
Injectable Biodegradable Silica Depot: Two Months of Sustained Release of the Blood Glucose Lowering Peptide, Pramlintide

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Silica Microparticles
2.3. Preparation of Pramlintide-Silica Microparticle Silica Hydrogel Depot Formulation
2.4. In Vitro Dissolution Test
2.5. HPLC Analysis of PRAMLINTIDE
2.6. Total Silica Content Measurement
2.7. Total Pramlintide Content Measurement
2.8. Characterization of Pramlintide-silica Microparticles and Depot Formulation
2.9. Endotoxin Analysis
2.10. Pharmacokinetic and Pharmacodynamic Study in Rat
3. Results and Discussion
3.1. Preparation of Pramlintide Loaded Silica Microparticles
3.2. SEM Analysis and Particle Size Distribution of the Microparticles
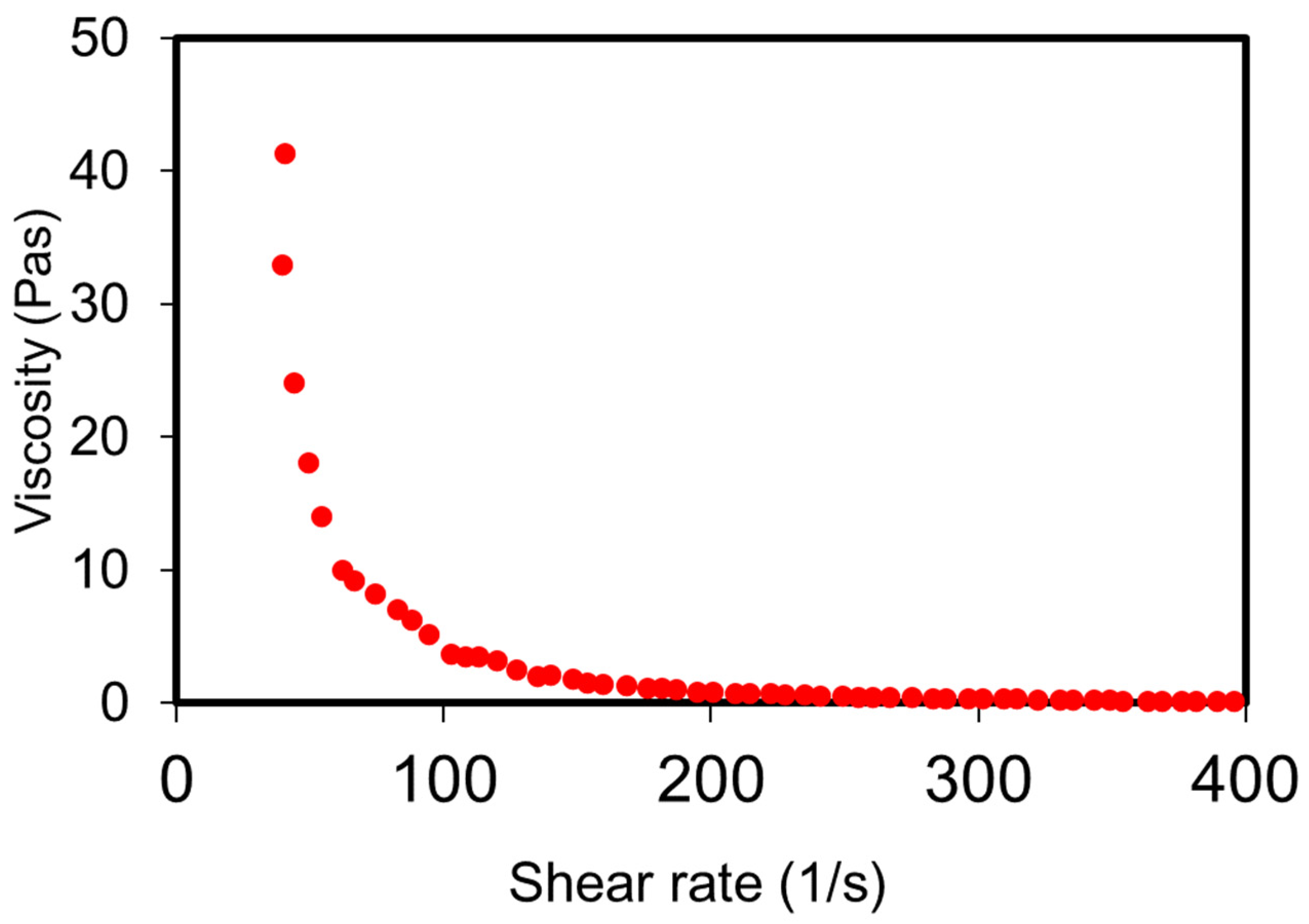
3.3. Rheology of the Depot Formulation
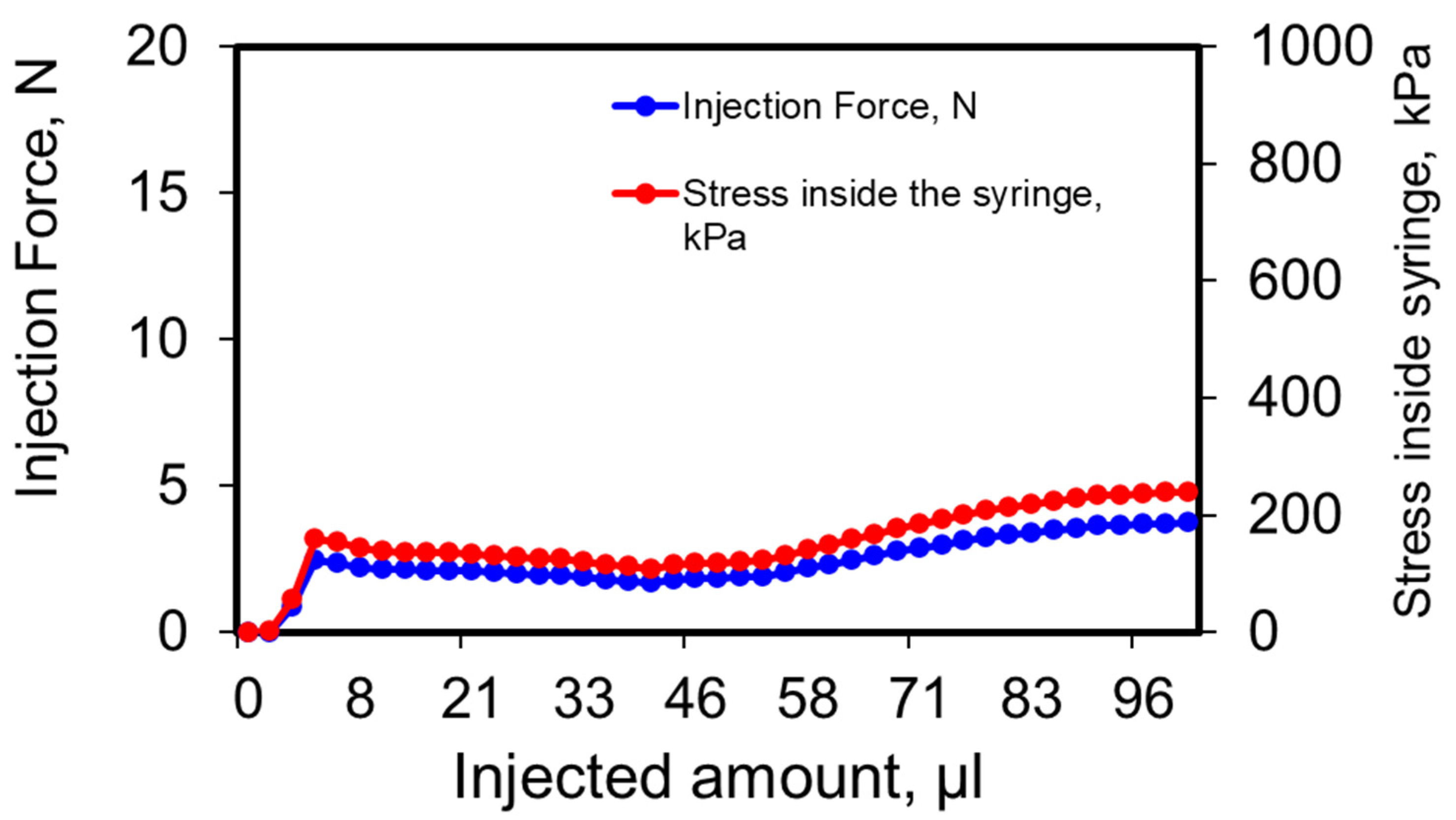
3.4. Injectability of Depot Formulation
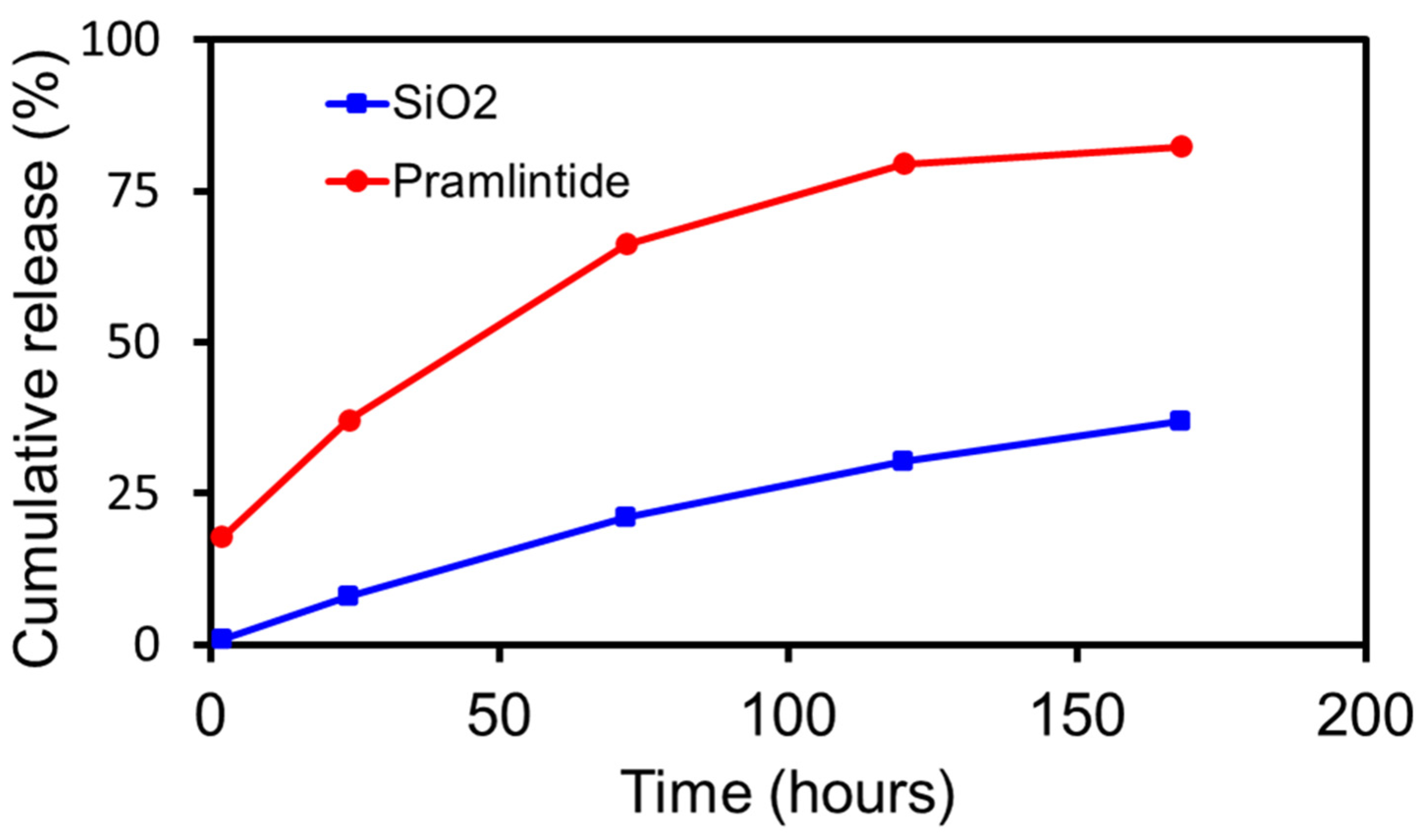
3.5. In Vitro Dissolution
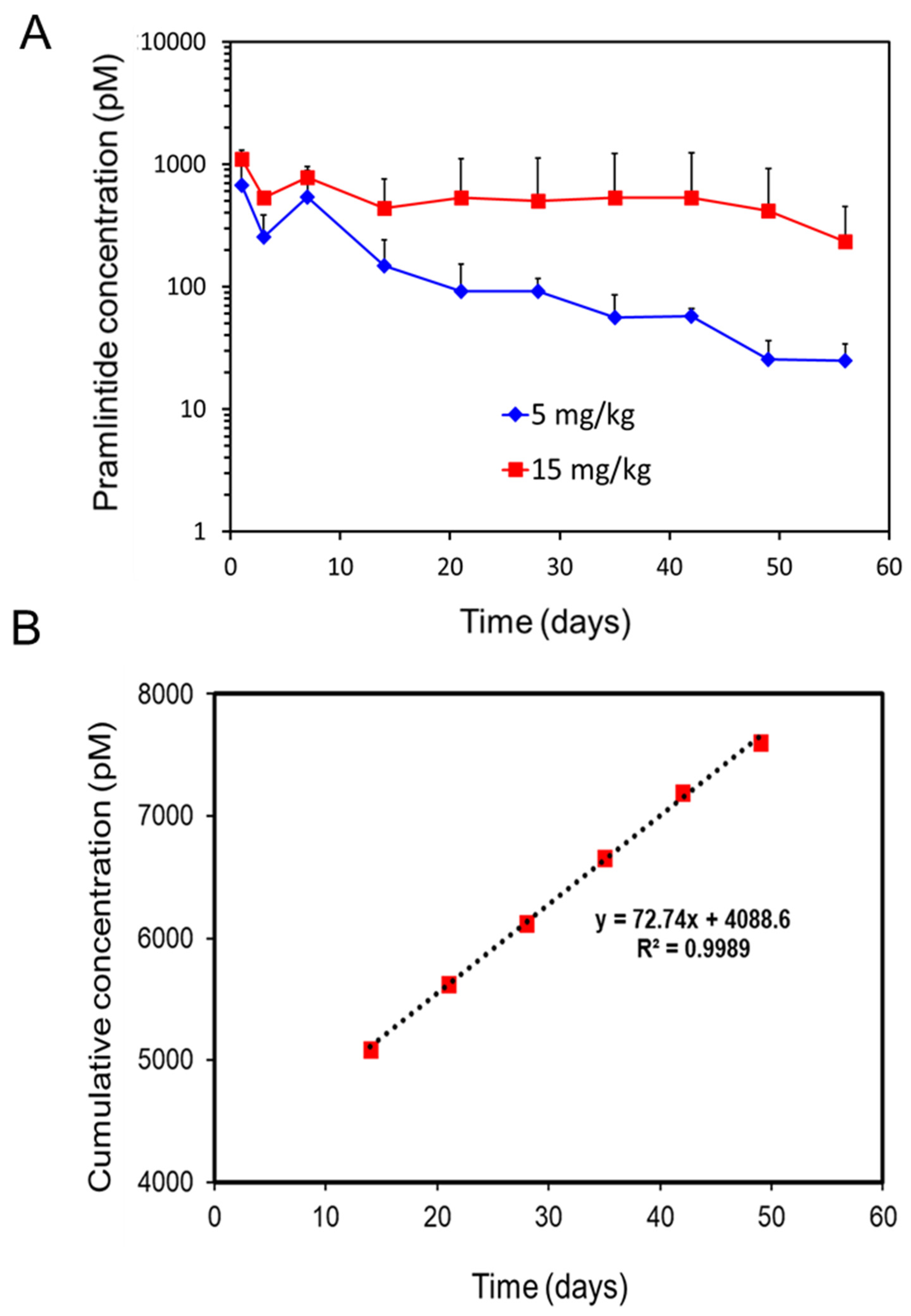
