. [Google Scholar]
- Guo, Z.; Dai, B.; Jiang, T.; Xu, K.; Xie, Y.; Kim, O.; Nesheiwat, I.; Kong, X.; Melamed, J.; Handratta, V.D.; et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006, 10, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 Is a RING Finger-dependent Ubiquitin Protein Ligase for Itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Pungaliya, P.; Li, X.; Uquillas, C.; Mutton, L.N.; Rubin, E.H.; Bieberich, C. Ubiquitination by TOPORS Regulates the Prostate Tumor Suppressor NKX3.1. J. Biol. Chem. 2008, 283, 4834–4840. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, K.; Linn, D.E.; Yang, X.; Guo, Z.; Shimelis, H.; Nakanishi, T.; Ross, D.D.; Chen, H.; Fazli, L.; et al. The 44-kDa Pim-1 Kinase Phosphorylates BCRP/ABCG2 and Thereby Promotes Its Multimerization and Drug-resistant Activity in Human Prostate Cancer Cells. J. Biol. Chem. 2008, 283, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Choo, Y.S.; Zhang, Z. Detection of Protein Ubiquitination. J. Vis. Exp. 2009, 30, e1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, P.; Horton, W.A. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2009, 2, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Faltermeier, C.M.; Mendelsohn, L.; Porter, P.L.; Clurman, B.E.; Roberts, J.M. Mislocalization of p27 to the cytoplasm of breast cancer cells confers resistance to anti-HER2 targeted therapy. Oncotarget 2014, 5, 12704–12714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Besser, A.H.; Wander, S.A.; Sun, J.; Zhou, W.; Wang, B.; Ince, T.; Durante, M.; Guo, W.; Mills, G.; et al. Cytoplasmic p27 promotes epithelial–mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene 2015, 34, 5447–5459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.-H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef]
- Deng, X.; Mercer, S.E.; Shah, S.; Ewton, D.Z.; Friedman, E. The Cyclin-dependent Kinase Inhibitor p27Kip1 Is Stabilized in G0 by Mirk/dyrk1B Kinase. J. Biol. Chem. 2004, 279, 22498–22504. [Google Scholar] [CrossRef] [Green Version]
- Hotte, S.J.; Saad, F. Current Management of Castrate-Resistant Prostate Cancer. Curr. Oncol. 2010, 17 (Suppl. 2), 72–79. [Google Scholar] [CrossRef]
- Freedland, S.J.; de Gregorio, F.; Sacoolidge, J.C.; Elshimali, Y.I.E.; Csathy, G.S.; Elashoff, D.A.; Reiter, R.E.; Aronson, W.J. Predicting biochemical recurrence after radical prostatectomy for patients with organ-confined disease using p27 expression. Urology 2003, 61, 1187–1192. [Google Scholar] [CrossRef]
- Hara, T.; Kamura, T.; Nakayama, K.; Oshikawa, K.; Hatakeyama, S.; Nakayama, K. Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J. Biol. Chem. 2001, 276, 48937–48943. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jonasch, E.; Alexander, A.; Short, J.D.; Cai, S.; Wen, S.; Tsavachidou, D.; Tamboli, P.; Czerniak, B.A.; Do, K.A.; et al. Cytoplasmic Sequestration of p27 via AKT Phosphorylation in Renal Cell Carcinoma. Clin. Cancer Res. 2008, 15, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Liu, Y.; Ford, O.H., III; Mohler, J.L.; Whang, Y.E. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate 2006, 66, 1292–1301. [Google Scholar] [CrossRef]
- Piva, R.; Cancelli, I.; Cavalla, P.; Bortolotto, S.; Dominguez, J.; Draetta, G.F.; Schiffer, D. Proteasome-dependent degradation of p27/kip1 in gliomas. J. Neuropathol. Exp. Neurol. 1999, 58, 691. [Google Scholar] [CrossRef] [Green Version]
- Hidaka, T.; Hama, S.; Shrestha, P.; Saito, T.; Kajiwara, Y.; Yamasaki, F.; Sugiyama, K.; Kurisu, K. The combination of low cytoplasmic and high nuclear expression of p27 predicts a better prognosis in high-grade astrocytoma. Anticancer Res. 2009, 29, 597–603. [Google Scholar]
- Hong, F.; Larrea, M.D.; Doughty, C.; Kwiatkowski, D.J.; Squillace, R.; Slingerland, J.M. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol. Cell 2008, 30, 701. [Google Scholar] [CrossRef]
- Schiappacassi, M.; Lovisa, S.; Lovat, F.; Fabris, L.; Colombatti, A.; Belletti, B.; Baldassarre, G. Role of T198 Modification in the Regulation of p27Kip1 Protein Stability and Function. PLoS ONE 2011, 6, e17673. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar] [CrossRef]
- McAllister, S.S.; Becker-Hapak, M.; Pintucci, G.; Pagano, M.; Dowdy, S.F. Novel p27 kip1 C-Terminal Scatter Domain Mediates Rac-Dependent Cell Migration Independent of Cell Cycle Arrest Functions. Mol. Cell. Biol. 2003, 23, 216–228. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.K.; Kotchetkov, R.; Cariou, S.; Resch, A.; Lupetti, R.; Beniston, R.G.; Melchior, F.; Hengst, L.; Slingerland, J.M. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol. Biol. Cell 2003, 14, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Kamura, T.; Hara, T.; Matsumoto, M.; Ishida, N.; Okumura, F.; Hatakeyama, S.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27Kip1 at G1 phase. Nat. Cell Biol. 2004, 6, 1229–1235. [Google Scholar] [CrossRef]
- Tsvetkov, L.M.; Yeh, K.H.; Lee, S.J.; Sun, H.; Zhang, H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999, 9, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Sequences |
|---|---|
| P27T187A | GTTCTGTGGAGCAGGCGCCCAAGAAGCCTG sense |
| CAGGCTTCTTGGGCGCCTGCTCCACAGAAC antisense | |
| RNF6ΔKIL | TGAAACTGGAACACTACCCAAAATCTGTAGTGTTTGTA sense |
| TACAAACACTACAGATTTTGGGTAGTGTTCCAGTTTCA antisense |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshmukh, D.; Xu, J.; Yang, X.; Shimelis, H.; Fang, S.; Qiu, Y. Regulation of p27 (Kip1) by Ubiquitin E3 Ligase RNF6. Pharmaceutics 2022, 14, 802. https://doi.org/10.3390/pharmaceutics14040802
Deshmukh D, Xu J, Yang X, Shimelis H, Fang S, Qiu Y. Regulation of p27 (Kip1) by Ubiquitin E3 Ligase RNF6. Pharmaceutics. 2022; 14(4):802. https://doi.org/10.3390/pharmaceutics14040802
Chicago/Turabian StyleDeshmukh, Dhanraj, Jin Xu, Xi Yang, Hermela Shimelis, Shengyun Fang, and Yun Qiu. 2022. "Regulation of p27 (Kip1) by Ubiquitin E3 Ligase RNF6" Pharmaceutics 14, no. 4: 802. https://doi.org/10.3390/pharmaceutics14040802