
Synthesis and Properties of α-Mangostin and Vadimezan Conjugates with Glucoheptoamidated and Biotinylated 3rd Generation Poly(amidoamine) Dendrimer, and Conjugation Effect on Their Anticancer and Anti-Nematode Activities


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Biochemical Reagents, Cell Lines and Materials
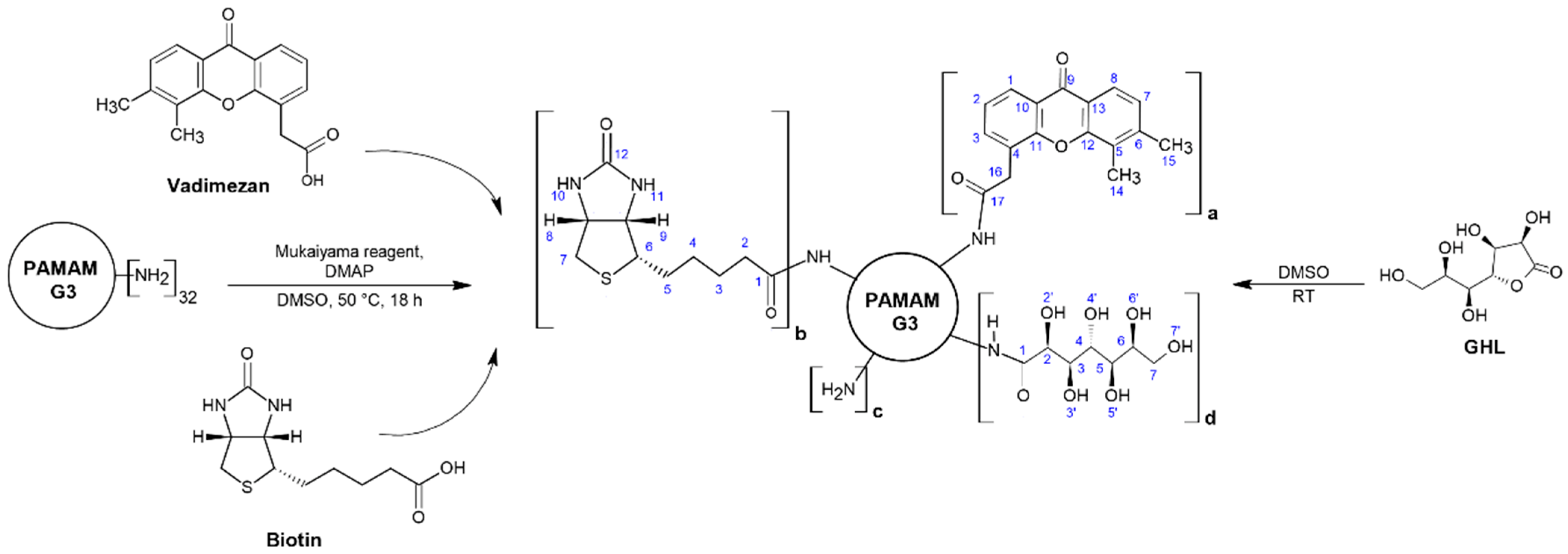
2.3. Syntheses of Dendrimer Conjugates with Ester-Bonded Biotin and α-Mangostin or Vadimezan
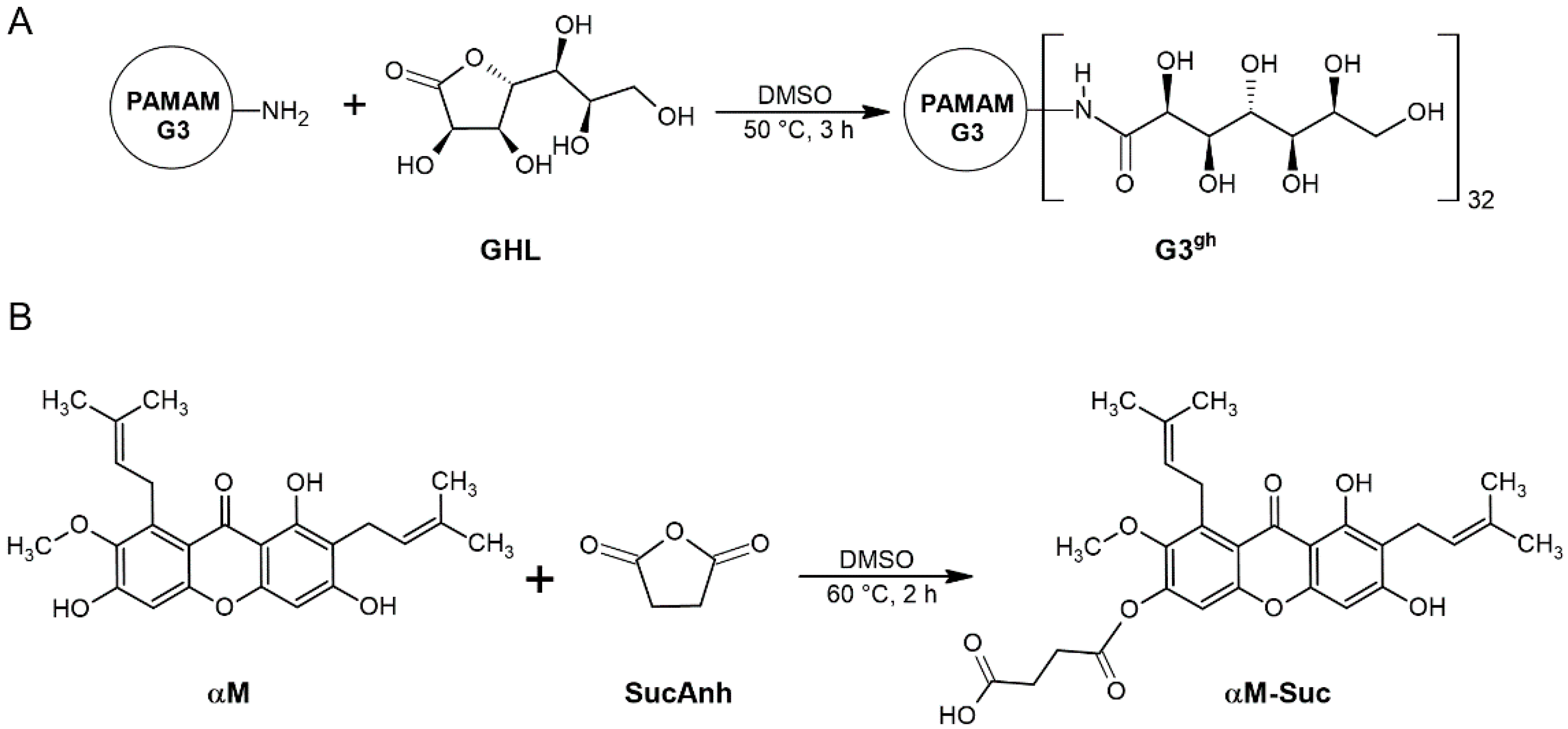
2.3.1. Synthesis of Fully Glucoheptoamidated PAMAM G3
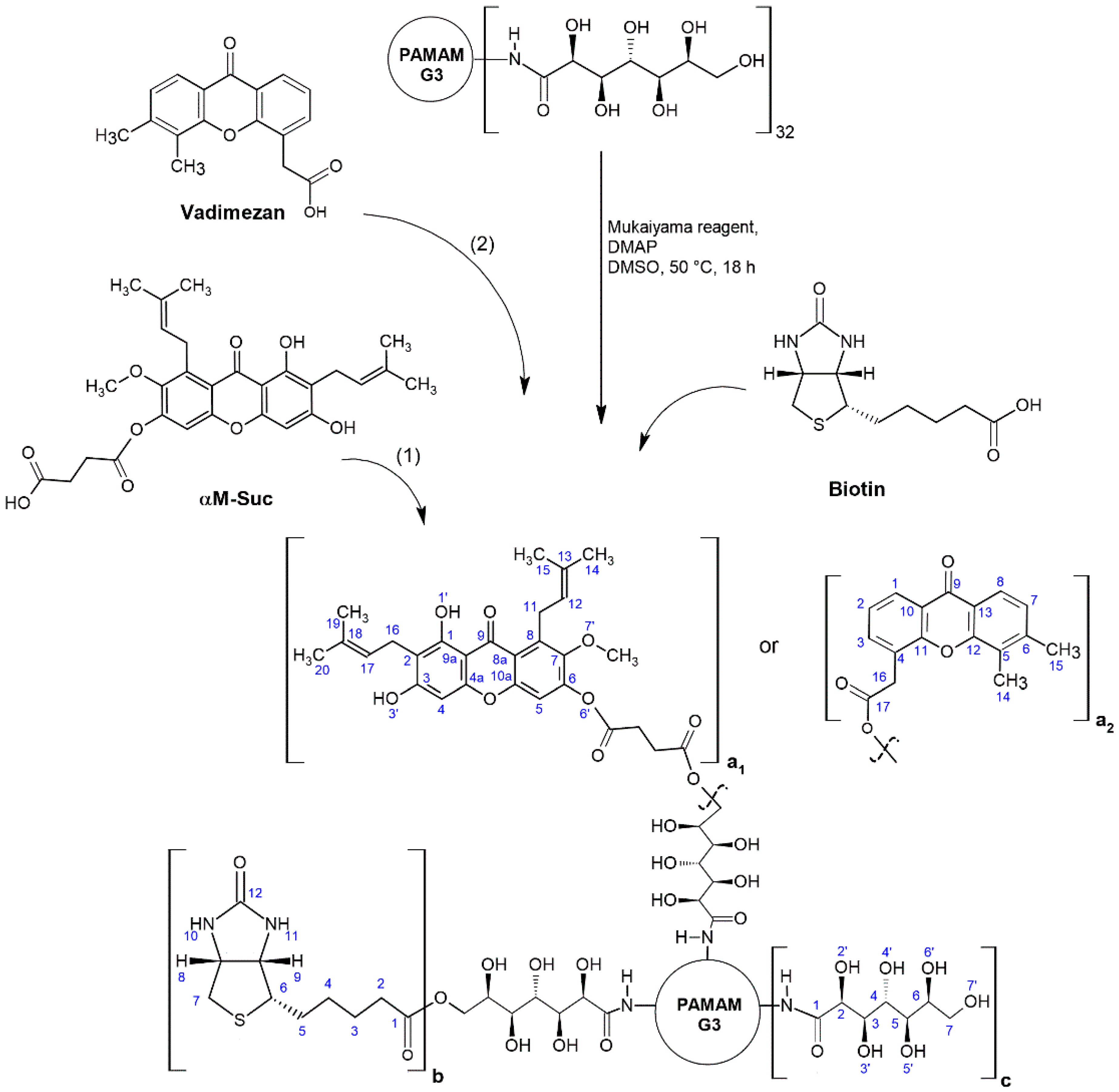
2.3.2. Synthesis of Biotinylated G3gh—α-Mangostin Conjugate
2.3.3. Synthesis of Biotinylated G3gh—Vadimezan Conjugate
2.4. Syntheses of Dendrimer Conjugates with Amide-Bonded Biotin, GHL and α-Mangostin or Vadimezan
Synthesis of Biotinylated and Half-Glucoheptoamidated G3 with Vadimezan
2.5. NMR Spectroscopy
2.6. Conjugates Size and ζ Potential Measurements
2.7. Biological Studies
2.7.1. Cell Cultures
2.7.2. Cytotoxicity
2.7.3. Proliferation
2.7.4. Toxicity to Caenorhabditis elegans and the Worm Survival Analysis
2.7.5. Statistical Analysis
3. Results and Discussion
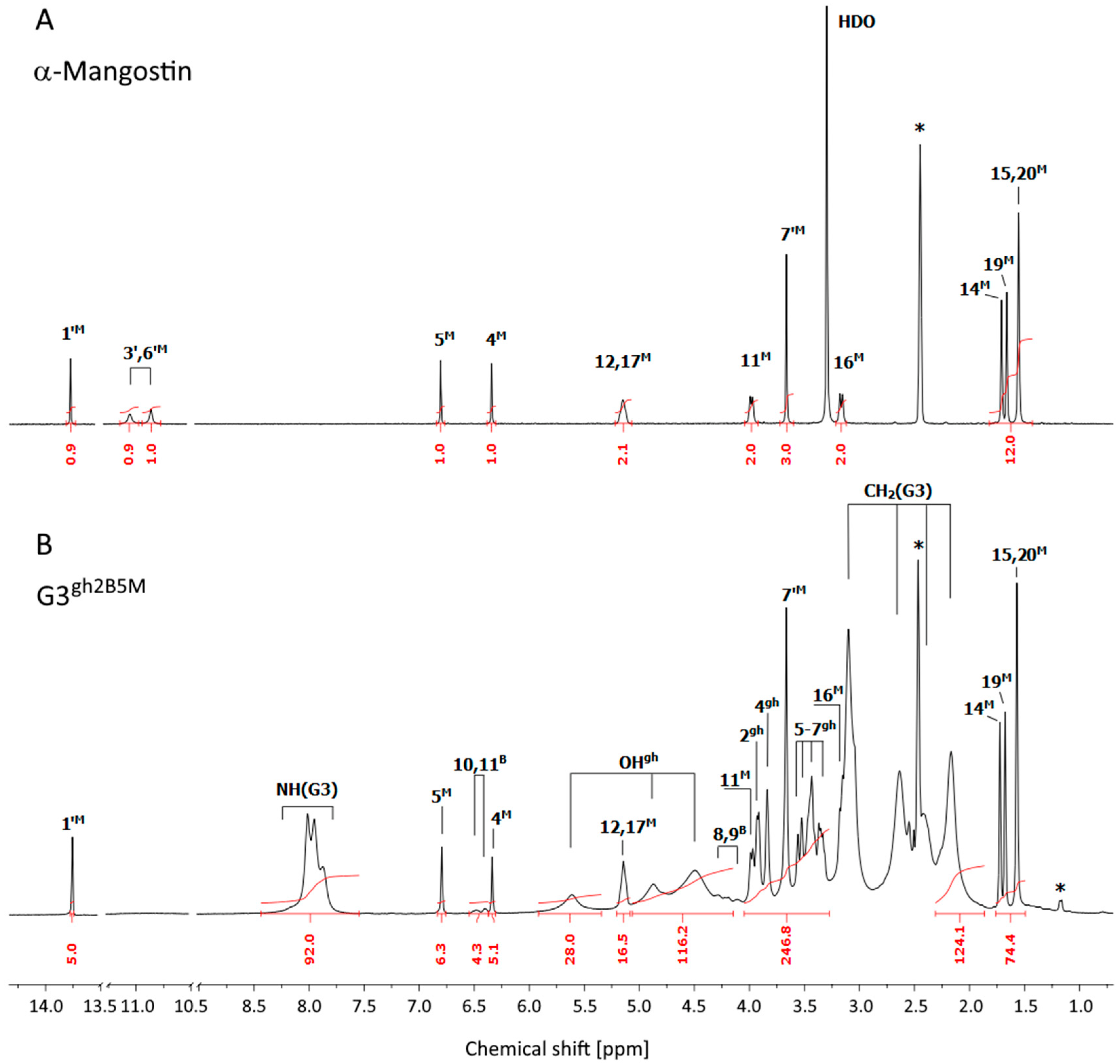
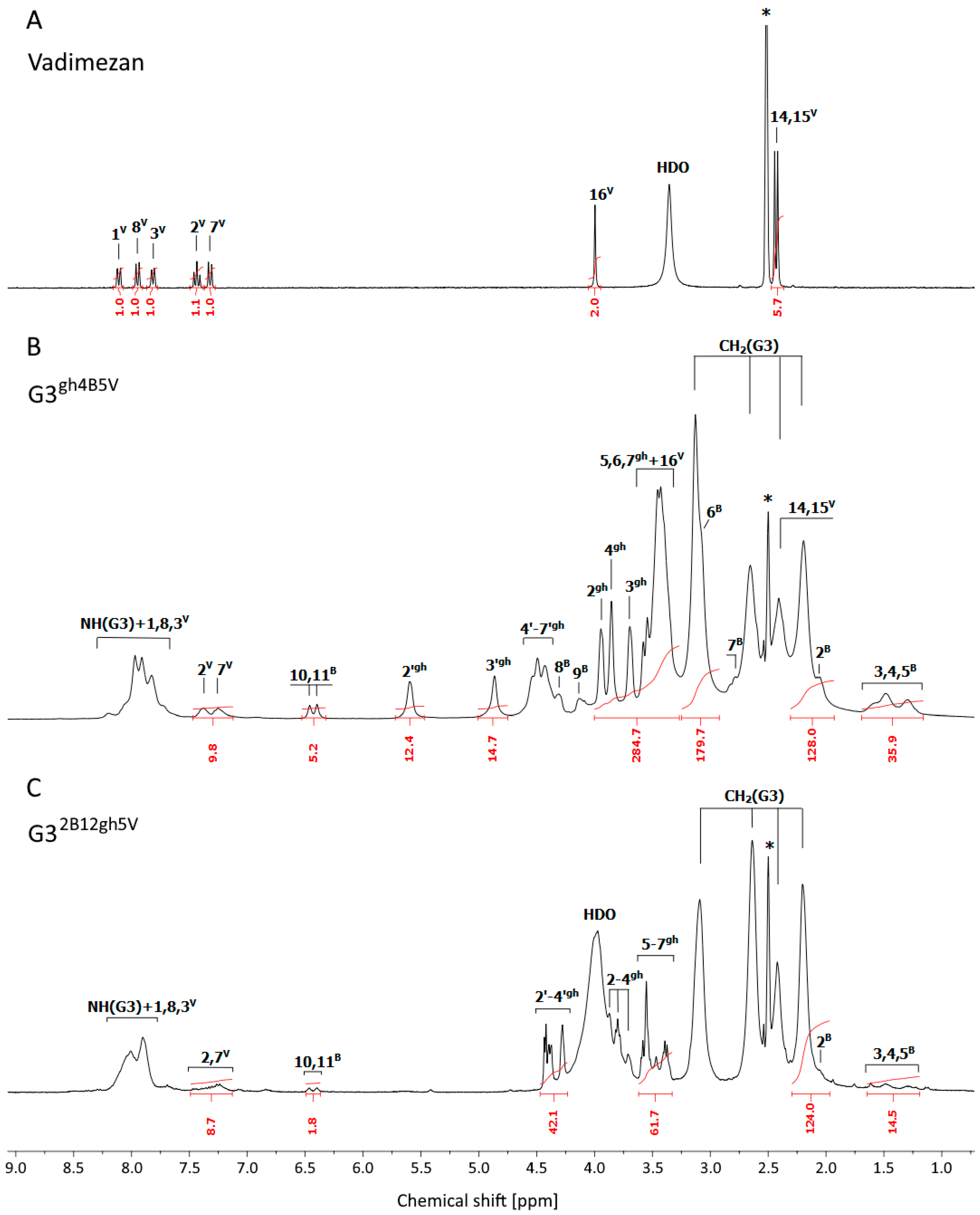
3.1. Syntheses and Characterization of Dendrimer Conjugates


3.2. Size and ζ Potential of Dendrimer Conjugates
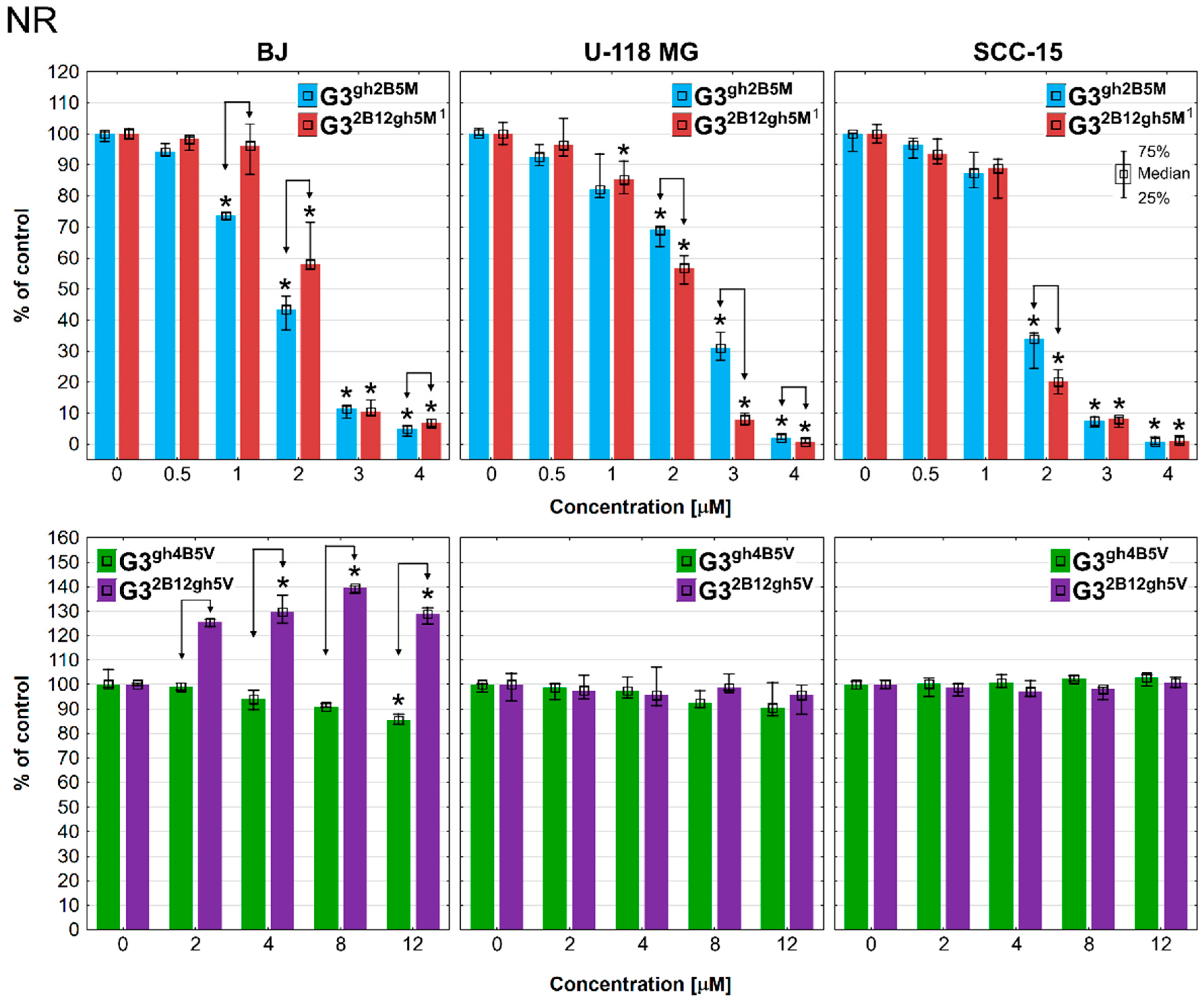
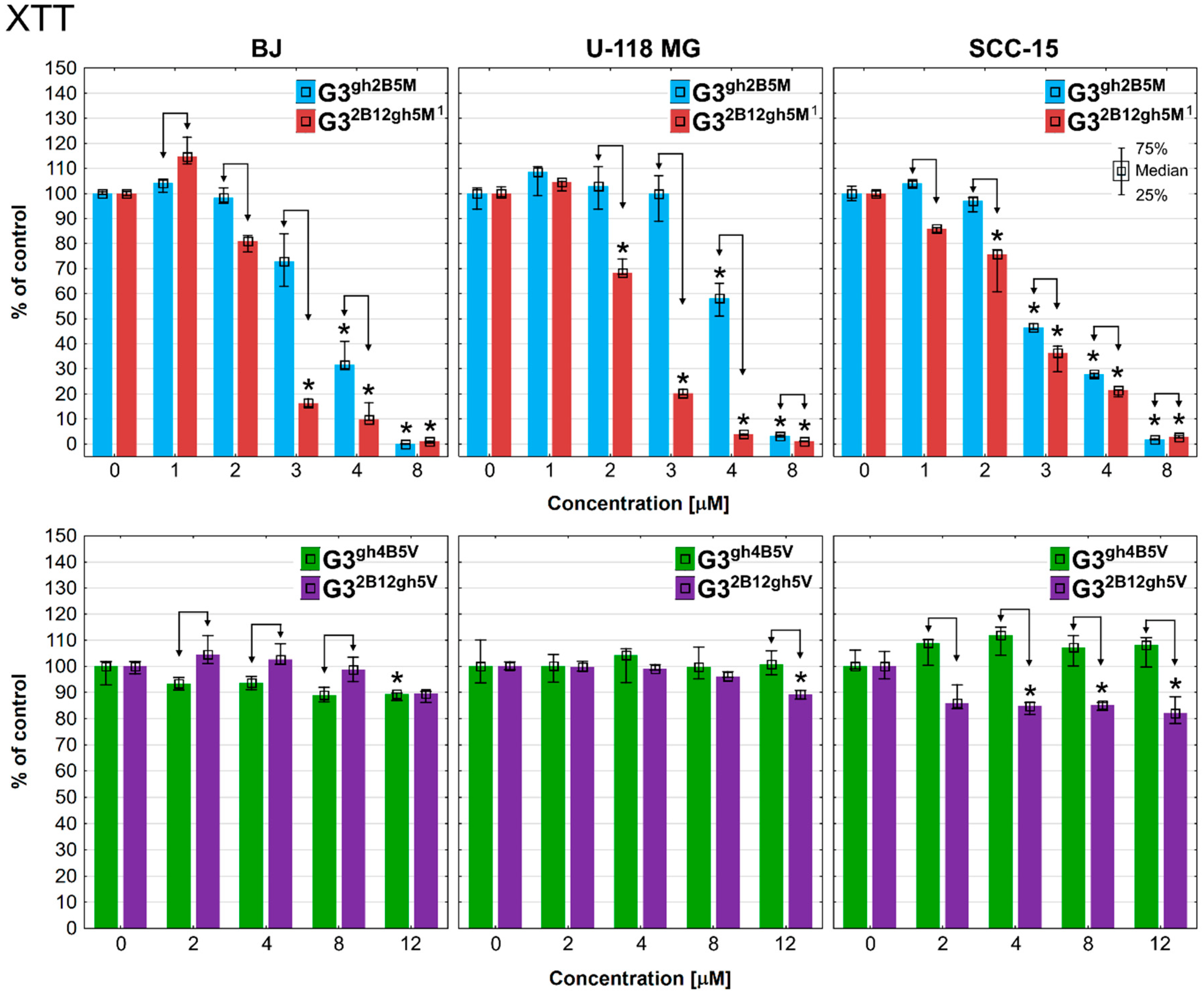
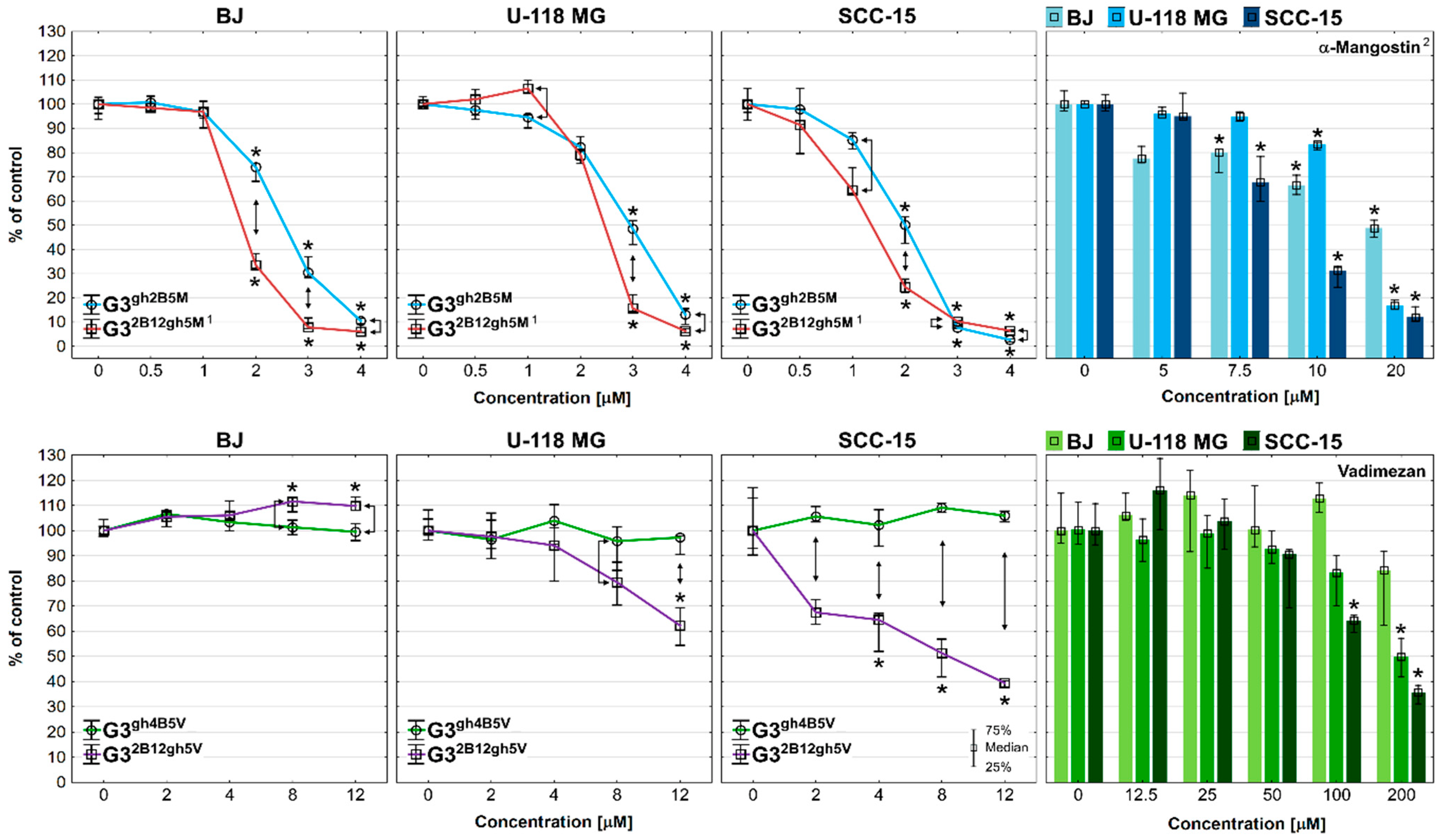
3.3. Cytotoxicity
3.4. Proliferation
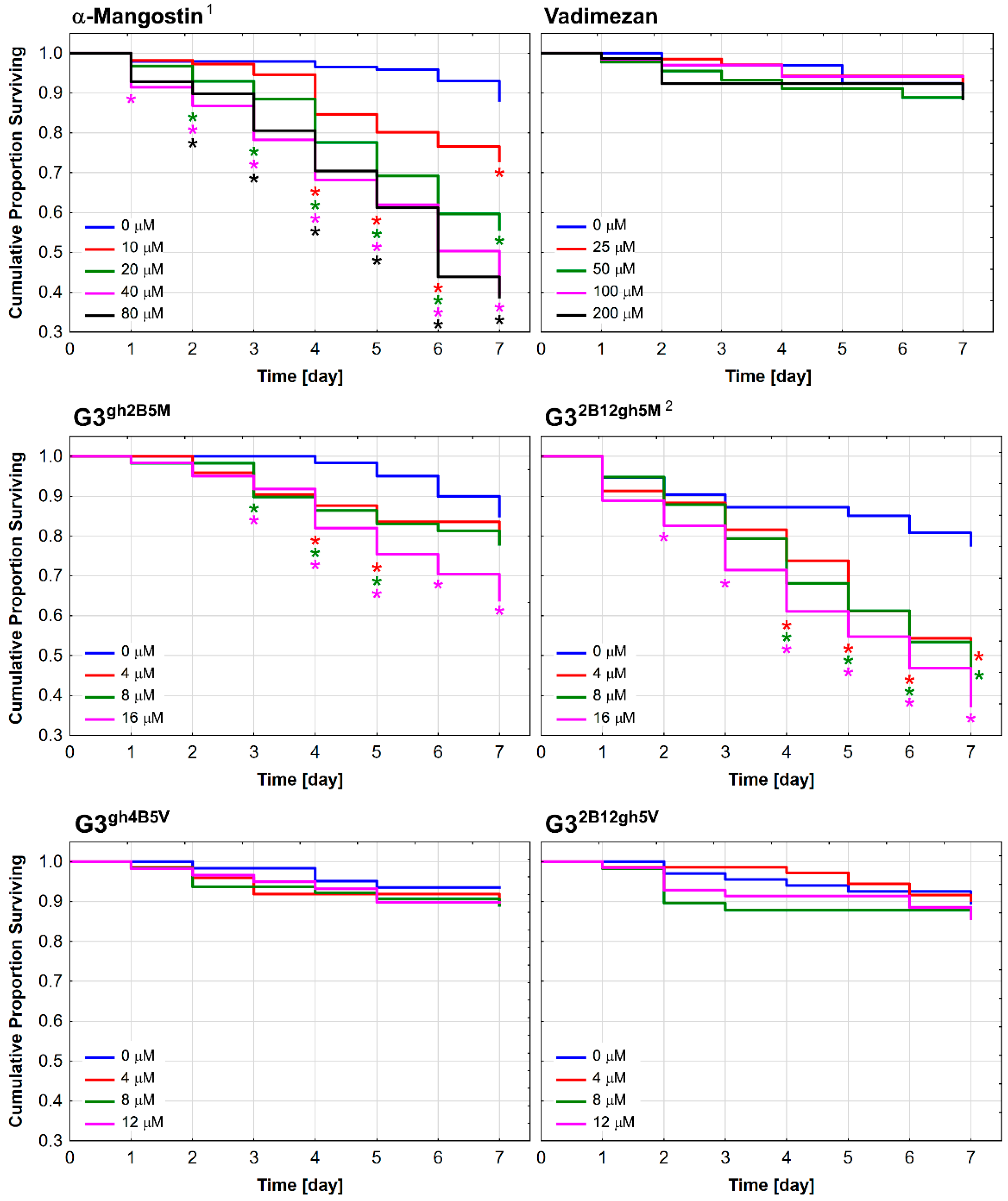
3.5. Toxicity against C. elegans and the Effect on the Worm Survival
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ho, L.Y.; Lim, Y.Y.; Tan, C.P.; Siow, L.F. Comparison of physicochemical properties and aqueous solubility of xanthone prepared via oil-in-water emulsion and complex coacervation techniques. Int. J. Food Prop. 2018, 21, 784–798. [Google Scholar] [CrossRef]
- Markowicz, J.; Uram, Ł.; Sobich, J.; Mangiardi, L.; Maj, P.; Rode, W. Antitumor and anti-nematode activities of α-mangostin. Eur. J. Pharmacol. 2019, 863, 172678. [Google Scholar] [CrossRef] [PubMed]
