
Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate



Abstract
:1. Introduction
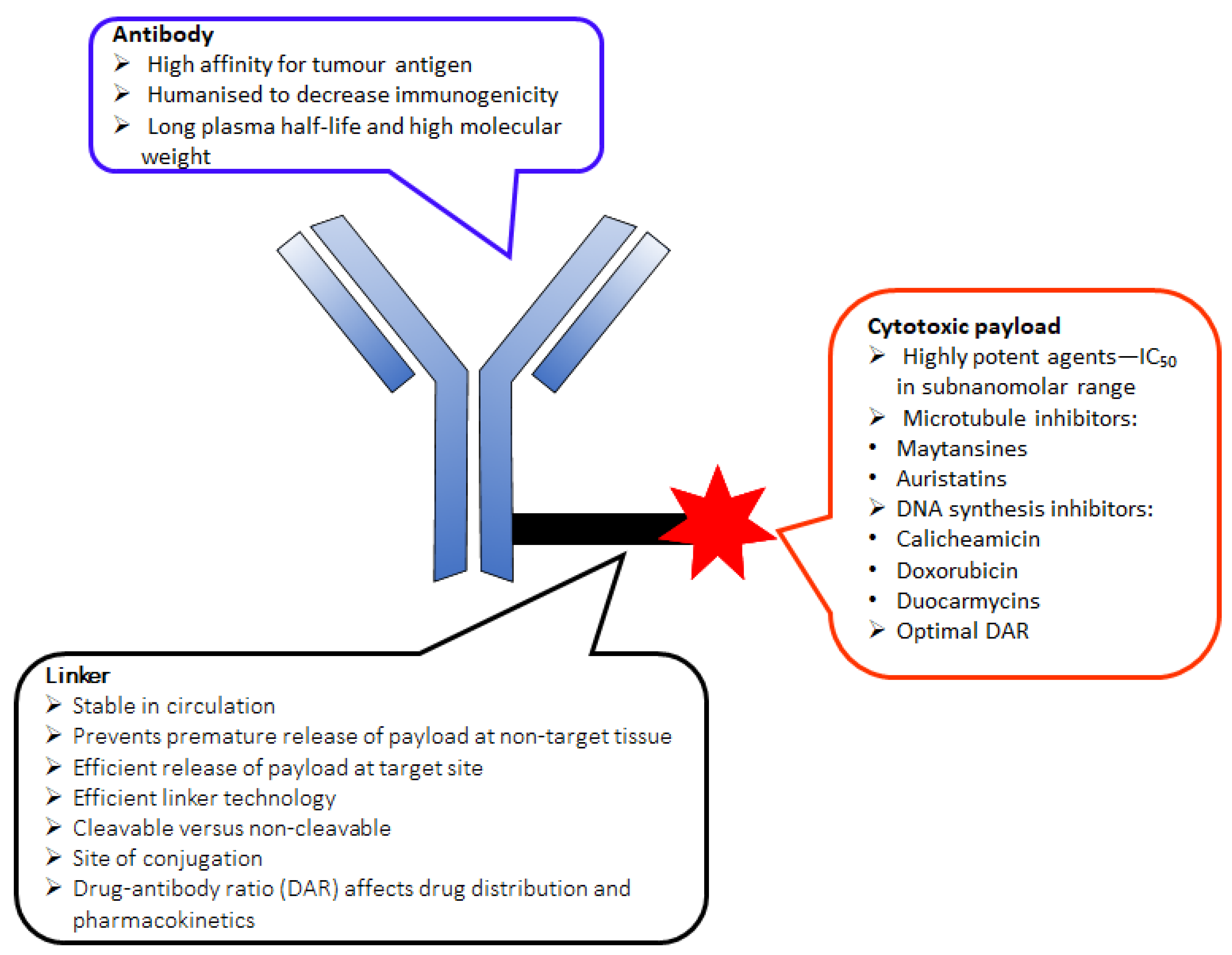