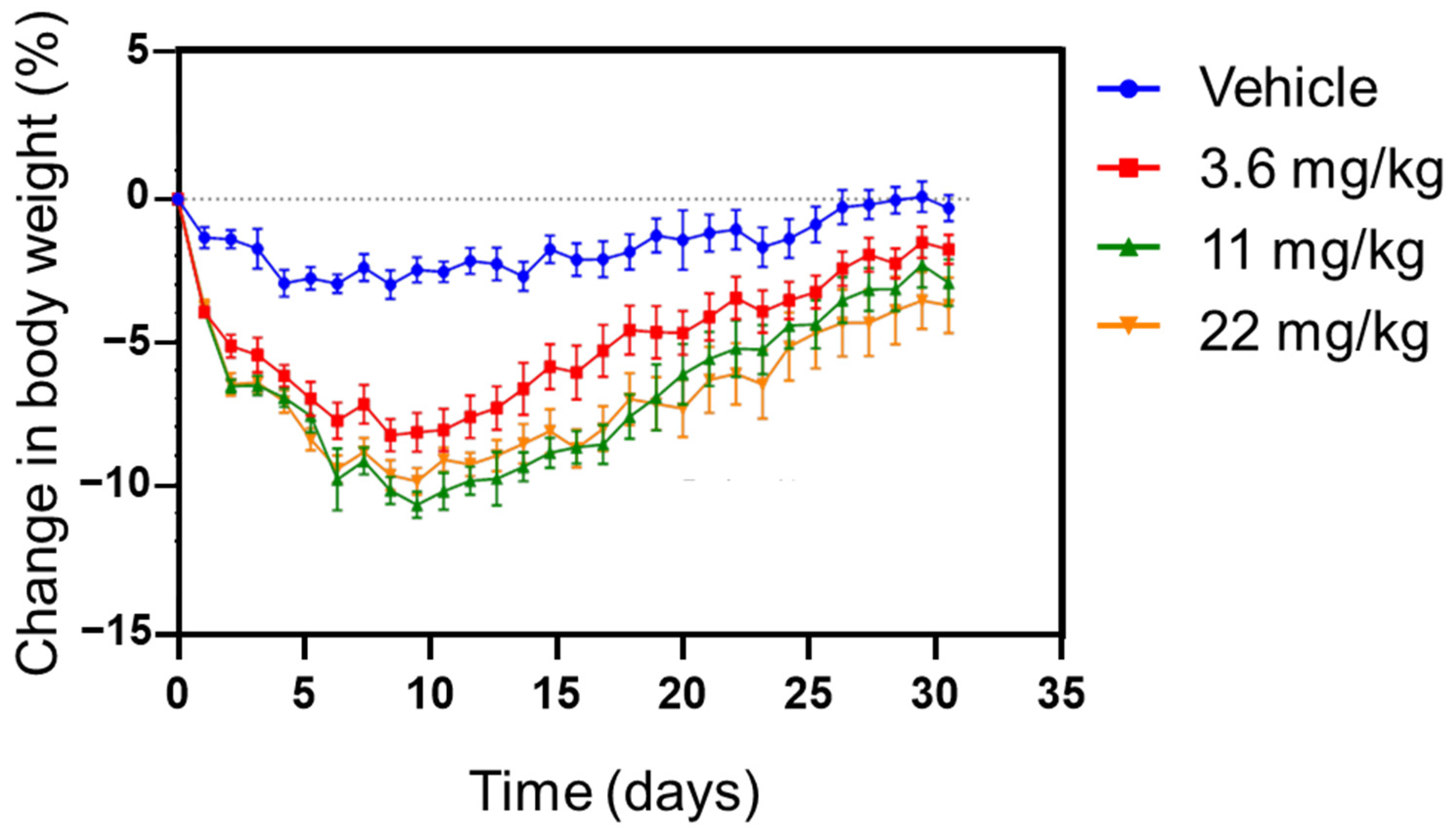
3.6. Pharmacokinetic and Pharmcodynamic Study in Rat
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diabetes Facts & Figures. Available online: http://www.idf.org (accessed on 22 February 2022).