- Aukkanimart, R.; Boonmars, T.; Sriraj, P.; Sripan, P.; Songsri, J.; Ratanasuwan, P.; Laummaunwai, P.; Boueroy, P.; Khueangchaingkhwang, S.; Pumhirunroj, B.; et al. In Vitro and in Vivo Inhibitory Effects of α-Mangostin on Cholangiocarcinoma Cells and Allografts. Asian Pac. J. Cancer Prev. 2017, 18, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Beninati, S.; Oliverio, S.; Cordella, M.; Rossi, S.; Senatore, C.; Liguori, I.; Lentini, A.; Piredda, L.; Tabolacci, C. Inhibition of cell proliferation, migration and invasion of B16-F10 melanoma cells by α-mangostin. Biochem. Biophys. Res. Commun. 2014, 450, 1512–1517. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, S.-C.; Huang, M.-H.; Cheng, C.-W.; Hung, J.-H.; Yang, S.-F.; Hsieh, Y.-H. α-Mangostin induces mitochondrial dependent apoptosis in human hepatoma SK-Hep-1 cells through inhibition of p38 MAPK pathway. Apoptosis 2013, 18, 1548–1560. [Google Scholar] [CrossRef]
- Shibata, M.-A.; Iinuma, M.; Morimoto, J.; Kurose, H.; Akamatsu, K.; Okuno, Y.; Akao, Y.; Otsuki, Y. α-Mangostin extracted from the pericarp of the mangosteen (Garcinia mangostana Linn) reduces tumor growth and lymph node metastasis in an immunocompetent xenograft model of metastatic mammary cancer carrying a p53 mutation. BMC Med. 2011, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.-C.; Hsu, Y.-L.; Liu, C.-K.; Kuo, P.-L. α-Mangostin, a Dietary Xanthone, Induces Autophagic Cell Death by Activating the AMP-Activated Protein Kinase Pathway in Glioblastoma Cells. J. Agric. Food Chem. 2011, 59, 2086–2096. [Google Scholar] [CrossRef]
- Bin Hafeez, B.; Mustafa, A.; Fischer, J.W.; Singh, A.; Zhong, W.; Shekhani, M.O.; Meske, L.; Havighurst, T.; Kim, K.; Verma, A.K. α-Mangostin: A Dietary Antioxidant Derived from the Pericarp of Garcinia mangostana L. Inhibits Pancreatic Tumor Growth in Xenograft Mouse Model. Antioxid. Redox Signal. 2014, 21, 682–699. [Google Scholar] [CrossRef] [Green Version]
- Cai, N.; Xie, S.-J.; Qiu, D.-B.; Jia, C.-C.; Du, C.; Liu, W.; Chen, J.-J.; Zhang, Q. Potential effects of α-mangostin in the prevention and treatment of hepatocellular carcinoma. J. Funct. Foods 2016, 26, 309–318. [Google Scholar] [CrossRef]
- Lee, C.-H.; Ying, T.-H.; Chiou, H.-L.; Hsieh, S.-C.; Wen, S.-H.; Chou, R.-H.; Hsieh, Y.-H. Alpha-mangostin induces apoptosis through activation of reactive oxygen species and ASK1/p38 signaling pathway in cervical cancer cells. Oncotarget 2017, 8, 47425–47439. [Google Scholar] [CrossRef]
- Johnson, J.J.; Petiwala, S.M.; Syed, D.N.; Rasmussen, J.T.; Adhami, V.M.; Siddiqui, I.A.; Kohl, A.M.; Mukhtar, H. α-Mangostin, a xanthone from mangosteen fruit, promotes cell cycle arrest in prostate cancer and decreases xenograft tumor growth. Carcinogenesis 2011, 33, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Chitchumroonchokchai, C.; Thomas-Ahner, J.M.; Li, J.; Riedl, K.M.; Nontakham, J.; Suksumrarn, S.; Clinton, S.K.; Kinghorn, A.D.; Failla, M.L. Anti-tumorigenicity of dietary α-mangostin in an HT-29 colon cell xenograft model and the tissue distribution of xanthones and their phase II metabolites. Mol. Nutr. Food Res. 2013, 57, 203–211. [Google Scholar] [CrossRef]