2. The Key Components of an ADC
2.1. Monoclonal Antibody (mAb)
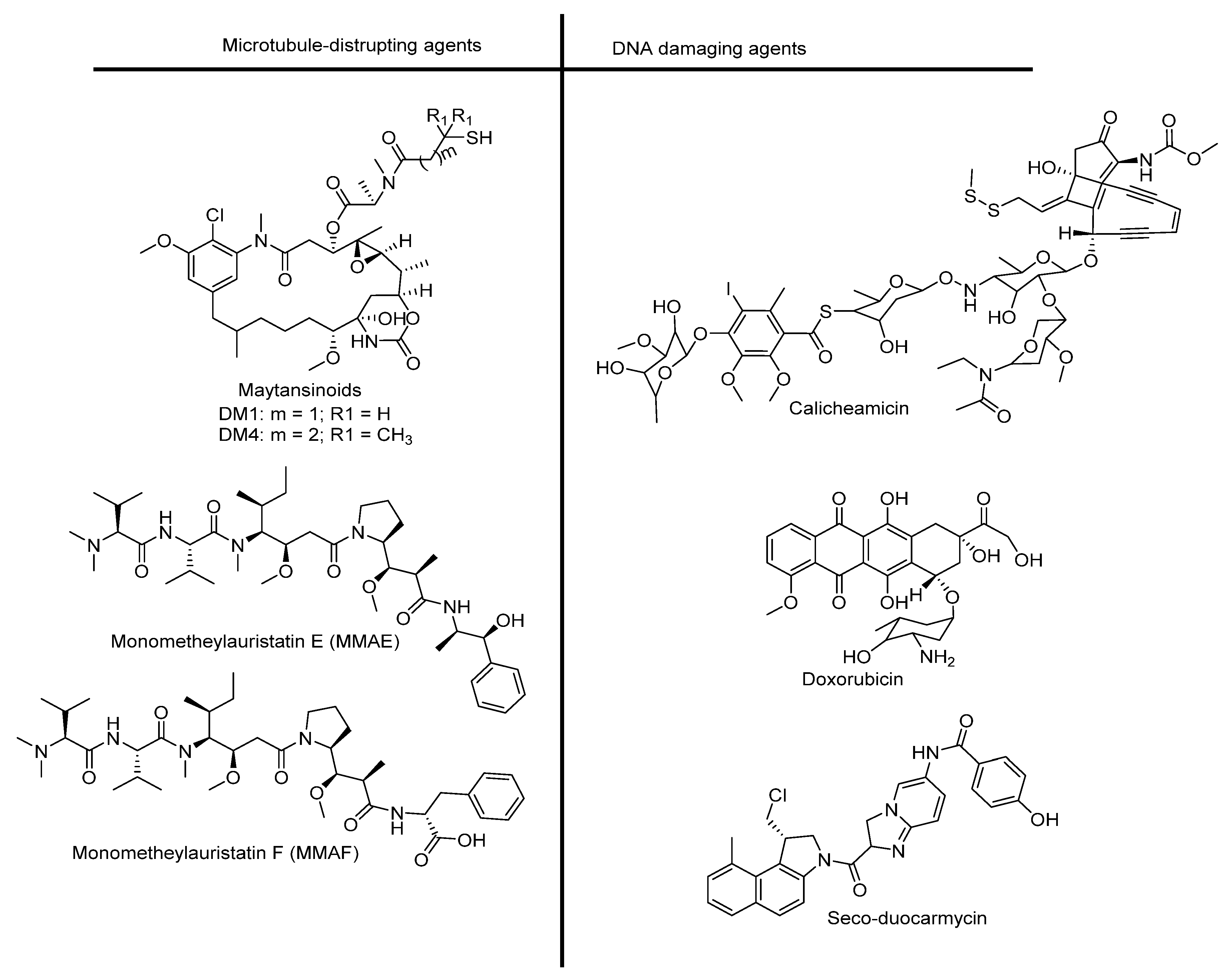