- National Diabetes Data Group. Classification and Diagnosis of Diabetes Mellitus and Other Categories of Glucose Intolerance. Diabetes 1979, 28, 1039–1057. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.K.; Srivastava, A.K. Diabetes mellitus: Complications and therapeutics. Med. Sci. Monit. 2006, 12, RA130–RA147. [Google Scholar] [PubMed]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.C.; Cull, C.A.; Frighi, V.; Holman, R.R.; for the UK Prospective Diabetes Study (UKPDS) Group. Glycemic Control With Diet, Sulfonylurea, Metformin, or Insulin in Patients With Type 2 Diabetes Mellitus Progressive Requirement for Multiple Therapies (UKPDS 49). JAMA 1999, 281, 2005–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groop, L. Bringing diabetes therapeutics to the big screen. Nat. Biotechnol. 2003, 21, 240–241. [Google Scholar] [CrossRef]
- Valla, V. Therapeutics of Diabetes Mellitus: Focus on Insulin Analogues and Insulin Pumps. Exp. Diabetes Res. 2010, 2010, 178372. [Google Scholar] [CrossRef] [Green Version]
- Veiseh, O.; Tang, B.C.; Whitehead, K.A.; Anderson, D.G.; Langer, R. Managing diabetes with nanomedicine: Challenges and opportunities. Nat. Rev. Drug Discov. 2015, 14, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.R.; Monnier, L.; Barnett, A.H. Future challenges and therapeutic opportunities in type 2 diabetes: Changing the paradigm of current therapy. Diabetes Obes. Metab. 2017, 19, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Walsh, G. Biopharmaceuticals: Recent approvals and likely directions. Trends Biotechnol. 2005, 23, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Wu-Pong, S.; Rojanasakul, Y. (Eds.) Biopharmaceutical Drug Design and Development; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Walsh, G. Biopharmaceuticals: Biochemistry and Biotechnology; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Shire, S.J. Formulation and manufacturability of biologics. Curr. Opin. Biotechnol. 2009, 20, 708–714. [Google Scholar] [CrossRef]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, O.; Brock, B.; Rungby, J. Amylin Agonists: A Novel Approach in the Treatment of Diabetes. Diabetes 2004, 53, S233–S238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQueen, J. Pramlintide acetate. Am. J. Health-Syst. Pharm. 2005, 62, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.; Jobe, L.; Briscoe, T. Review of pramlintide as adjunctive therapy in treatment of type 1 and type 2 diabetes. Drug Des. Dev. Ther. 2008, 2, 203–214. [Google Scholar] [CrossRef] [Green Version]
- McCoy, R.G.; Van Houten, H.K.; Ziegenfuss, J.Y.; Shah, N.D.; Wermers, R.A.; Smith, S.A.; Gruden, G.; Barutta, F.; Chaturvedi, N.; Schalkwijk, C.; et al. Increased Mortality of Patients With Diabetes Reporting Severe Hypoglycemia. Diabetes Care 2012, 35, 1897–1901. [Google Scholar] [CrossRef] [Green Version]
- Bode, B.W.; Steed, R.D.; Davidson, P.C. Reduction in Severe Hypoglycemia With Long-Term Continuous Subcutaneous Insulin Infusion in Type I Diabetes. Diabetes Care 1996, 19, 324–327. [Google Scholar] [CrossRef]
- Huffman, D.M.; McLean, G.W.; Seagrove, M.A. Continuous Subcutaneous Pramlintide Infusion Therapy In Patients With Type 1 Diabetes: Observations From A Pilot Study. Endocr. Pract. 2009, 15, 689–695. [Google Scholar] [CrossRef]
- LinBit and Linplant Product Instructions. Available online: http://www.linshincanada.com (accessed on 22 February 2022).
