
Toxicokinetics of β-Amanitin in Mice and In Vitro Drug–Drug Interaction Potential

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Toxicokinetic Study of β-Amanitin in Male ICR Mice
2.2.1. Toxicokinetics Study
2.2.2. Tissue Distribution Experiments
2.2.3. Excretion Experiments
2.2.4. Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS) of β-Amanitin in Mouse Plasma, Tissue, Urine, and Feces Samples
2.2.5. Toxicokinetic Parameters and Statistical Analysis
2.3. Transport Studies of α- and β-Amanitin
2.4. Inhibitory Effects of β-Amanitin on the Major Drug-Metabolizing Enzymes and Transporters
3. Results
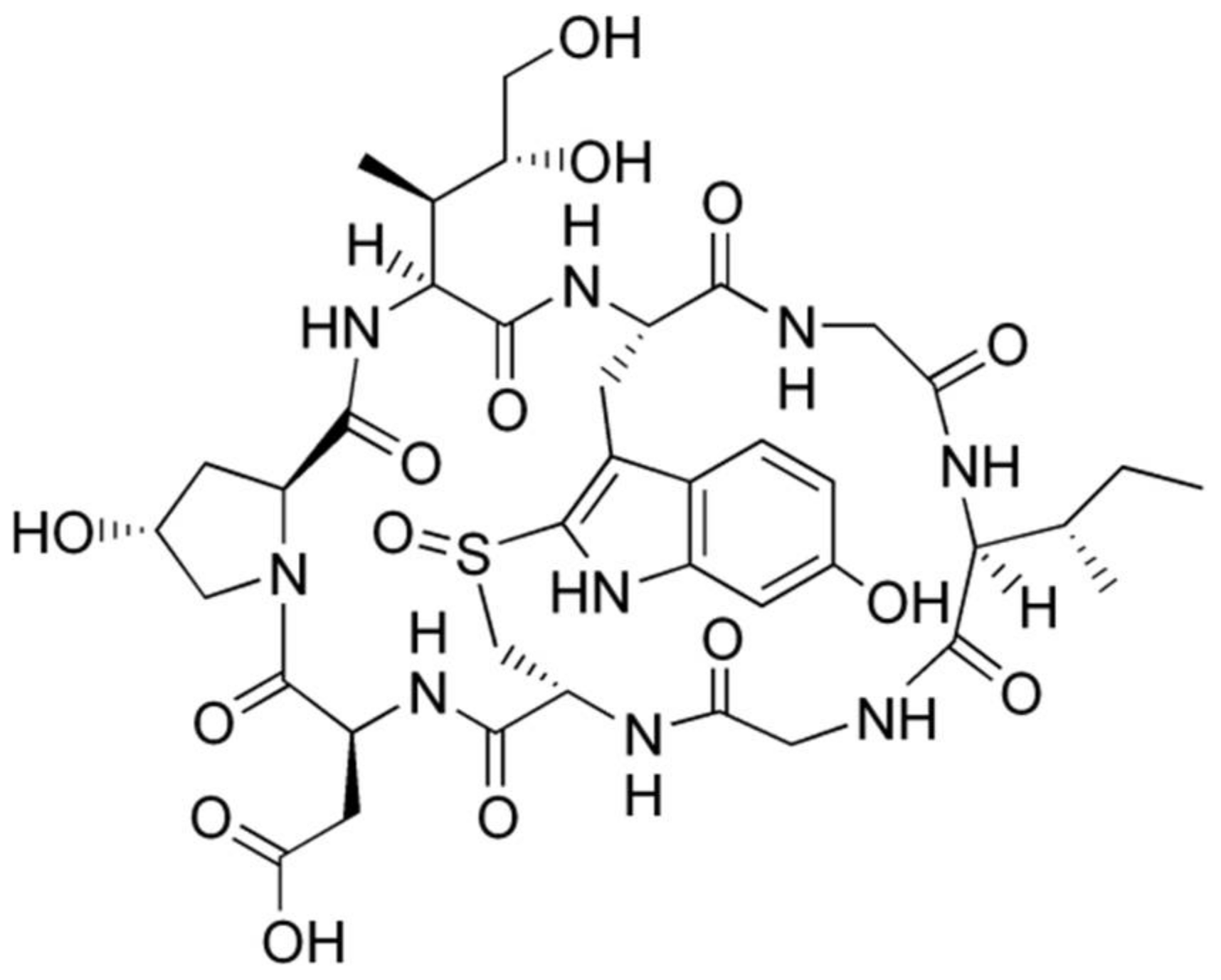
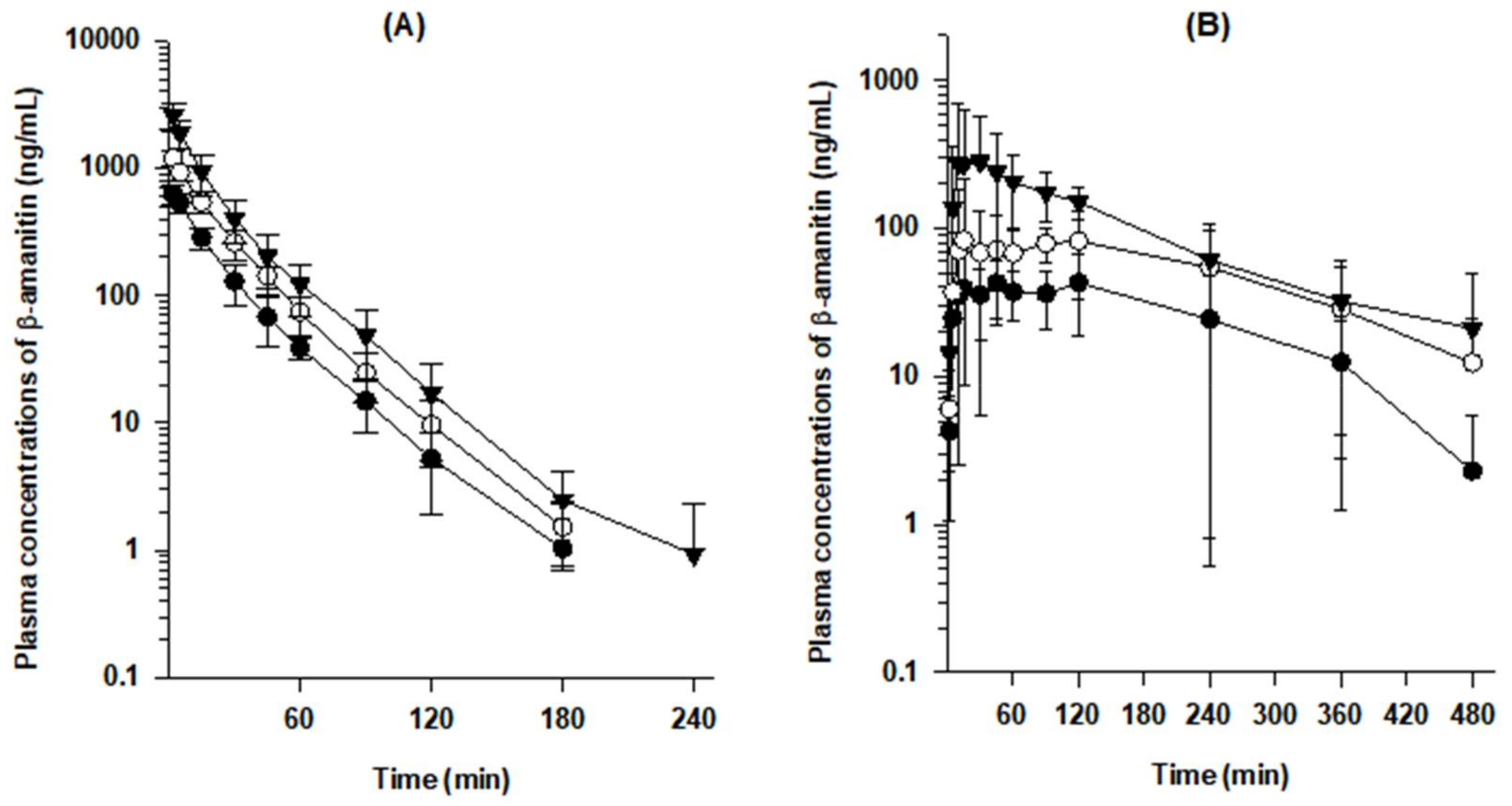
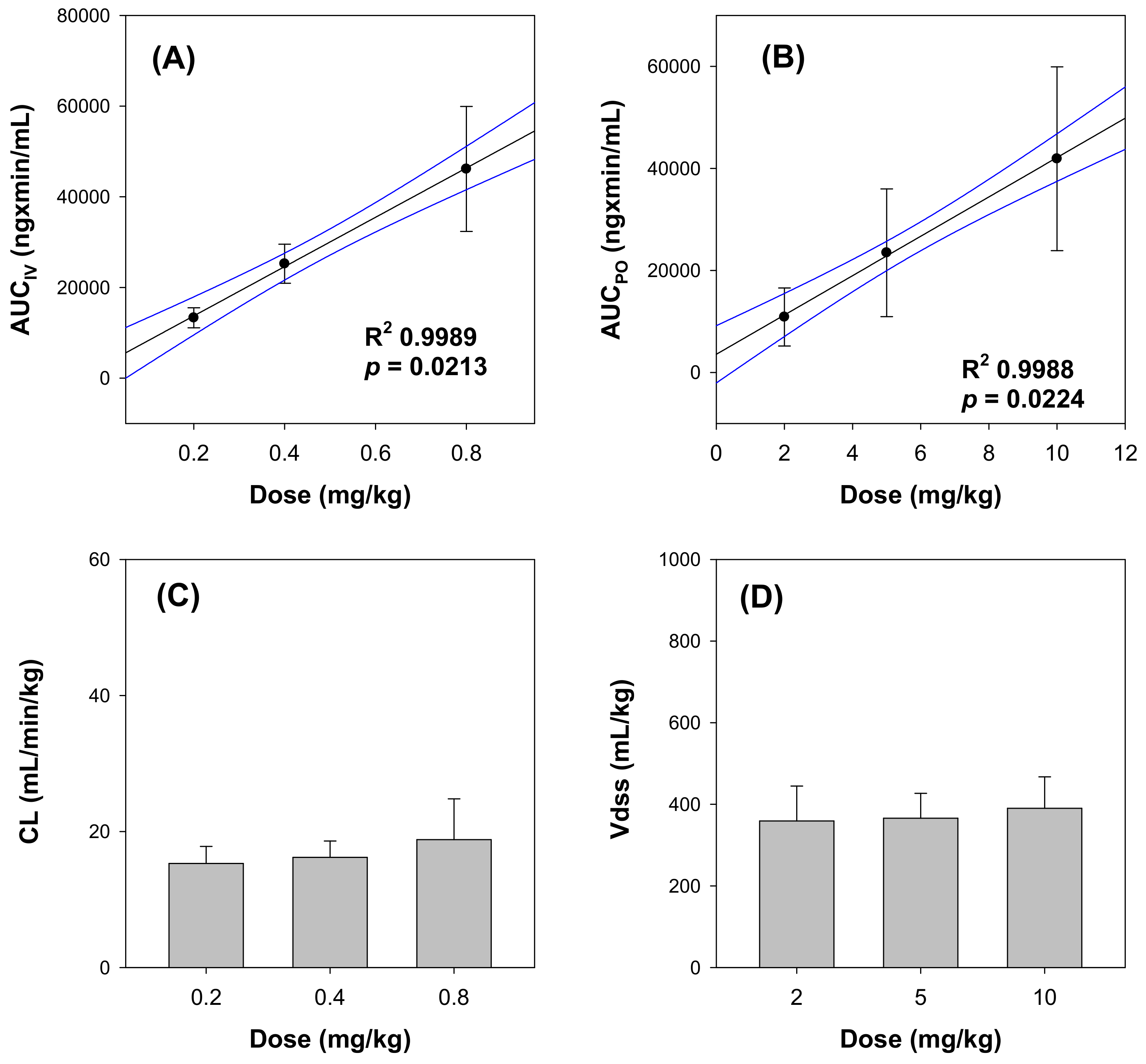
3.1. Toxicokinetics of β-Amanitin in Mice
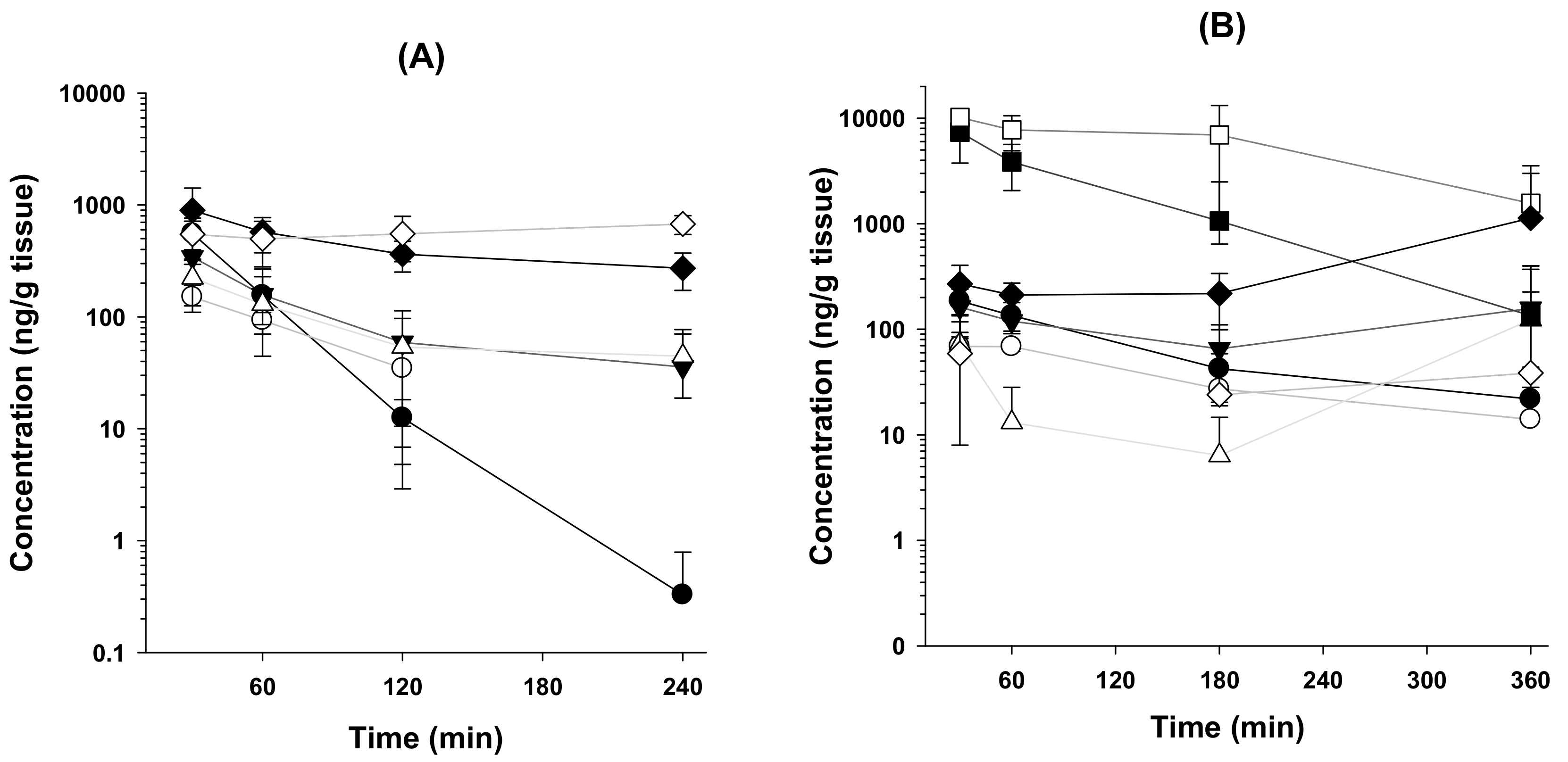
3.2. Tissue Distribution of β-Amanitin in Mice
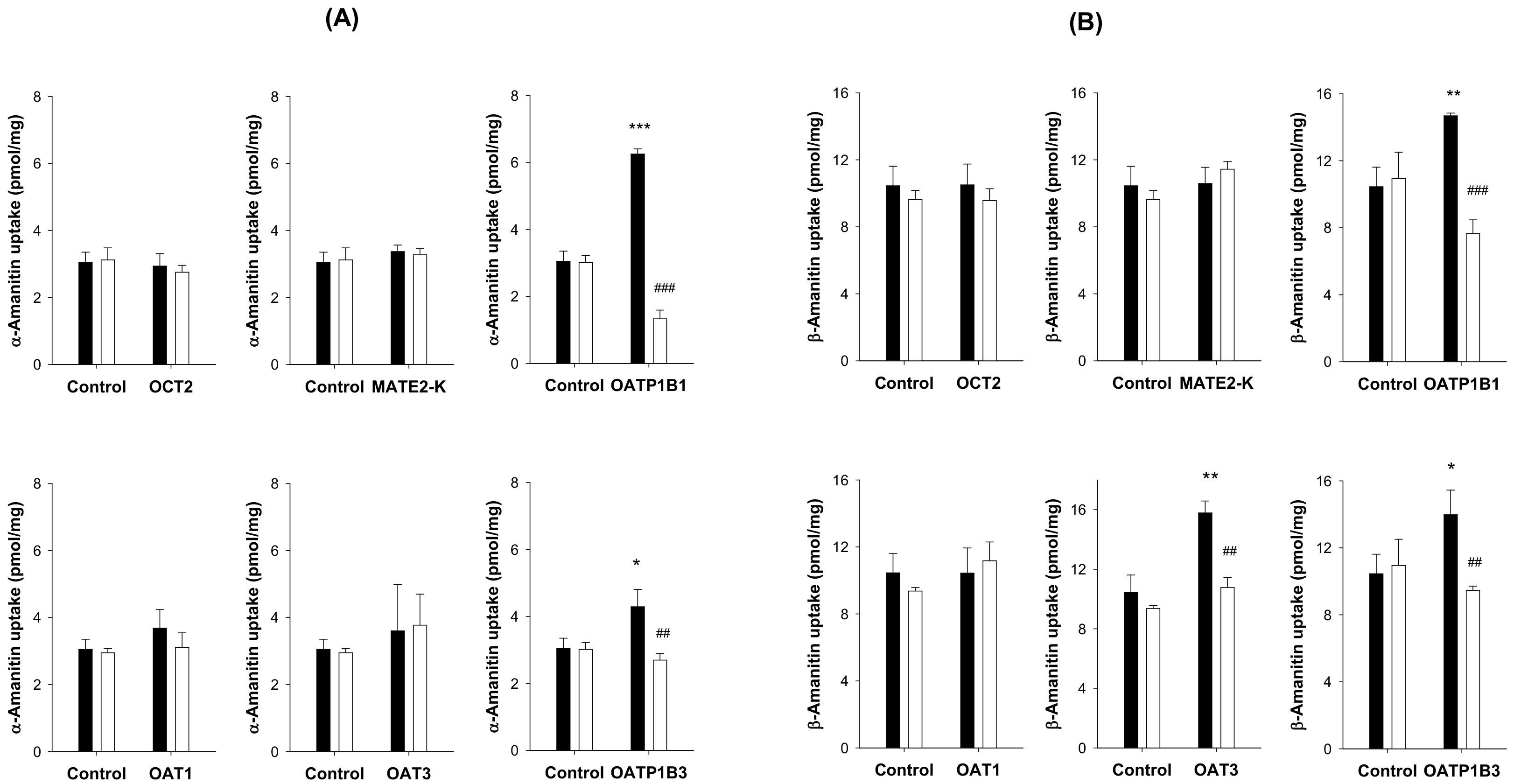
3.3. Transport Substrate of α- and β-Amanitin for OATP1B1 and OATP1B3
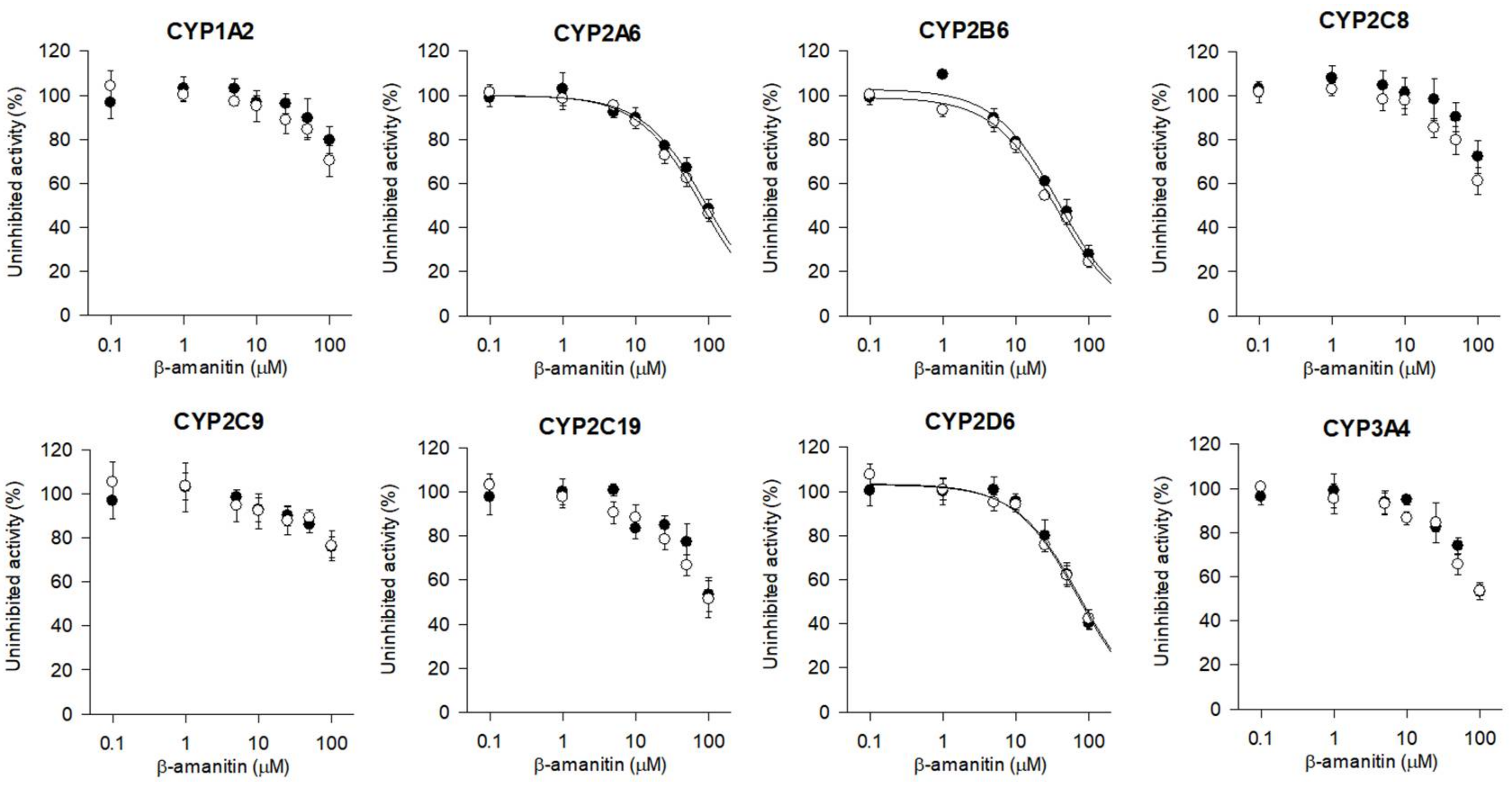
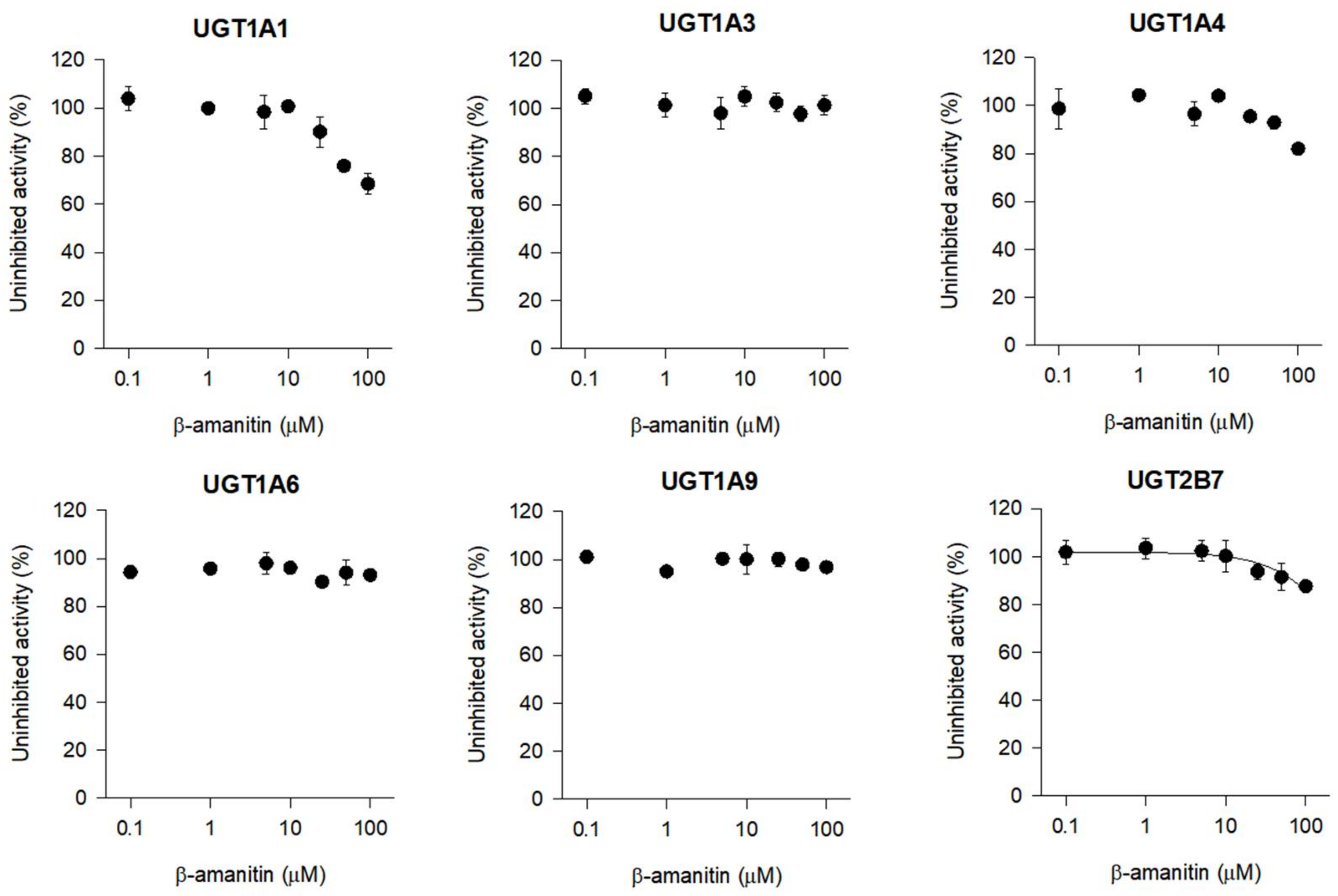
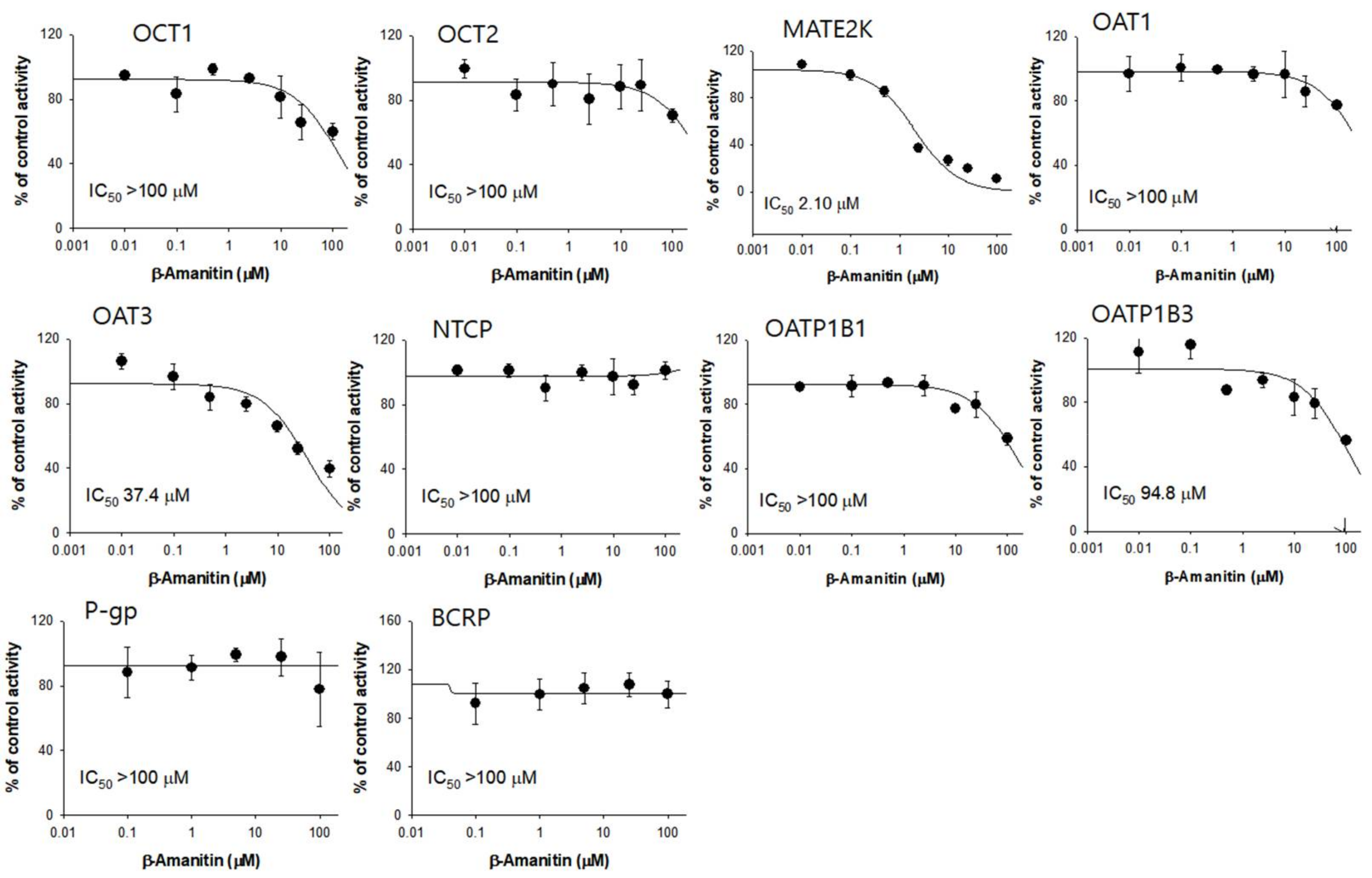
3.4. The Inhibitory Effects of β-Amanitin on the Major Drug-Metabolizing Enzymes and Transporters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, K.J.; Guss, D.A. Mycotoxins revisited: Part I. J. Emerg. Med. 2005, 28, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Costa, V.M.; Carvalho, A.; Baptista, P.; de Pinho, P.G.; Bastos, M.D.; Carvalho, F. Amanita phalloides poisoning: Mechanisms of toxicity and treatment. Food Chem. Toxicol. 2015, 86, 41–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, M.; Suzuki, Y.; Kaneko, R.; Ogawa, T.; Hattori, H.; Seno, H.; Ishii, A. Simple and rapid analysis of amatoxins using UPLC–MS–MS. Forensic Toxicol. 2012, 30, 185–192. [Google Scholar] [CrossRef]
- Mas, A. Mushrooms, amatoxins and the liver. J. Hepatol. 2005, 42, 166–169. [Google Scholar] [CrossRef]