2.2. Cytotoxic Drug
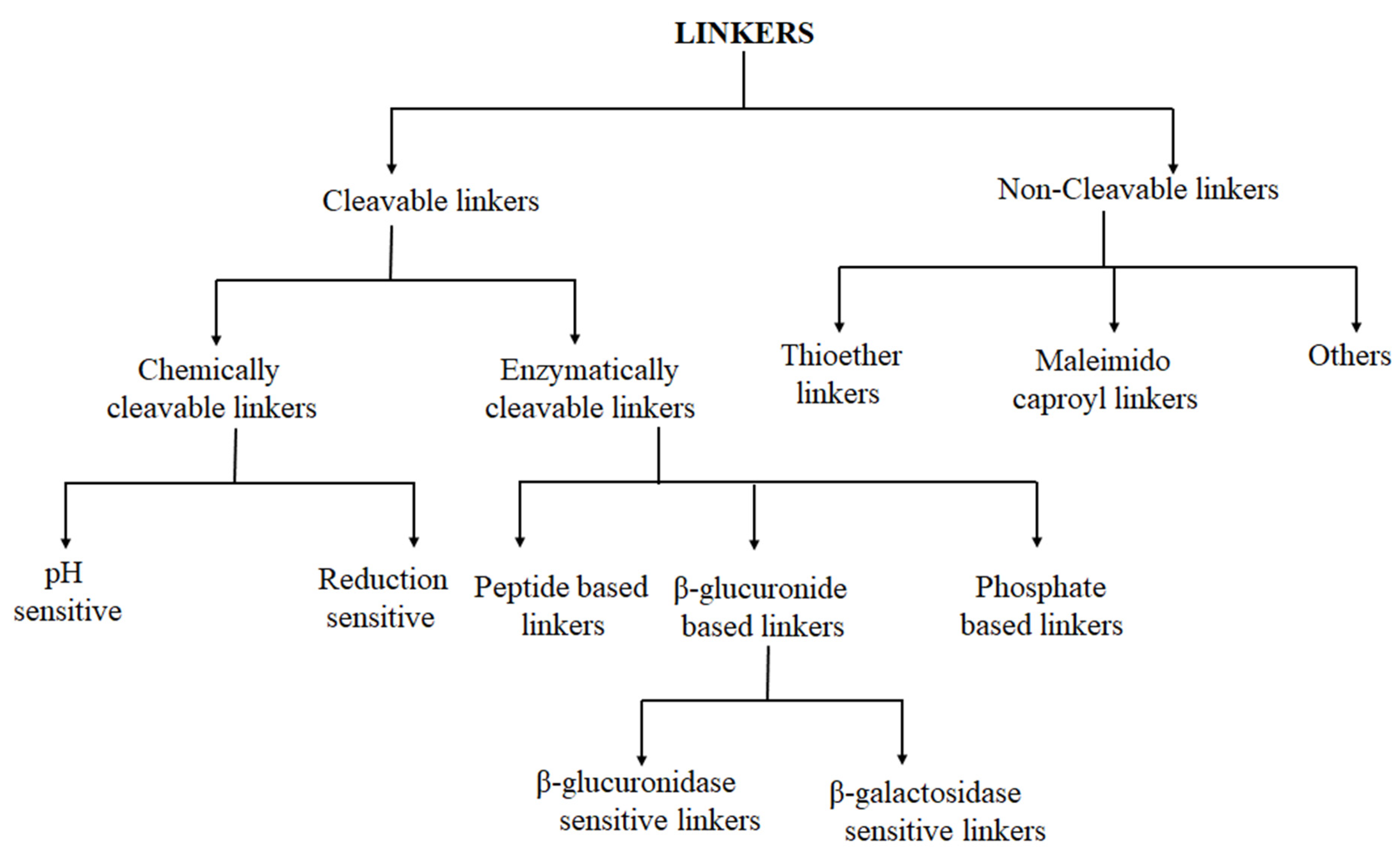
3. Linker Chemistry and Conjugation to Antibody