- Vilsbøll, T.; Agersø, H.; Krarup, T.; Holst, J.J. Similar Elimination Rates of Glucagon-Like Peptide-1 in Obese Type 2 Diabetic Patients and Healthy Subjects. J. Clin. Endocrinol. Metab. 2003, 88, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur. J. Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef]
- Choi, S.; Baudys, M.; Kim, S.W. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm. Res. 2004, 21, 827–831. [Google Scholar] [CrossRef]
- Crotts, G.; Park, T.G. Protein delivery from poly(lactic-co-glycolic acid) biodegradable microspheres: Release kinetics and stability issues. J. Microencapsul. 1998, 15, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, R.; Babu, R.J.; Palakurthi, S. Nanomedicine Scale-up Technologies: Feasibilities and Challenges. AAPS PharmSciTech 2014, 15, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luginbuhl, K.M.; Schaal, J.; Umstead, B.; Mastria, E.; Li, X.; Banskota, S.; Arnold, S.; Feinglos, M.; D’Alessio, D.; Chilkoti, A. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng. 2017, 1, 78. [Google Scholar] [CrossRef] [PubMed]
- Kortesuo, P.; Ahola, M.; Kangas, M.; Kangasniemi, I.; Yli-Urpo, A.; Kiesvaara, J. In vitro evaluation of sol–gel processed spray dried silica gel microspheres as carrier in controlled drug delivery. Int. J. Pharm. 2000, 200, 223–229. [Google Scholar] [CrossRef]
- Kortesuo, P.; Ahola, M.; Karlsson, S.; Kangasniemi, I.; Yli-Urpo, A.; Kiesvaara, J. Silica xerogel as an implantable carrier for controlled drug delivery—evaluation of drug distribution and tissue effects after implantation. Biomaterials 1999, 21, 193–198. [Google Scholar] [CrossRef]
- Viitala, R.; Jokinen, M.; Tuusa, S.; Rosenholm, J.B.; Jalonen, H. Adjustably biodegradable sol-gel derived SiO2 matrices for protein release. J. Sol-Gel Sci. Tech. 2005, 36, 147–156. [Google Scholar] [CrossRef]
- Viitala, R.; Jokinen, M.; Maunu, S.L.; Jalonen, H.; Rosenholm, J.B. Chemical characterization of bioresorbable sol–gel derived SiO2 matrices prepared at protein-compatible pH. J. Non-Cryst. Solids 2005, 351, 3225–3234. [Google Scholar] [CrossRef]
- Coradin, T.; Eglin, D.; Livage, J. The silicomolybdic acid spectrophotometric method and its application to silicate/biopolymer interaction studies. Spectroscopy 2004, 18, 567–576. [Google Scholar] [CrossRef]
- Czuryszkiewicz, T.; Areva, S.; Honkanen, M.; Lindén, M. Synthesis of sol gel silica materials providing a slow release of biphosphonate. Colloids Surf. A Physicochem. Eng. Asp. 2005, 254, 69–74. [Google Scholar] [CrossRef]
- Jokinen, M.; Koskinen, M.; Areva, S. Rationale of Using Conventional Sol-Gel Derived SiO2 for Delivery of Biologically Active Agents. Key Eng. Mater. 2008, 377, 195–210. [Google Scholar] [CrossRef]
- Nayak, U.; Queiroga, J. Pinch grip, power grip and wrist twisting strengths of healthy older adults. Gerontechnology 2004, 3, 77–88. [Google Scholar] [CrossRef]