- Bissoli, I.; Muscari, C. Doxorubicin and α-Mangostin oppositely affect luminal breast cancer cell stemness evaluated by a new retinaldehyde-dependent ALDH assay in MCF-7 tumor spheroids. Biomed. Pharmacother. 2020, 124, 109927. [Google Scholar] [CrossRef]
- Adli, A.D.F.; Jahanban-Esfahlan, R.; Seidi, K.; Samandari-Rad, S.; Zarghami, N. An overview on Vadimezan (DMXAA): The vascular disrupting agent. Chem. Biol. Drug Des. 2018, 91, 996–1006. [Google Scholar] [CrossRef]
- Baguley, B.C.; McKeage, M.J. ASA404: A tumor vascular-disrupting agent with broad potential for cancer therapy. Future Oncol. 2010, 6, 1537–1543. [Google Scholar] [CrossRef]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. OncoImmunology 2020, 9, 1777624. [Google Scholar] [CrossRef]
- Shih, A.Y.; Damm-Ganamet, K.L.; Mirzadegan, T. Dynamic Structural Differences between Human and Mouse STING Lead to Differing Sensitivity to DMXAA. Biophys. J. 2018, 114, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Kim, G.B.; Krump, N.A.; Zhou, Y.; Riley, J.L.; You, J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 13730–13739. [Google Scholar] [CrossRef]
- Chen, D.; Yu, Q.; Huang, X.; Dai, H.; Luo, T.; Shao, J.; Chen, J.; Huang, W.; Dong, X. A Highly-Efficient Type I Photosensitizer with Robust Vascular-Disruption Activity for Hypoxic-and-Metastatic Tumor Specific Photodynamic Therapy. Small 2020, 16, e2001059. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Ambekar, R.S.; Choudhary, M.; Kandasubramanian, B. Recent advances in dendrimer-based nanoplatform for cancer treatment: A review. Eur. Polym. J. 2020, 126, 109546. [Google Scholar] [CrossRef]
- Tunki, L.; Kulhari, H.; Sistla, R.; Pooja, D. 5—Dendrimer-Based Targeted Drug Delivery. In Pharmaceutical Applications of Dendrimers; Chauhan, A., Kulhari, H., Eds.; Micro and Nano Technologies; Elsevier: Amsterdam, The Netherlands, 2020; pp. 107–129. ISBN 978-0-12-814527-2. [Google Scholar]