3.1. Cleavable Linkers
3.1.1. Chemically Labile Linkers
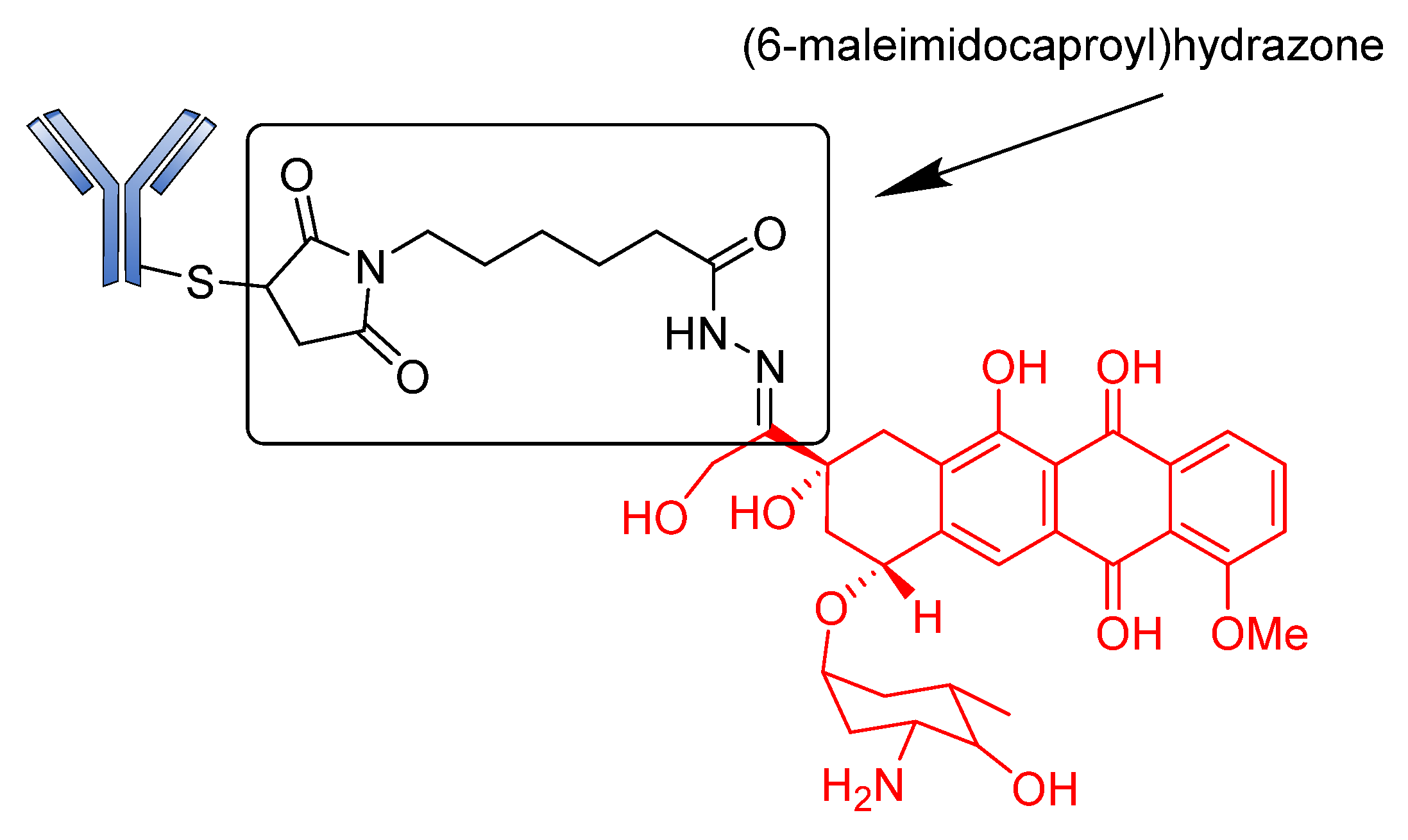