- Kaya, E.; Karahan, S.; Bayram, R.; Yaykasli, K.O.; Colakoglu, S.; Saritas, A. Amatoxin and phallotoxin concentration in Amanita phalloides spores and tissues. Toxicol. Ind. Health 2015, 31, 1172–1177. [Google Scholar] [CrossRef]
- Kaya, E.; Surmen, M.G.; Yaykasli, K.O.; Karahan, S.; Oktay, M.; Turan, H.; Colakoglu, S.; Erdem, H. Dermal absorption and toxicity of alpha amanitin in mice. Cutan. Ocul. Toxicol. 2014, 33, 154–160. [Google Scholar] [CrossRef]
- Yilmaz, I.; Ermis, F.; Akata, I.; Kaya, E. A case study: What doses of Amanita phalloides and amatoxins are lethal to humans? Wilderness Environ. Med. 2015, 26, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, I.; Kaya, E.; Sinirlioglu, Z.A.; Bayram, R.; Surmen, M.G.; Colakoglu, S. Clinical importance of toxin concentration in Amanita verna mushroom. Toxicon 2014, 87, 68–75. [Google Scholar] [CrossRef]
- Sun, J.; Niu, Y.-M.; Zhang, Y.-T.; Li, H.-J.; Yin, Y.; Zhang, Y.-Z.; Ma, P.-B.; Zhou, J.; Huang, L.; Zhang, H.-S. Toxicity and toxicokinetics of Amanita exitialis in beagle dogs. Toxicon 2018, 143, 59–67. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.; Na, A.-Y.; Sohn, C.H.; Lee, S.; Lee, H.S. Identification of decrease in TRiC proteins as novel targets of alpha-amanitin-derived hepatotoxicity by comparative proteomic analysis in vitro. Toxins 2021, 13, 197. [Google Scholar] [CrossRef]
- Le Dare, B.; Ferron, P.-J.; Gicquel, T. Toxic effects of amanitins: Repurposing toxicities toward new therapeutics. Toxins 2021, 13, 417. [Google Scholar] [CrossRef]
- Kaya, E.; Hancı, M.; Karahan, S.; Bayram, S.; Yaykaşlı, K.O.; Sürmen, M.G. Thermostability of alpha amanitin in water and methanol. Eur. J. Basic Med. Sci. 2012, 2, 106–111. [Google Scholar] [CrossRef]
- Karlson-Stiber, C.; Persson, H. Cytotoxic fungi—An overview. Toxicon 2003, 42, 339–349. [Google Scholar] [CrossRef]
