
NMS-873 Leads to Dysfunctional Glycometabolism in A p97-Independent Manner in HCT116 Colon Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Lines and Reagents
2.2. Lactate and Glucose Concentration Measurement
2.3. OCR and ECAR Measurement
2.4. Cell Viability Measurement
2.5. Western Blot Assay
2.6. TMT Labeling Proteomics
2.7. PISA T Assay
2.8. In Silico Docking Analysis of NMS-873 with NDUFAF5
2.9. Quantification and Statistical Analysis
3. Results
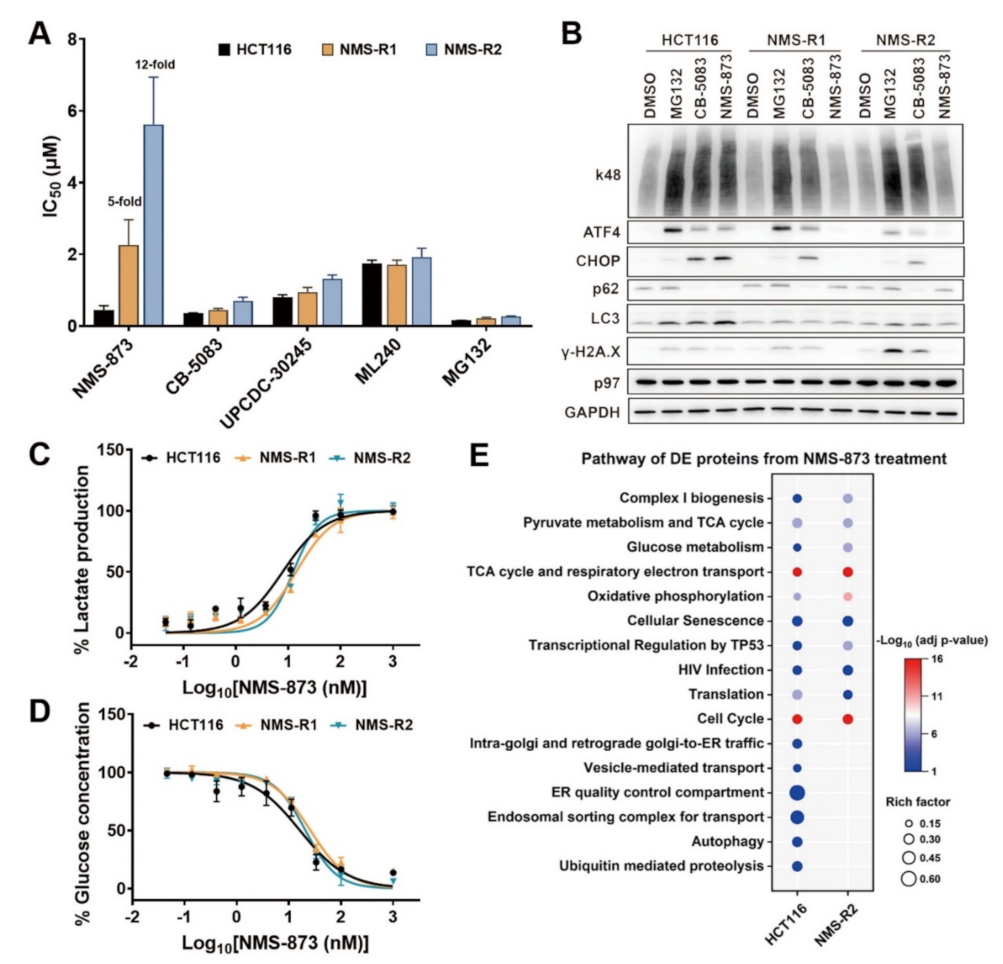
3.1. An Allosteric p97 Inhibitor, NMS-873, Regulates Glycometabolism
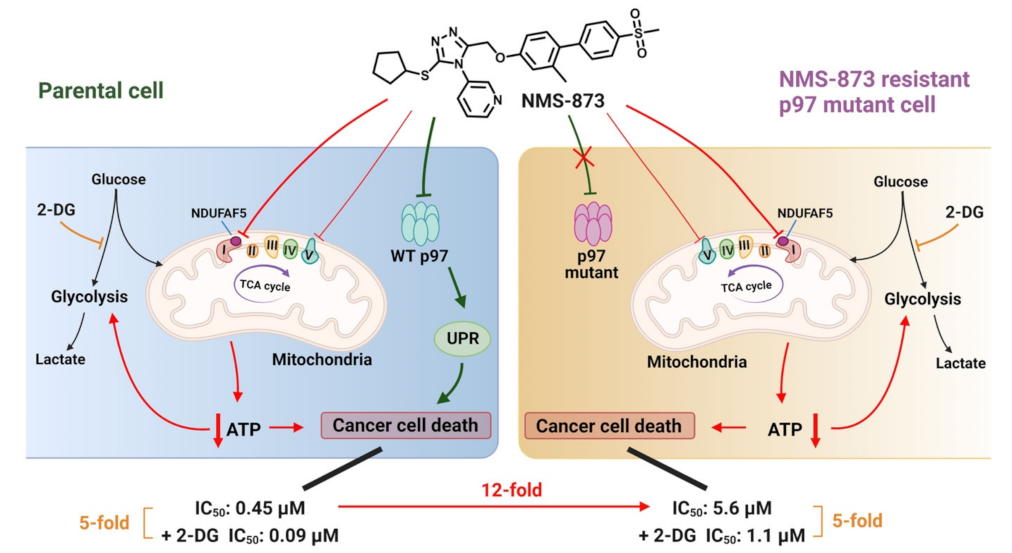
3.2. 2-DG Blocks NMS-873-Induced Glycolysis and Enhances Its Anti-Proliferative Activity
3.3. NMS-873 Interfered with Glycometabolism in a p97-Independent Manner
3.4. Quantitative Proteomics to Identify p97 Dependence of the NMS-873 Affected Pathways
3.5. NMS-873 Affects the Thermal Stability of Mitochondrial Complex I and ATP Synthase
3.6. Overexpression of NDUFAF5 Rescues NMS-873 Induced Lactate Production
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fessart, D.; Marza, E.; Taouji, S.; Delom, F.; Chevet, E. P97/CDC-48: Proteostasis control in tumor cell biology. Cancer Lett. 2013, 337, 26–34. [Google Scholar] [CrossRef]
- Li, C.; Huang, Y.S.; Fan, Q.Q.; Quan, H.Y.; Dong, Y.Q.; Nie, M.; Wang, J.Q.; Xie, F.C.; Ji, J.; Zhou, L.; et al. p97/VCP is highly expressed in the stem-like cells of breast cancer and controls cancer stemness partly through the unfolded protein response. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Tomita, Y.; Hoshida, Y.; Kono, T.; Oka, T.; Yamamoto, S.; Nonomura, N.; Okuyama, A.; Aozasa, K. Elevated expression of valosin-containing protein (p97) is associated with poor prognosis of prostate cancer. Clin. Cancer Res. 2004, 10, 3007–3012. [Google Scholar] [CrossRef] [Green Version]
- Huryn, D.M.; Kornfilt, D.J.P.; Wipf, P. p97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections. J. Med. Chem. 2020, 63, 1892–1907. [Google Scholar] [CrossRef]
- Zhang, G.; Li, S.; Cheng, K.W.; Chou, T.F. AAA ATPases as therapeutic targets: Structure, functions, and small-molecule inhibitors. Eur. J. Med. Chem. 2021, 219, 113446. [Google Scholar] [CrossRef]
- Sui, X.; Pan, M.; Li, Y.M. Insights into the Design of p97-targeting Small Molecules from Structural Studies on p97 Functional Mechanism. Curr. Med. Chem. 2020, 27, 298–316. [Google Scholar] [CrossRef]
- Lan, B.; Chai, S.; Wang, P.; Wang, K. VCP/p97/Cdc48, A Linking of Protein Homeostasis and Cancer Therapy. Curr. Mol. Med. 2017, 17, 608–618. [Google Scholar] [CrossRef]