- Mahesh, S.; Tang, K.-C.; Raj, M. Amide Bond Activation of Biological Molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.-F.; Chen, W.-H.; Liu, Y.; Lei, Q.; Zhuo, R.-X.; Zhang, X.-Z. Multifunctional Enveloped Mesoporous Silica Nanoparticles for Subcellular Co-delivery of Drug and Therapeutic Peptide. Sci. Rep. 2014, 4, 6064. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, P.R.; Wilson, I.D.; Gill, R.U.; Butler, P.J.; Dilworth, C.; Athersuch, T.J. Metabolic Hydrolysis of Aromatic Amides in Selected Rat, Minipig, and Human In Vitro Systems. Sci. Rep. 2018, 8, 2405. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Majumder, M.M.; Lievonen, J.; Silvennoinen, R.; Anttila, P.; Nupponen, N.N.; Lehmann, F.; Heckman, C.A. Prognostic significance of esterase gene expression in multiple myeloma. Br. J. Cancer 2021, 124, 1428–1436. [Google Scholar] [CrossRef]
- Dong, H.; Pang, L.; Cong, H.; Shen, Y.; Yu, B. Application and design of esterase-responsive nanoparticles for cancer therapy. Drug Deliv. 2019, 26, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Kurtoglu, Y.E.; Mishra, M.K.; Kannan, S.; Kannan, R.M. Drug release characteristics of PAMAM dendrimer–drug conjugates with different linkers. Int. J. Pharm. 2010, 384, 189–194. [Google Scholar] [CrossRef]
- Markowicz, J.; Uram, Ł.; Wołowiec, S.; Rode, W. Biotin Transport-Targeting Polysaccharide-Modified PAMAM G3 Dendrimer as System Delivering α-Mangostin into Cancer Cells and C. elegans Worms. Int. J. Mol. Sci. 2021, 22, 12925. [Google Scholar] [CrossRef]
- Perumal, D.; Golla, M.; Pillai, K.S.; Raj, G.; Krishna, P.K.A.; Varghese, R. Biotin-decorated NIR-absorbing nanosheets for targeted photodynamic cancer therapy. Org. Biomol. Chem. 2021, 19, 2804–2810. [Google Scholar] [CrossRef]
- Uram, Ł.; Szuster, M.; Filipowicz, A.; Zaręba, M.; Wałajtys-Rode, E.; Wolowiec, S. Cellular uptake of glucoheptoamidated poly(amidoamine) PAMAM G3 dendrimer with amide-conjugated biotin, a potential carrier of anticancer drugs. Bioorg. Med. Chem. 2017, 25, 706–713. [Google Scholar] [CrossRef]
- Yang, W.; Cheng, Y.; Xu, T.; Wang, X.; Wen, L.-P. Targeting cancer cells with biotin–dendrimer conjugates. Eur. J. Med. Chem. 2009, 44, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Yellepeddi, V.; Kumar, A.; Palakurthi, S. Biotinylated poly(amido)amine (PAMAM) dendrimers as carriers for drug delivery to ovarian cancer cells in vitro. Anticancer Res. 2009, 29, 2933–2943. [Google Scholar] [PubMed]