- Park, R.; Choi, W.-G.; Lee, M.S.; Cho, Y.-Y.; Lee, J.Y.; Kang, H.C.; Sohn, C.H.; Song, I.-S.; Lee, H.S. Pharmacokinetics of α-amanitin in mice using liquid chromatography-high resolution mass spectrometry and in vitro drug–drug interaction potentials. J. Toxicol. Environ. Health A 2021, 84, 821–835. [Google Scholar] [CrossRef]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef]
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally absorbed cyclic peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef]
- Mydlík, M.; Derzsiová, K. Liver and kidney damage in acute poisonings. BANTAO J. 2006, 4, 30–33. [Google Scholar]
- Li, C.; Qian, H.; Bao, T.; Yang, G.; Wang, S.; Liu, X. Simultaneous identification and characterization of amanita toxins using liquid chromatography-photodiode array detection-ion trap and time-of-flight mass spectrometry and its applications. Toxicol. Lett. 2018, 296, 95–104. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.-T.; Niu, Y.-M.; Li, H.-J.; Yin, Y.; Zhang, Y.-Z.; Ma, P.-B.; Zhou, J.; Lu, J.-J.; Zhang, H.-S. Effect of biliary drainage on the toxicity and toxicokinetics of Amanita exitialis in beagles. Toxins 2018, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Costa, V.M.; Baptista, P.; Bastos Mde, L.; Carvalho, F. Quantification of alpha-amanitin in biological samples by HPLC using simultaneous UV- diode array and electrochemical detection. J. Chromatogr. B 2015, 997, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wei, F.; Muhammad, S.; Yang, G.; Wang, S.; Liu, X. A cost-effective LC–MS/MS method for identification and quantification of α-amanitin in rat plasma: Application to toxicokinetic study. J. Chromatogr. B 2017, 1064, 36–39. [Google Scholar] [CrossRef]
- Bang, Y.Y.; Lee, M.S.; Lim, C.H.; Lee, H.S. Simultaneous determination of α-amanitin and β-amanitin in mouse plasma using liquid chromatography-high resolution mass spectrometry. Mass Spectro. Lett. 2021, 12, 112–117. [Google Scholar] [CrossRef]