- Chou, T.F.; Brown, S.J.; Minond, D.; Nordin, B.E.; Li, K.L.; Jones, A.C.; Chase, P.; Porubsky, P.R.; Stoltz, B.M.; Schoenen, F.J.; et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 4834–4839. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.F.; Li, K.L.; Frankowski, K.J.; Schoenen, F.J.; Deshaies, R.J. Structure-Activity Relationship Study Reveals ML240 and ML241 as Potent and Selective Inhibitors of p97 ATPase. Chemmedchem 2013, 8, 297–312. [Google Scholar] [CrossRef]
- Magnaghi, P.; D’Alessio, R.; Valsasina, B.; Avanzi, N.; Rizzi, S.; Asa, D.; Gasparri, F.; Cozzi, L.; Cucchi, U.; Orrenius, C.; et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9, 548–556. [Google Scholar] [CrossRef]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.H.; Yao, B.; Valle, E.; von Soly, S.K.; et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPorte, M.G.; Burnett, J.C.; Colombo, R.; Bulfer, S.L.; Alverez, C.; Chou, T.F.; Neitz, R.J.; Green, N.; Moore, W.J.; Yue, Z.; et al. Optimization of Phenyl Indole Inhibitors of the AAA+ ATPase p97. ACS Med. Chem. Lett. 2018, 9, 1075–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, R.; Zhang, T.; Wilson, D.J.; Xie, J.; Williams, J.; Xu, Y.; Ye, Y.; Chen, L. Discovery of Irreversible p97 Inhibitors. J. Med. Chem. 2019, 62, 2814–2829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, S.; Wang, F.; Jones, A.C.; Goldberg, A.F.G.; Lin, B.; Virgil, S.; Stoltz, B.M.; Deshaies, R.J.; Chou, T.F. A covalent p97/VCP ATPase inhibitor can overcome resistance to CB-5083 and NMS-873 in colorectal cancer cells. Eur. J. Med. Chem. 2021, 213, 113148. [Google Scholar] [CrossRef]
- Nishimura, N.; Radwan, M.O.; Amano, M.; Endo, S.; Fujii, E.; Hayashi, H.; Ueno, S.; Ueno, N.; Tatetsu, H.; Hata, H.; et al. Novel p97/VCP inhibitor induces endoplasmic reticulum stress and apoptosis in both bortezomib-sensitive and -resistant multiple myeloma cells. Cancer Sci. 2019, 110, 3275–3287. [Google Scholar] [CrossRef] [Green Version]
- Roux, B.; Vaganay, C.; Vargas, J.D.; Alexe, G.; Benaksas, C.; Pardieu, B.; Fenouille, N.; Ellegast, J.M.; Malolepsza, E.; Ling, F.; et al. Targeting acute myeloid leukemia dependency on VCP-mediated DNA repair through a selective second-generation small-molecule inhibitor. Sci. Transl. Med. 2021, 13, eabg1168. [Google Scholar] [CrossRef]
- Wang, F.; Li, S.; Gan, T.; Stott, G.M.; Flint, A.; Chou, T.F. Allosteric p97 Inhibitors Can Overcome Resistance to ATP-Competitive p97 Inhibitors for Potential Anticancer Therapy. ChemMedChem 2020, 15, 685–694. [Google Scholar] [CrossRef]
- Bouwer, M.F.; Hamilton, K.E.; Jonker, P.B.; Kuiper, S.R.; Louters, L.L.; Looyenga, B.D. NMS-873 functions as a dual inhibitor of mitochondrial oxidative phosphorylation. Biochimie 2021, 185, 33–42. [Google Scholar] [CrossRef]