- Ma, J.; Yao, H. Dendrimer-paclitaxel complexes for efficient treatment in ovarian cancer: Study on OVCAR-3 and HEK293T cells. Acta Biochim. Pol. 2018, 65, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, R.; Hall, A.; Spaulding, R.; Rossow, B.; Hester, M.; Caroway, M.; Haskamp, A.; Wall, S.; Bullen, H.A.; Morris, C.; et al. Analysis of Biotinylated Generation 4 Poly(amidoamine) (PAMAM) Dendrimer Distribution in the Rat Brain and Toxicity in a Cellular Model of the Blood-Brain Barrier. Molecules 2013, 18, 11537–11552. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.E.S.; Brender, J.R.; Dürr, U.H.N.; Xu, J.; Mullen, D.G.; Holl, M.M.B.; Ramamoorthy, A. Solid-State NMR Reveals the Hydrophobic-Core Location of Poly(amidoamine) Dendrimers in Biomembranes. J. Am. Chem. Soc. 2010, 132, 8087–8097. [Google Scholar] [CrossRef] [Green Version]
- Honnen, S. Caenorhabditis elegans as a powerful alternative model organism to promote research in genetic toxicology and biomedicine. Arch. Toxicol. 2017, 91, 2029–2044. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Czarnik-Kwaśniak, J.; Kwaśniak, K.; Tutaj, K.; Filiks, I.; Uram, Ł.; Stompor, M.; Wołowiec, S. Glucoheptoamidated polyamidoamine PAMAM G3 dendrimer as a vehicle for succinate linked doxorubicin; enhanced toxicity of DOX against grade IV glioblastoma U-118 MG cells. J. Drug Deliv. Sci. Technol. 2020, 55, 101424. [Google Scholar] [CrossRef]
- Stiernagle, T. Maintenance of C. elegans. In WormBook; The C. elegans Research Community, Ed.; WormBook: Pasadena, CA, USA, 2006. [Google Scholar] [CrossRef] [Green Version]
- Crosignani, S.; Gonzalez, A.J.; Swinnen, D. Polymer-Supported Mukaiyama Reagent: A Useful Coupling Reagent for the Synthesis of Esters and Amides. Org. Lett. 2004, 6, 4579–4582. [Google Scholar] [CrossRef]
- Czerniecka-Kubicka, A.; Tutka, P.; Pyda, M.; Walczak, M.; Uram, Ł.; Misiorek, M.; Chmiel, E.; Wołowiec, S. Stepwise Glucoheptoamidation of Poly(Amidoamine) Dendrimer G3 to Tune Physicochemical Properties of the Potential Drug Carrier: In Vitro Tests for Cytisine Conjugates. Pharmaceutics 2020, 12, 473. [Google Scholar] [CrossRef]
- Martinho, N.; Florindo, H.; Silva, L.; Brocchini, S.; Zloh, M.; Barata, T. Molecular Modeling to Study Dendrimers for Biomedical Applications. Molecules 2014, 19, 20424–20467. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-J.; Ding, Z.-S.; Jiang, F.-S.; Ge, Q.-F.; Guo, D.-W.; Li, H.-B.; Hu, W.-X. Synthesis, anticancer evaluation and docking study of vadimezan derivatives with carboxyl substitution. MedChemComm 2014, 5, 512–520. [Google Scholar] [CrossRef]
- Lv, S.; Tang, Z.; Song, W.; Zhang, D.; Li, M.; Liu, H.; Cheng, J.; Zhong, W.; Chen, X. Inhibiting Solid Tumor Growth In Vivo by Non-Tumor-Penetrating Nanomedicine. Small 2017, 13, 1600954. [Google Scholar] [CrossRef]