- Maurer, H.H.; Schmitt, C.J.; Weber, A.A.; Kraemer, T. Validated electrospray liquid chromatographic–mass spectrometric assay for the determination of the mushroom toxins α-and β-amanitin in urine after immunoaffinity extraction. J. Chromatogr. B 2000, 748, 125–135. [Google Scholar] [CrossRef]
- Bambauer, T.P.; Wagmann, L.; Weber, A.A.; Meyer, M.R. Analysis of α- and β-amanitin in Human Plasma at Subnanogram per Milliliter Levels by Reversed Phase Ultra-High Performance Liquid Chromatography Coupled to Orbitrap Mass Spectrometry. Toxins 2020, 12, 671. [Google Scholar] [CrossRef]
- Xu, X.M.; Meng, Z.; Zhang, J.S.; Chen, Q.; Han, J.L. Analytical method development for α-amanitin and β-amanitin in plasma at ultra-trace level by online solid phase extraction-high performance liquid chromatography-triple quadrupole mass spectrometry and its application in poisoning events. J. Pharm. Biomed. Anal. 2020, 190, 113523. [Google Scholar] [CrossRef]
- Jaeger, A.; Jehl, F.; Flesch, F.; Sauder, P.; Kopferschmitt, J. Kinetics of amatoxins in human poisoning: Therapeutic implications. J. Toxicol. Clin. Toxicol. 1993, 31, 63–80. [Google Scholar] [CrossRef]
- Choi, M.K.; Song, I.S. Interactions of ginseng with therapeutic drugs. Arch. Pharm. Res. 2019, 42, 862–878. [Google Scholar] [CrossRef]
- Kang, Y.J.; Lee, C.H.; Park, S.J.; Lee, H.S.; Choi, M.K.; Song, I.S. Involvement of organic anion transporters in the pharmacokinetics and drug interaction of rosmarinic acid. Pharmaceutics 2021, 13, 83. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Letschert, K.; Faulstich, H.; Keller, D.; Keppler, D. Molecular characterization and inhibition of amanitin uptake into human hepatocytes. Toxicol. Sci. 2006, 91, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Choi, W.-G.; Kwon, M.; Lee, S.; Cho, Y.-Y.; Lee, J.Y.; Kang, H.C.; Song, I.