- Nalbandian, A.; Llewellyn, K.J.; Gomez, A.; Walker, N.; Su, H.; Dunnigan, A.; Chwa, M.; Vesa, J.; Kenney, M.C.; Kimonis, V.E. In vitro studies in VCP-associated multisystem proteinopathy suggest altered mitochondrial bioenergetics. Mitochondrion 2015, 22, 1–8. [Google Scholar] [CrossRef]
- Parzych, K.; Saavedra-Garcia, P.; Valbuena, G.N.; Al-Sadah, H.A.; Robinson, M.E.; Penfold, L.; Kuzeva, D.M.; Ruiz-Tellez, A.; Loaiza, S.; Holzmann, V.; et al. The coordinated action of VCP/p97 and GCN2 regulates cancer cell metabolism and proteostasis during nutrient limitation. Oncogene 2019, 38, 3216–3231. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley, M.; Natarajan, S.K.; Mondal, J.; Best, D.; Goldman, D.; Shanthappa, B.; Pellowe, M.; Dash, C.; Saha, T.; Khiste, S.; et al. Nanoengineered Disruption of Heat Shock Protein 90 Targets Drug-Induced Resistance and Relieves Natural Killer Cell Suppression in Breast Cancer. Cancer Res. 2020, 80, 5355–5366. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.V.S.; Portilho, A.J.D.; Da Silva, E.L.; Sales, L.D.; Maues, J.H.D.; De Moraes, M.E.A.; Moreira-Nunes, C.A. Establishment of Drug-resistant Cell Lines as a Model in Experimental Oncology: A Review. Anticancer Res. 2019, 39, 6443–6455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, L.M.; O’Toole, S.A.; Spillane, C.D.; Martin, C.M.; Gallagher, M.F.; Stordal, B.; Blackshields, G.; Sheils, O.; O’Leary, J.J. Identifying novel hypoxia-associated markers of chemoresistance in ovarian cancer. BMC Cancer 2015, 15, 547. [Google Scholar] [CrossRef] [Green Version]
- Alimbetov, D.; Askarova, S.; Umbayev, B.; Davis, T.; Kipling, D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. Int. J. Mol. Sci. 2018, 19, 61690. [Google Scholar] [CrossRef] [Green Version]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Nordlund, P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for In Situ Drug Target Engagement and Mechanistic Biomarker Studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–161. [Google Scholar] [CrossRef]
- Franken, H.; Mathieson, T.; Childs, D.; Sweetman, G.M.A.; Werner, T.; Togel, I.; Doce, C.; Gade, S.; Bantscheff, M.; Drewes, G.; et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. [Google Scholar] [CrossRef]
- Gaetani, M.; Sabatier, P.; Saei, A.A.; Beusch, C.M.; Yang, Z.; Lundstrom, S.L.; Zubarev, R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019, 18, 4027–4037. [Google Scholar] [CrossRef]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903.e815. [Google Scholar] [CrossRef] [Green Version]