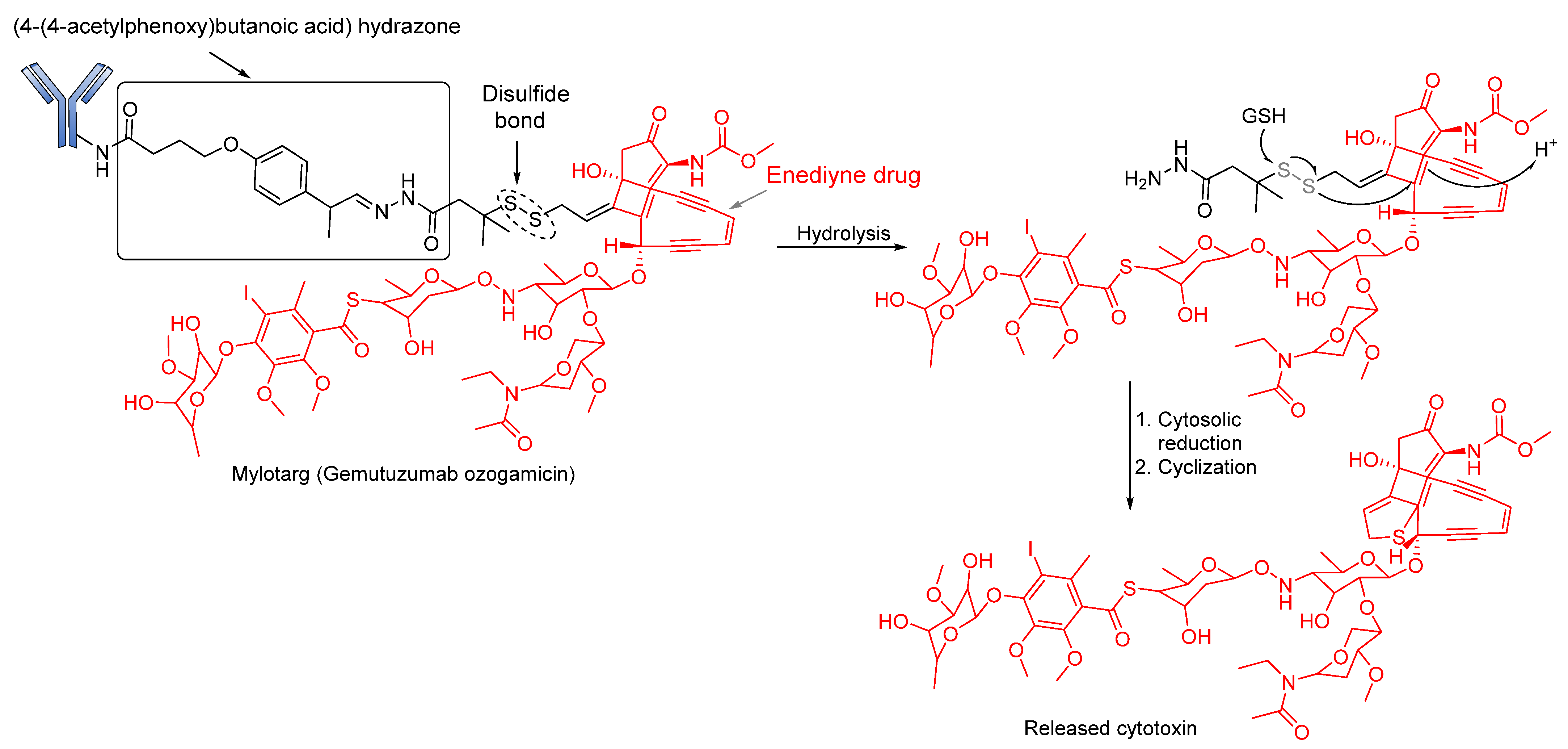
Acid-Cleavable/pH-Sensitive Linkers
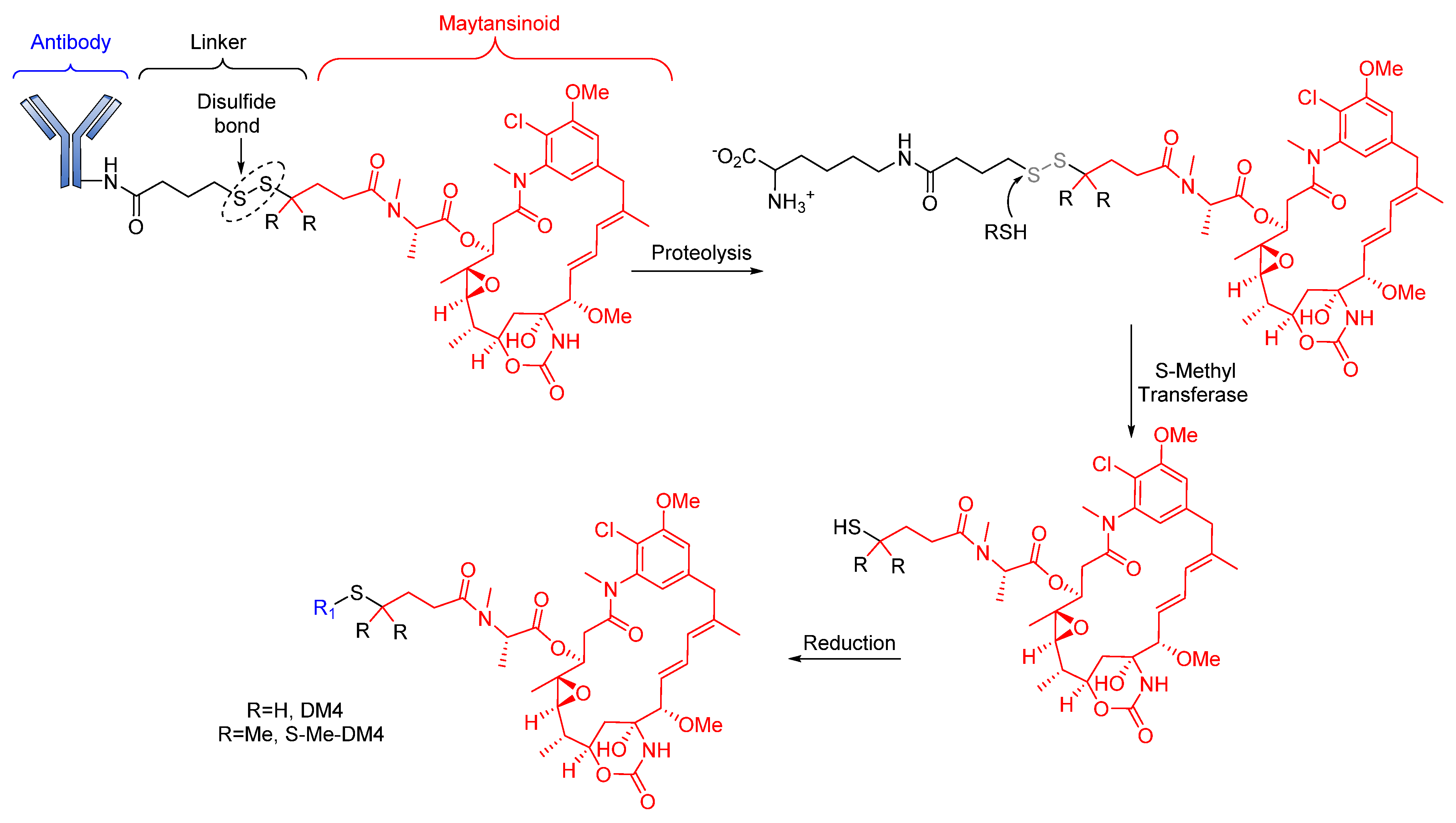