-S.; Lee, H.S. In vitro inhibitory effects of APINACA on human major Cytochrome P450, UDP-Glucuronosyltransferase enzymes, and drug transporters. Molecules 2019, 24, 3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Park, R.; Jeon, J.H.; Cho, Y.Y.; Lee, J.Y.; Kang, H.C.; Song, I.S.; Lee, H.S. Inhibitory effect of AB-PINACA, indazole carboxamide synthetic cannabinoid, on human major drug-metabolizing enzymes and transporters. Pharmaceutics 2020, 12, 1036. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Jeong, H.U.; Choi, M.K.; Kwon, M.; Shin, Y.; Kim, J.H.; Lee, H.S. Interactions between cyazofamid and human drug transporters. J. Biochem. Mol. Toxicol. 2020, 34, e22459. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvez, M.M.; Jung, J.A.; Shin, H.J.; Kim, D.H.; Shin, J.-G. Characterization of 22 antituberculosis drugs for inhibitory interaction potential on organic anionic transporter polypeptide (OATP)-mediated uptake. Antimicrob. Agents Chemother. 2016, 60, 3096–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathialagan, S.; Piotrowski, M.A.; Tess, D.A.; Feng, B.; Litchfield, J.; Varma, M.V. Quantitative prediction of human renal clearance and drug-drug interactions of organic anion transporter substrates using in vitro transport data: A relative activity factor approach. Drug Metab. Dispos. 2017, 45, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, E.; Bayram, R.; Yaykaşli, K.O.; Yilmaz, I.; Bayram, S.; Yaykaşli, E.; Yavuz, M.Z.; Gepdiremen, A.A. Evaluation and comparison of alpha-and beta-amanitin toxicity on MCF-7 cell line. Turk. J. Med. Sci. 2014, 44, 728–732. [Google Scholar] [CrossRef]
- Vetter, J. Toxins of Amanita phalloides. Toxicon 1998, 36, 13–24. [Google Scholar] [CrossRef]
- Faulstich, H.; Kirchner, K.; Derenzini, M. Strongly enhanced toxicity of the mushroom toxin α-amanitin by an amatoxin-specific Fab or monoclonal antibody. Toxicon 1988, 26, 491–499. [Google Scholar] [CrossRef]
- Le Daré, B.; Ferron, P.-J.; Couette, A.; Ribault, C.; Morel, I.; Gicquel, T. In vivo and in vitro α-amanitin metabolism studies using molecular networking. Toxicol. Lett. 2021, 346, 1–6. [Google Scholar] [CrossRef]