- AAT Bioquest. Available online: https://www.aatbio.com/tools/ic50-calculator (accessed on 6 January 2022).
- Kurniawan, Y.S.; Priyangga, K.T.A.; Jumina; Pranowo, H.D.; Sholikhah, E.N.; Zulkarnain, A.K.; Fatimi, H.A.; Julianus, J. An Update on the Anticancer Activity of Xanthone Derivatives: A Review. Pharmaceuticals 2021, 14, 1144. [Google Scholar] [CrossRef]
- Castanheiro, R.A.P.; Silva, A.M.S.; Campos, N.A.N.; Nascimento, M.S.J.; Pinto, M.M.M. Antitumor Activity of Some Prenylated Xanthones. Pharmaceuticals 2009, 2, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Teh, S.S.; Ee, G.C.L.; Mah, S.H.; Lim, Y.M.; Ahmad, Z. Cytotoxicity and Structure-Activity Relationships of Xanthone Derivatives from Mesua beccariana, Mesua ferrea and Mesua congestiflora towards Nine Human Cancer Cell Lines. Molecules 2013, 18, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Jain, K.; Mehra, N.K.; Jain, N.K. Dendrimers in anticancer drug delivery: Mechanism of interaction of drug and dendrimers. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1626–1634. [Google Scholar] [CrossRef]
- Zeng, Y.; Kurokawa, Y.; Win-Shwe, T.-T.; Zeng, Q.; Hirano, S.; Zhang, Z.; Sone, H. Effects of PAMAM dendrimers with various surface functional groups and multiple generations on cytotoxicity and neuronal differentiation using human neural progenitor cells. J. Toxicol. Sci. 2016, 41, 351–370. [Google Scholar] [CrossRef] [Green Version]
- Janaszewska, A.; Lazniewska, J.; Trzepiński, P.; Marcinkowska, M.; Klajnert-Maculewicz, B. Cytotoxicity of Dendrimers. Biomolecules 2019, 9, 330. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Raczka, E.; Piehler, L.; Lee, I.; Myc, A.; Majoros, I.; Patri, A.K.; Thomas, T.P.; Mulé, J.; Baker, J.R., Jr. Design and Function of a Dendrimer-Based Therapeutic Nanodevice Targeted to Tumor Cells Through the Folate Receptor. Pharm. Res. 2002, 19, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.P.; Majoros, I.J.; Kotlyar, A.; Kukowska-Latallo, J.F.; Bielinska, A.; Myc, A.A.; Baker, J.J.R. Targeting and Inhibition of Cell Growth by an Engineered Dendritic Nanodevice. J. Med. Chem. 2005, 48, 3729–3735. [Google Scholar] [CrossRef]
- de Almeida, M.S.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397–5434. [Google Scholar] [CrossRef]