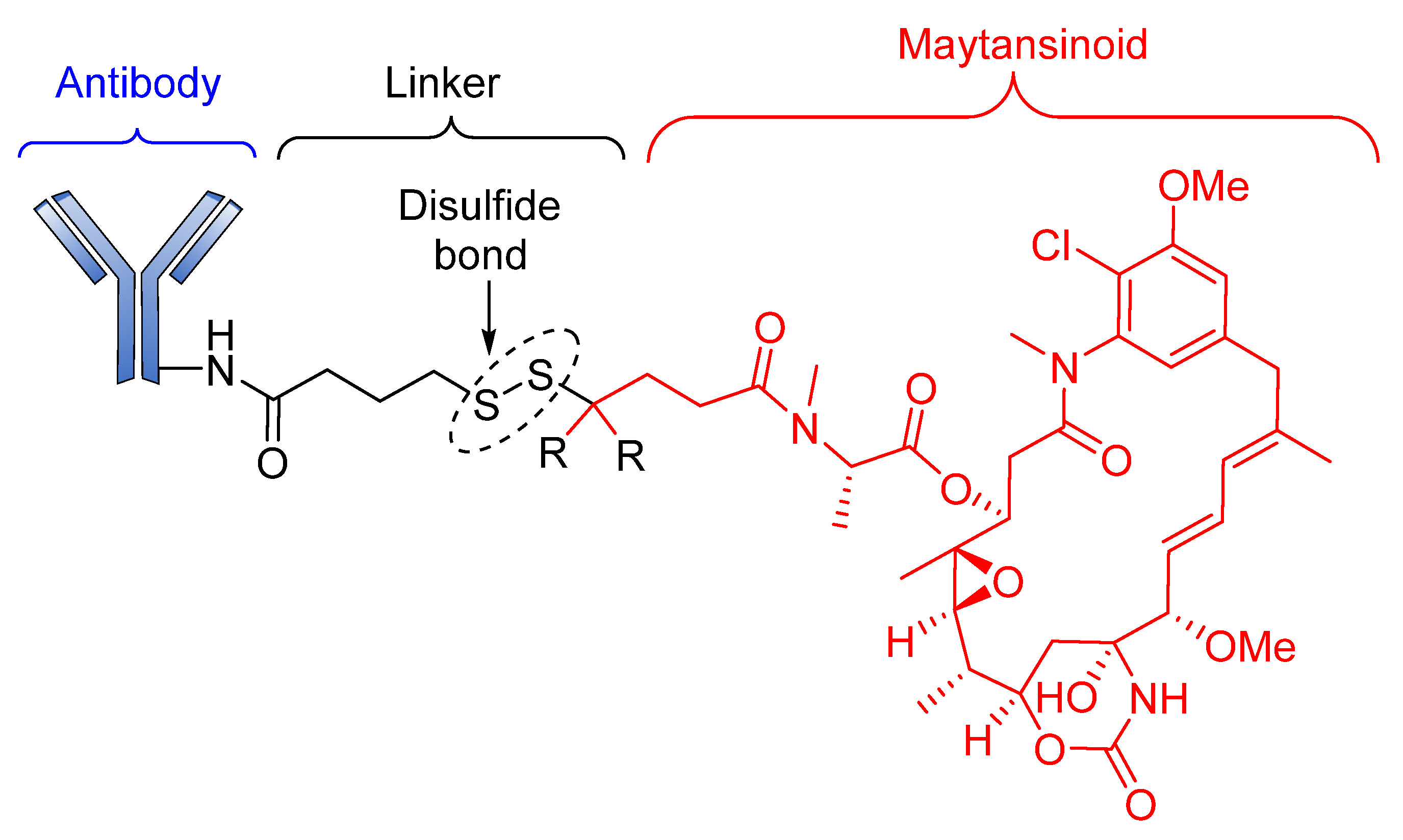
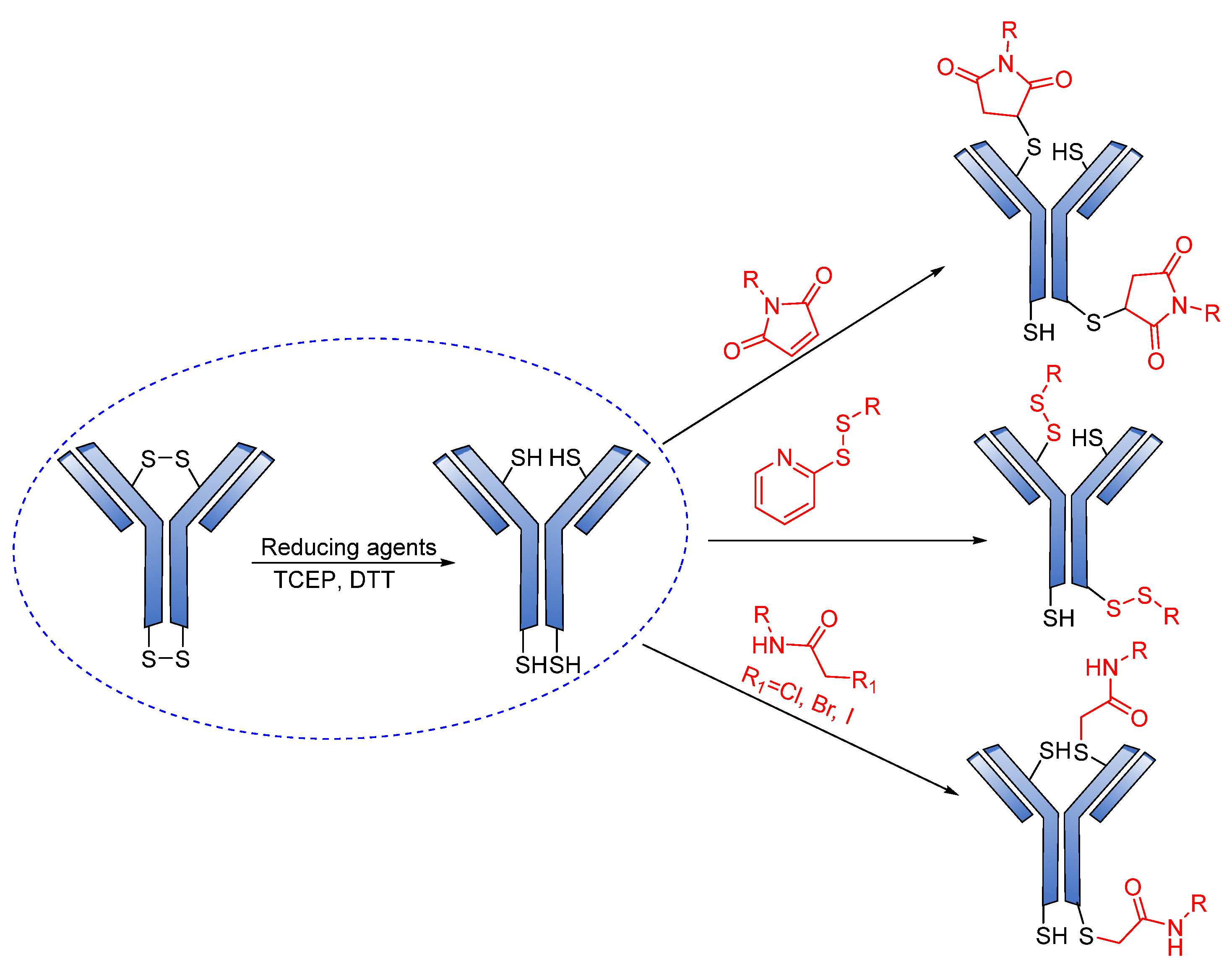
Reducible Linkers
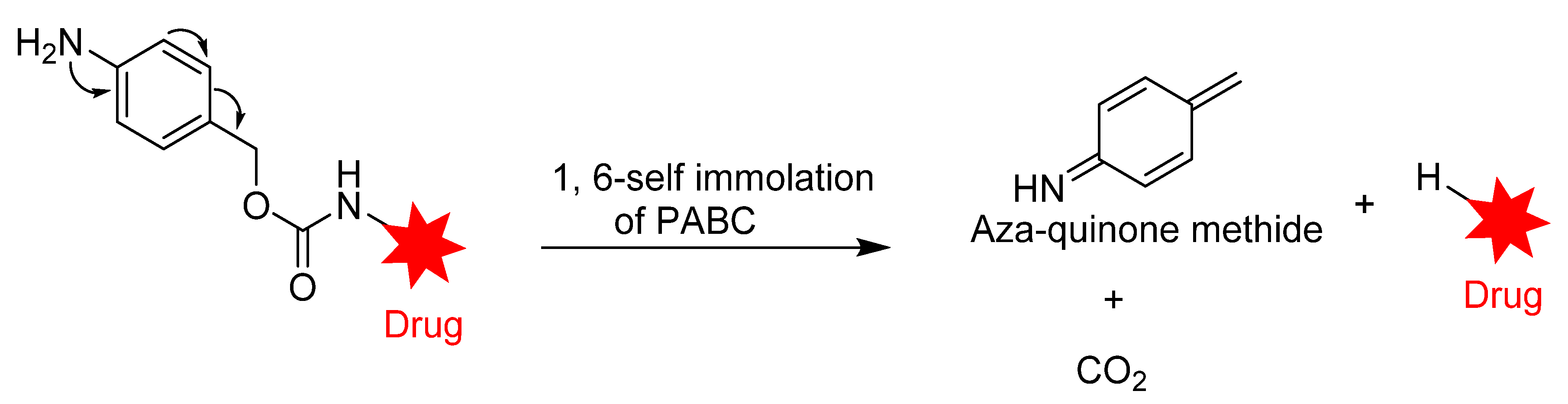