- Stump, A.D.; Jablonski, S.E.; Bouton, L.; Wilder, J.A. Distribution and mechanism of α-amanitin tolerance in mycophagous drosophila (Diptera: Drosophilidae). Environ. Entomol. 2011, 40, 1604–1612. [Google Scholar] [CrossRef]
- Becker, C.E.; Tong, T.G.; Boerner, U.; Roe, R.L.; Sco, T.A.; MacQuarrie, M.B.; Bartter, F. Diagnosis and treatment of Amanita phalloides-type mushroom poisoning: Use of thioctic acid. West J. Med. 1976, 125, 100–109. [Google Scholar]
- Kroncke, K.D.; Fricker, G.; Meier, P.J.; Gerok, W.; Wieland, T.; Kurz, G. alphaAmanitin uptake into hepatocytes. Identification of hepatic membrane transport systems used by amatoxins. J. Biol. Chem. 1986, 261, 12562–12567. [Google Scholar] [CrossRef]
- Wlcek, K.; Koller, F.; Ferenci, P.; Stieger, B. Hepatocellular organic aniontransporting polypeptides (OATPs) and multidrug resistance-associated protein 2 (MRP2) are inhibited by silibinin. Drug Metab. Dispos. 2013, 41, 1522–1528. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.-H.; Lee, J.; Park, J.-H.; Lee, C.-H.; Choi, M.-K.; Song, I.-S. Effect of lactic acid bacteria on the pharmacokinetics and metabolism of ginsenosides in mice. Pharmaceutics 2021, 13, 1496. [Google Scholar] [CrossRef]
- Dufour, P.; Courtois, J.; Seynaeve, Y.; Peeters, S.; Le Goff, C.; Cavalier, E.; Ponthier, J. Development and validation of a liquid chromatography coupled to mass spectrometer (LC-MS) method for the simultaneous quantification of estrone-3-sulfate, progesterone, estrone and estradiol in serum of mares and American bisons. Res. Vet. Sci. 2021, 136, 343–350. [Google Scholar] [CrossRef]
- Wada, K.; Takeuchi, A.; Saiki, K.; Sutomo, R.; Van Rostenberghe, H.; Yusoff, N.M.; Laosombat, V.; Sadewa, A.H.; Talib, N.A.; Yusoff, S.; et al. Evaluation of mutation effects on UGT1A1 activity toward 17β-estradiol using liquid chromatography–tandem mass spectrometry. J. Chromatogr. B. 2006, 838, 9–14. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Meaning | Calculation Method |