- Bastola, P.; Wang, F.; Schaich, M.A.; Gan, T.; Freudenthal, B.D.; Chou, T.F.; Chien, J. Specific mutations in the D1-D2 linker region of VCP/p97 enhance ATPase activity and confer resistance to VCP inhibitors. Cell Death Discov. 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- STRING Database. Available online: http://www.string-db.org/ (accessed on 20 October 2021).
- Wang, F.; Li, S.; Hourebi, N.; Chou, T.-F. Temporal proteomics reveal specific cell cycle oncoprotein downregulation by p97/VCP inhibition. Cell Chem. Biol. 2021, 29, 517–529. [Google Scholar] [CrossRef]
- Alverez, C.; Arkin, M.R.; Bulfer, S.L.; Colombo, R.; Kovaliov, M.; LaPorte, M.G.; Lim, C.; Liang, M.; Moore, W.J.; Neitz, R.J.; et al. Structure-Activity Study of Bioisosteric Trifluoromethyl and Pentafluorosulfanyl Indole Inhibitors of the AAA ATPase p97. ACS Med. Chem. Lett. 2015, 6, 1225–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachsberger, P.R.; Gressen, E.L.; Bhala, A.; Bobyock, S.B.; Storck, C.; Coss, R.A.; Berd, D.; Leeper, D.B. Variability in glucose transporter-1 levels and hexokinase activity in human melanoma. Melanoma Res. 2002, 12, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Siwiak, E.; Soltyka, M.; Priebe, A.; Zielinski, R.; Fokt, I.; Ziemniak, M.; Jaskiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 10234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.L. Development of cancer metabolism as a therapeutic target: New pathways, patient studies, stratification and combination therapy. Brit. J. Cancer 2020, 122, 1–3. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [Green Version]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-d-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Tagg, S.L.; Foster, P.A.; Leese, M.P.; Potter, B.V.; Reed, M.J.; Purohit, A.; Newman, S.P. 2-Methoxyoestradiol-3,17-o,o-bis-sulphamate and 2-deoxy-d-glucose in combination: A potential treatment for breast and prostate cancer. Br. J. Cancer 2008, 99, 1842–1848. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Diao, D.; Zhang, H.; Guo, Q.; Wu, X.; Song, Y.; Dang, C. High glucose-induced resistance to 5-fluorouracil in pancreatic cancer cells alleviated by 2-deoxy-d-glucose. Biomed. Rep. 2014, 2, 188–192. [Google Scholar] [CrossRef]
- Cao, S.S.; Zhen, Y.S. Potentiation of antimetabolite antitumor activity in vivo by dipyridamole and amphotericin B. Cancer Chemother. Pharmacol. 1989, 24, 181–186. [Google Scholar] [CrossRef]