3.1.2. Enzyme-Cleavable Linkers
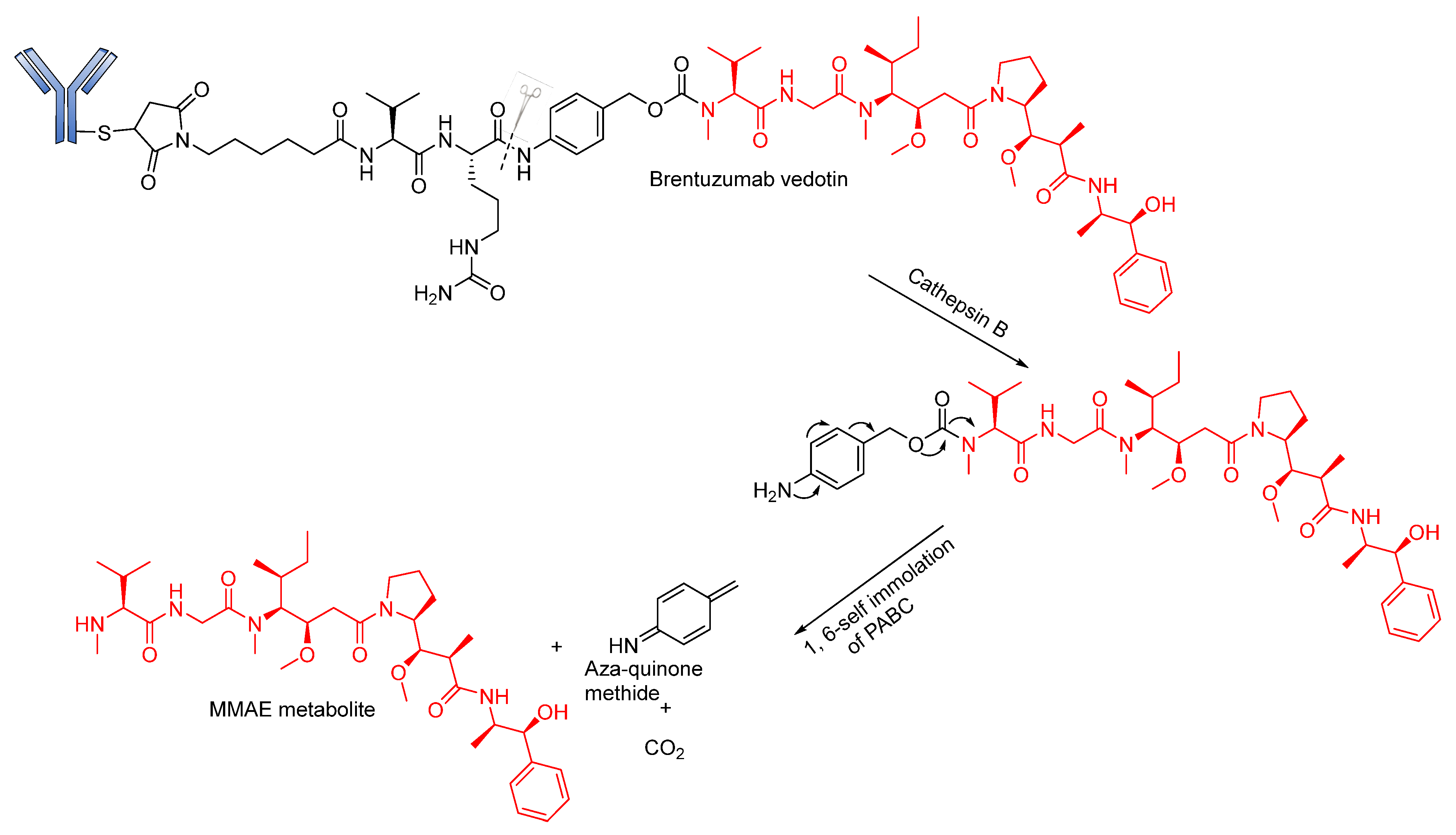
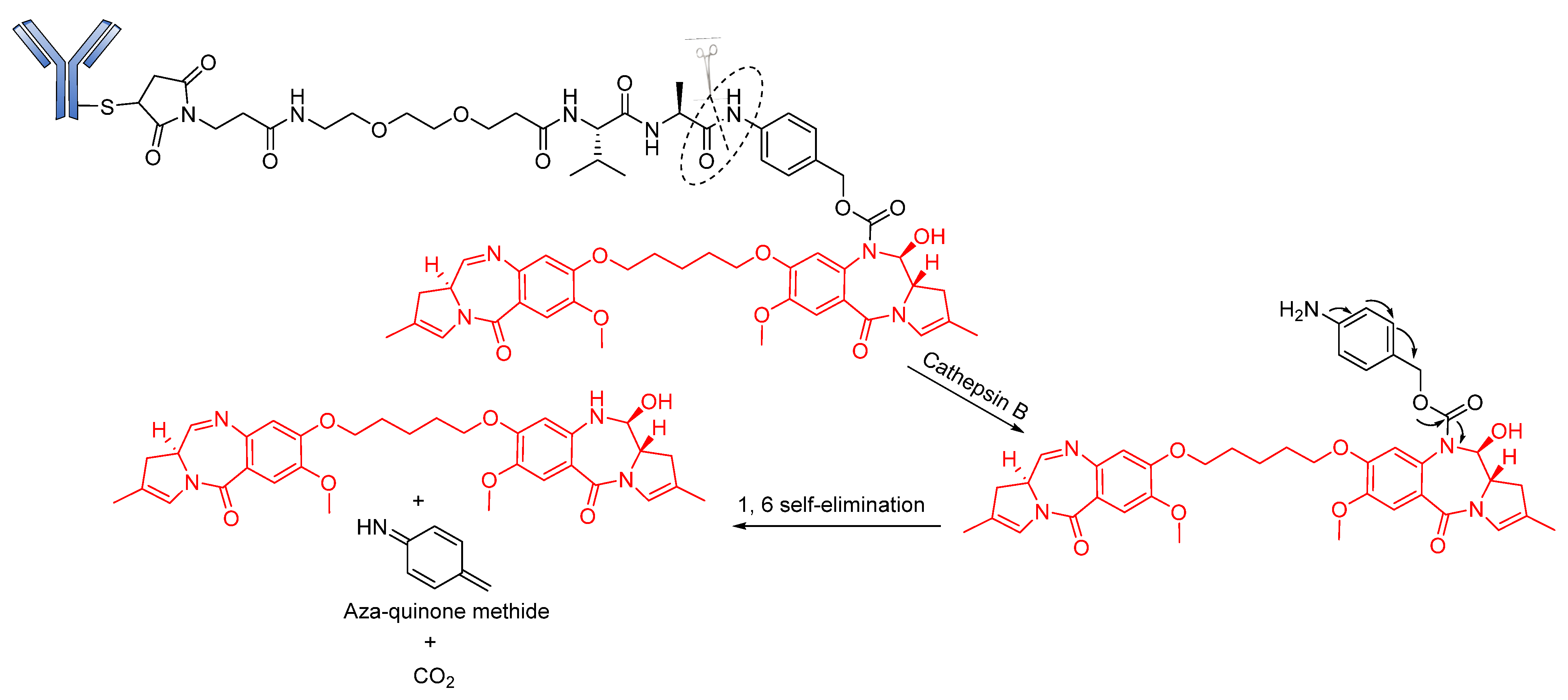