|---|---|---|
| Co | Initial plasma concentration estimated at time zero following iv injection | Intercept of the plasma concentration–time curve |
| K | Elimination constant | Slope of the terminal phase of the plasma concentration–time curve |
| AUC | Area under the plasma concentration–time curve | Linear trapezoidal method ) |
| T1/2 | Elimination half-life | 0.693/K |
| MRT | Mean residence time | AUMC (Area under the moment curve)/AUC |
| CL | Clearance | Dose/AUC |
| Vss | Volume of distribution at steady-state | CLxMRT |
| Cmax | Maximum plasma concentration | Graphical observation of the highest plasma concentration |
| Tmax | Time to reach Cmax | Graphical observation of time that showing the highest plasma concentration |
| F | Oral bioavailability | (Dose normalized AUCPO/Dose normalized AUCIV) × 100 |
| Parameters | 0.2 mg/kg (n = 8) | 0.4 mg/kg (n = 6) | 0.8 mg/kg (n = 7) |
|---|---|---|---|
| C0 (ng/mL) | 711.9 ± 182.3 | 1435.7 ± 324.2 | 3249.6 ± 903.6 |
| AUClast (ng⋅min/mL) | 13,304.2 ± 2216.8 | 25,229.5 ± 4298.3 | 46,135.8 ± 13,772.3 |
| AUCinf (ng⋅min/mL) | 13,366.7 ± 2203.5 | 25,281.4 ± 4285.2 | 46,253.2 ± 13,839.7 |
| CL (mL/min/kg) | 15.3 ± 2.5 | 16.2 ± 2.4 | 18.8 ± 6.0 |
| Vss (mL/kg) | 359.3 ± 85.5 | 365.9 ± 61.0 | 390.1 ±77.1 |
| t1/2 (min) | 21.8 ± 8.3 | 18.3 ± 3.1 | 33.6 ± 22.4 |
| MRT (min) | 22.8 ± 3.5 | 22.4 ± 2.7 | 20.7 ± 3.5 |
| Parameters | 2 mg/kg (n = 7) | 5 mg/kg (n = 9) | 10 mg/kg (n = 7) |
|---|---|---|---|
| Cmax (ng/mL) | 67.1 ± 18.6 | 140.1 ± 115.8 | 360.1 ± 385.0 |
| Tmax a (min) | 60 (10–120) | 90 (15–120) | 60 (10–120) |
| AUClast (ng⋅min/mL) | 10,875 ± 5688 | 23,467 ± 12,517 | 41,896 ± 18,014 |
| AUCinf (ng⋅min/mL) | 11,433 ± 5770 | 26,266 ± 14,447 | 48,620 ± 23,655 |
| t1/2 (min) | 105.0 ± 94.1 | 132.0 ± 50.3 | 130.2 ± 78.5 |
| F (%) | 9.4 ± 4.9 | 8.1 ± 4.3 | 7.3 ± 3.1 |