- Adli, A.D.F.; Jahanban-Esfahlan, R.; Seidi, K.; Farajzadeh, D.; Behzadi, R.; Zarghami, N. Co-Administration of Vadimezan and Recombinant Coagulase-NGR Inhibits Growth of Melanoma Tumor in Mice. Adv. Pharm. Bull. 2021, 11, 385–392. [Google Scholar] [CrossRef]
- van Dongen, M.A.; Vaidyanathan, S.; Holl, M.M.B. PAMAM dendrimers as quantized building blocks for novel nanostructures. Soft Matter 2013, 9, 11188–11196. [Google Scholar] [CrossRef] [Green Version]
- Uram, Ł.; Szuster, M.; Misiorek, M.; Filipowicz, A.; Wołowiec, S.; Wałajtys-Rode, E. The effect of G3 PAMAM dendrimer conjugated with B-group vitamins on cell morphology, motility and ATP level in normal and cancer cells. Eur. J. Pharm. Sci. 2017, 102, 275–283. [Google Scholar] [CrossRef]
- Kurose, H.; Shibata, M.-A.; Iinuma, M.; Otsuki, Y. Alterations in Cell Cycle and Induction of Apoptotic Cell Death in Breast Cancer Cells Treated with α-Mangostin Extracted from Mangosteen Pericarp. J. Biomed. Biotechnol. 2012, 2012, 672428. [Google Scholar] [CrossRef] [Green Version]
- Kwak, H.-H.; Kim, I.-R.; Kim, H.-J.; Park, B.-S.; Yu, S.-B. α-Mangostin Induces Apoptosis and Cell Cycle Arrest in Oral Squamous Cell Carcinoma Cell. Evid.-Based Complement. Altern. Med. 2016, 2016, 5352412. [Google Scholar] [CrossRef] [Green Version]
- Lucio, D.; Martínez-Ohárriz, M.C.; Gu, Z.; He, Y.; Aranaz, P.; Vizmanos, J.L.; Irache, J.M. Cyclodextrin-grafted poly(anhydride) nanoparticles for oral glibenclamide administration. In vivo evaluation using C. elegans. Int. J. Pharm. 2018, 547, 97–105. [Google Scholar] [CrossRef]
- Martínez-López, A.L.; González-Navarro, C.J.; Aranaz, P.; Vizmanos, J.L.; Irache, J.M. In vivo testing of mucus-permeating nanoparticles for oral insulin delivery using Caenorhabditis elegans as a model under hyperglycemic conditions. Acta Pharm. Sin. B 2021, 11, 989–1002. [Google Scholar] [CrossRef]
- Ma, H.; Bertsch, P.M.; Glenn, T.C.; Kabengi, N.; Williams, P.L. Toxicity of manufactured zinc oxide nanoparticles in the nematode Caenorhabditis elegans. Environ. Toxicol. Chem. 2009, 28, 1324–1330. [Google Scholar] [CrossRef]
- Marcelino, M.Y.; Borges, F.A.; Scorzoni, L.; Singulani, J.D.L.; Garms, B.C.; Niemeyer, J.C.; Guerra, N.B.; Brasil, G.S.P.; Mussagy, C.U.; Carvalho, F.A.D.O.; et al. Synthesis and characterization of gold nanoparticles and their toxicity in alternative methods to the use of mammals. J. Environ. Chem. Eng. 2021, 9, 106779. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Size [nm] | Zeta Potential [mV] | ||||
|---|---|---|---|---|---|---|
| pH 7 | pH 5 | pH 7 | pH 5 | |||