- Xu, S.P.; Sun, G.P.; Shen, Y.X.; Peng, W.R.; Wang, H.; Wei, W. Synergistic effect of combining paeonol and cisplatin on apoptotic induction of human hepatoma cell lines. Acta Pharmacol. Sin. 2007, 28, 869–878. [Google Scholar] [CrossRef]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R., Jr.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Gao, J.L.; Ji, J.W.; Gao, M.; Yin, Q.S.; Qiu, Q.L.; Wang, C.; Chen, S.Z.; Xu, J.; Liang, R.S.; et al. Cytotoxicity enhancement in MDA-MB-231 cells by the combination treatment of tetrahydropalmatine and berberine derived from Corydalis yanhusuo W. T. Wang. J. Intercult. Ethnopharmacol. 2014, 3, 68–72. [Google Scholar] [CrossRef]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.S.; Choi, J.Y. Synergistic anti-cancer effect of phenformin and oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Her, N.G.; Toth, J.I.; Ma, C.T.; Wei, Y.; Motamedchaboki, K.; Sergienko, E.; Petroski, M.D. p97 Composition Changes Caused by Allosteric Inhibition Are Suppressed by an On-Target Mechanism that Increases the Enzyme’s ATPase Activity. Cell Chem. Biol. 2016, 23, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Savitski, M.M.; Reinhard, F.B.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Martinez Molina, D.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.Y.; Bacanu, S.; Sreekumar, L.; Ramos, A.D.; Dai, L.; Michaelis, M.; Cinatl, J.; Seki, T.; Cao, Y.; Coffill, C.R.; et al. CETSA interaction proteomics define specific RNA-modification pathways as key components of fluorouracil-based cancer drug cytotoxicity. Cell Chem. Biol. 2021, 21, S2451. [Google Scholar] [CrossRef]
- Li, J.; Van Vranken, J.G.; Paulo, J.A.; Huttlin, E.L.; Gygi, S.P. Selection of Heating Temperatures Improves the Sensitivity of the Proteome Integral Solubility Alteration Assay. J. Proteome Res. 2020, 19, 2159–2166. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Antolin, A.A.; Clarke, P.A.; Collins, I.; Workman, P.; Al-Lazikani, B. Evolution of kinase polypharmacology across HSP90 drug discovery. Cell Chem. Biol. 2021, 28, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Wang, F.; Zhang, G.; Chou, T.-F. NMS-873 Leads to Dysfunctional Glycometabolism in A p97-Independent Manner in HCT116 Colon Cancer Cells. Pharmaceutics 2022, 14, 764. https://doi.org/10.3390/pharmaceutics14040764
Li S, Wang F, Zhang G, Chou T-F. NMS-873 Leads to Dysfunctional Glycometabolism in A p97-Independent Manner in HCT116 Colon Cancer Cells. Pharmaceutics. 2022; 14(4):764. https://doi.org/10.3390/pharmaceutics14040764
Chicago/Turabian StyleLi, Shan, Feng Wang, Gang Zhang, and Tsui-Fen Chou. 2022. "NMS-873 Leads to Dysfunctional Glycometabolism in A p97-Independent Manner in HCT116 Colon Cancer Cells" Pharmaceutics 14, no. 4: 764. https://doi.org/10.3390/pharmaceutics14040764