| Tissue | AUClast of β-Amanitin (ng⋅min/mL Plasma; ng⋅min/g Tissue) | Tissue to Plasma Ratio | ||
|---|---|---|---|---|
| iv (0.8 mg/kg) | po (10 mg/kg) | iv | po | |
| Plasma | 54,606 ± 3464 | 23,667 ± 2812 | - | - |
| Brain | - | - | - | - |
| Heart | 13,397 ± 908 | 12,563 ± 2696 | 0.2 | 0.5 |
| Lung | 35,970 ± 3070 | 37,831 ± 11,434 | 0.7 | 1.6 |
| Liver | 25,780 ± 2440 | 12,665 ± 11,731 | 0.5 | 0.5 |
| Stomach | - | 680,749 ± 137,691 | - | 28.8 |
| Intestine | - | 2,066,298 ± 492,689 | - | 87.3 |
| Spleen | 137,855 ± 11,949 | 42,409 ± 33,867 | 2.5 | 1.8 |
| Kidney | 142,023 ± 7510 | 158,677 ± 84,620 | 2.6 | 6.7 |
| CYPs | Enzyme Activities | IC50 (μM) | |
|---|---|---|---|
| without NADPH Preincubation | with NADPH Preincubation | ||
| 1A2 | Phenacetin O-deethylase | >100 | >100 |
| 2A6 | Coumarin 7-hydroxylase | 93.9 | 80.1 |
| 2B6 | Bupropion hydroxylase | 38.0 | 34.8 |
| 2C8 | Amodiaquine N-deethylase | >100 | >100 |
| 2C9 | Diclofenac 4′-hydroxylase | >100 | >100 |
| 2C19 | [S]-Mephenytoin 4′-hydroxylase | >100 | >100 |
| 2D6 | Bufuralol 1′-hydroxylase | 76.2 | 71.6 |
| 3A4 | Midazolam 1′-hydroxylase | >100 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bang, Y.Y.; Song, I.-S.; Lee, M.S.; Lim, C.H.; Cho, Y.-Y.; Lee, J.Y.; Kang, H.C.; Lee, H.S. Toxicokinetics of β-Amanitin in Mice and In Vitro Drug–Drug Interaction Potential. Pharmaceutics 2022, 14, 774. https://doi.org/10.3390/pharmaceutics14040774
Bang YY, Song I-S, Lee MS, Lim CH, Cho Y-Y, Lee JY, Kang HC, Lee HS. Toxicokinetics of β-Amanitin in Mice and In Vitro Drug–Drug Interaction Potential. Pharmaceutics. 2022; 14(4):774. https://doi.org/10.3390/pharmaceutics14040774
Chicago/Turabian StyleBang, Young Yoon, Im-Sook Song, Min Seo Lee, Chang Ho Lim, Yong-Yeon Cho, Joo Young Lee, Han Chang Kang, and Hye Suk Lee. 2022. "Toxicokinetics of β-Amanitin in Mice and In Vitro Drug–Drug Interaction Potential" Pharmaceutics 14, no. 4: 774. https://doi.org/10.3390/pharmaceutics14040774