| d(V) | d(N) | d(V) | d(N) | |||
| G3gh | 1.7 ± 0.44 | 1.5 ± 0.51 | 4.3 ± 0.34 | 3.6 ± 0.32 | 4.5 ± 0.58 | 5.3 ± 0.81 |
| G3gh2B5M | 155.1 ± 3.46 | 113 ± 5.89 | 212.7 ± 4.29 | 144.5 ± 6.49 | 10 ± 0.39 | 24.9 ± 1.78 |
| G32B12gh5M [29] | 1367 ± 245.7 | 1262 ± 196.4 | 178.3 ± 5.73 | 113.8 ± 7.34 | 22.7 ± 1.01 | 37.5 ± 2.32 |
| G3gh4B5V | 3.7 ± 0.23 | 2.9 ± 0.56 | 4.9 ± 0.14 | 4.2 ± 0.18 | 7.6 ± 4.35 | 6.2 ± 0.81 |
| G32B12gh5V | 1166 ± 153 | 970.4 ± 81.6 | 1090 ± 39.2 | 924.4 ± 39 | 22.8 ± 0.69 | 33.4 ± 2.82 |
| IC50 [μM] NR Assay | |||
| BJ | U-118 MG | SCC-15 | |
| α-mangostin [2] | 8.97 | 9.59 | 6.43 |
| G3gh2B5M | 2.02 | 2.52 | 1.7 |
| G32B12gh5M [29] | 2 | 1.83 | 1.41 |
| Vadimezan | >>200 | >>200 | >>200 |
| G3gh4B5V | >>24 | >>24 | >>24 |
| G32B12gh5V | >>12 | >>12 | >>12 |
| IC50 [μM] XTT Assay | |||
| BJ | U-118 MG | SCC-15 | |
| α-mangostin [2] | 18.58 | 18.15 | 7.72 |
| G3gh2B5M | 3.36 | 4.14 | 3.02 |
| G32B12gh5M [29] | 2.37 | 2.05 | 2.52 |
| Vadimezan | >>200 | >>200 | >200 |
| G3gh4B5V | >>24 | >>24 | >>24 |
| G32B12gh5V | >>12 | >>12 | >>12 |
| LC50 [μM] | 1st Quartile | 3rd Quartile | |
|---|---|---|---|
| α-Mangostin [2] | 18.74 | 18.64 | 20.23 |
| G3gh2B5M | >16 | >16 | >16 |
| G32B12gh5M [29] | 7.87 | 4.95 | 8.09 |
| Vadimezan | >>200 | >>200 | >>200 |
| G3gh4B5V | >>24 | >>24 | >>24’ |
| G32B12gh5V | >>12 | >>12 | >>12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markowicz, J.; Wołowiec, S.; Rode, W.; Uram, Ł. Synthesis and Properties of α-Mangostin and Vadimezan Conjugates with Glucoheptoamidated and Biotinylated 3rd Generation Poly(amidoamine) Dendrimer, and Conjugation Effect on Their Anticancer and Anti-Nematode Activities. Pharmaceutics 2022, 14, 606. https://doi.org/10.3390/pharmaceutics14030606
Markowicz J, Wołowiec S, Rode W, Uram Ł. Synthesis and Properties of α-Mangostin and Vadimezan Conjugates with Glucoheptoamidated and Biotinylated 3rd Generation Poly(amidoamine) Dendrimer, and Conjugation Effect on Their Anticancer and Anti-Nematode Activities. Pharmaceutics. 2022; 14(3):606. https://doi.org/10.3390/pharmaceutics14030606
Chicago/Turabian StyleMarkowicz, Joanna, Stanisław Wołowiec, Wojciech Rode, and Łukasz Uram. 2022. "Synthesis and Properties of α-Mangostin and Vadimezan Conjugates with Glucoheptoamidated and Biotinylated 3rd Generation Poly(amidoamine) Dendrimer, and Conjugation Effect on Their Anticancer and Anti-Nematode Activities" Pharmaceutics 14, no. 3: 606. https://doi.org/10.3390/pharmaceutics14030606