- Arendt-Nielsen, L.; Egekvist, H.; Bjerring, P. Pain following controlled cutaneous insertion of needles with different diameters. Somatosens. Mot. Res. 2006, 23, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Rungseevijitprapa, W.; Bodmeier, R. Injectability of biodegradable in situ forming microparticle systems (ISM). Eur. J. Pharm. Sci. 2009, 36, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef]
- Young, A.A.; Vine, W.; Gedulin, B.R.; Pittner, R.; Janes, S.; Gaeta, L.S.; Percy, A.; Moore, C.X.; Koda, J.E.; Rink, T.J.; et al. Preclinical pharmacology of pramlintide in the rat: Comparisons with human and rat amylin. Drug Dev. Res. 1996, 37, 231–248. [Google Scholar] [CrossRef]
- Dunican, K.C.; Adams, N.M.; Desilets, A.R. The Role of Pramlintide for Weight Loss. Ann. Pharmacother. 2010, 44, 538–545. [Google Scholar] [CrossRef]
- Roth, J.D.; Hughes, H.; Kendall, E.; Baron, A.D.; Anderson, C.M. Antiobesity Effects of the β-Cell Hormone Amylin in Diet-Induced Obese Rats: Effects on Food Intake, Body Weight, Composition, Energy Expenditure, and Gene Expression. Endocrinology 2006, 147, 5855–5864. [Google Scholar] [CrossRef] [Green Version]
- Arnelo, U.; Permert, J.; Adrian, T.E.; Larsson, J.; Westermark, P.; Reidelberger, R.D. Chronic infusion of islet amyloid polypeptide causes anorexia in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1996, 271, R1654–R1659. [Google Scholar] [CrossRef]
- Lutz, T.; Mollet, A.; Rushing, P.; Riediger, T.; Scharrer, E. The anorectic effect of a chronic peripheral infusion of amylin is abolished in area postrema/nucleus of the solitary tract (AP/NTS) lesioned rats. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Mack, C.; Wilson, J.; Athanacio, J.; Reynolds, J.; Laugero, K.; Guss, S.; Vu, C.; Roth, J.; Parkes, D. Pharmacological actions of the peptide hormone amylin in the long-term regulation of food intake, food preference, and body weight. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1855–R1863. [Google Scholar] [CrossRef] [Green Version]
- Young, A.A.; Cooper, G.J.; Carlo, P.A.; Rink, T.J.; Wang, M.W. Response to intravenous injections of amylin and glucagon in fasted, fed, and hypoglycemic rats. Am. J. Physiol. Endocrinol. Metab. 1993, 264, E943–E950. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, P.; Koskinen, M.; Mikkola, J.; Sarkhel, S.; Leino, L.; Seth, A.; Madalli, S.; Will, S.; Howard, V.G.; Brant, H.; et al. Injectable Biodegradable Silica Depot: Two Months of Sustained Release of the Blood Glucose Lowering Peptide, Pramlintide. Pharmaceutics 2022, 14, 553. https://doi.org/10.3390/pharmaceutics14030553
Tyagi P, Koskinen M, Mikkola J, Sarkhel S, Leino L, Seth A, Madalli S, Will S, Howard VG, Brant H, et al. Injectable Biodegradable Silica Depot: Two Months of Sustained Release of the Blood Glucose Lowering Peptide, Pramlintide. Pharmaceutics. 2022; 14(3):553. https://doi.org/10.3390/pharmaceutics14030553
Chicago/Turabian StyleTyagi, Puneet, Mika Koskinen, Jari Mikkola, Sanjay Sarkhel, Lasse Leino, Asha Seth, Shimona Madalli, Sarah Will, Victor G. Howard, Helen Brant, and et al. 2022. "Injectable Biodegradable Silica Depot: Two Months of Sustained Release of the Blood Glucose Lowering Peptide, Pramlintide" Pharmaceutics 14, no. 3: 553. https://doi.org/10.3390/pharmaceutics14030553