Peptide-Based Linkers
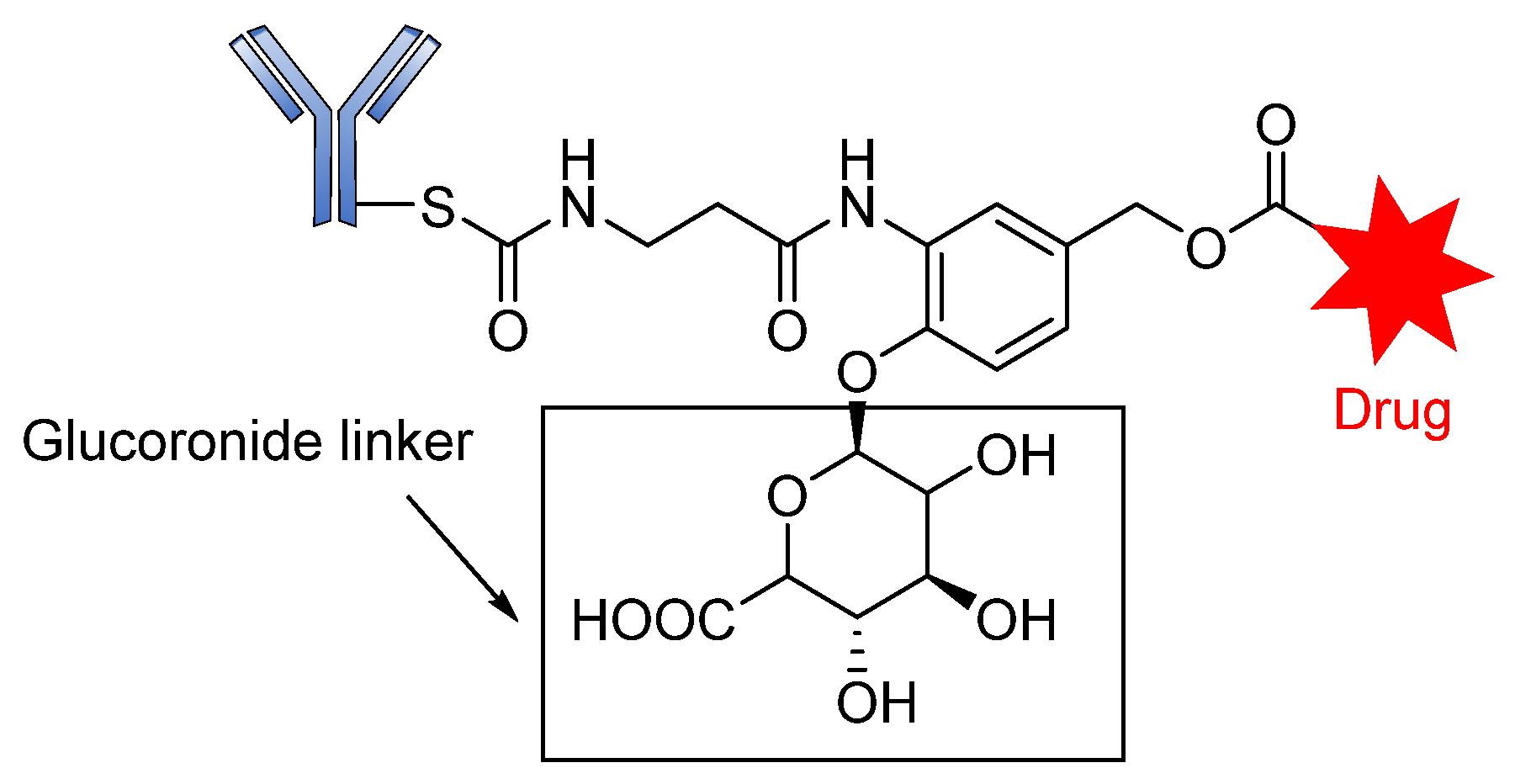
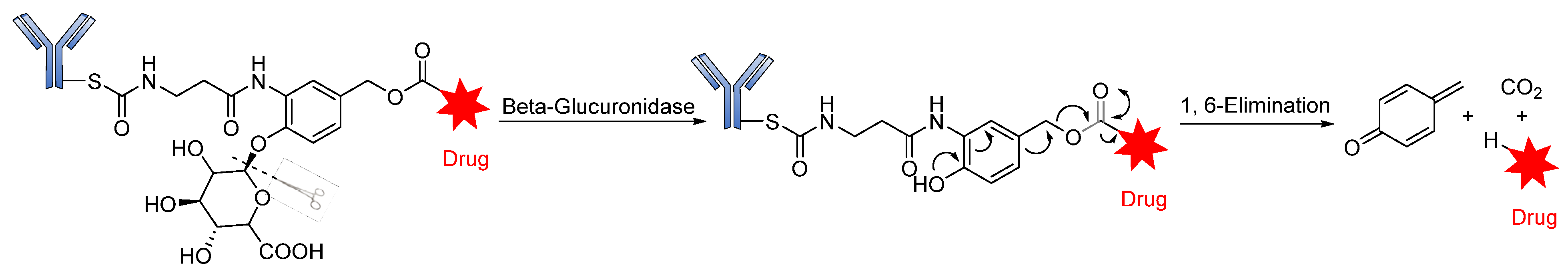
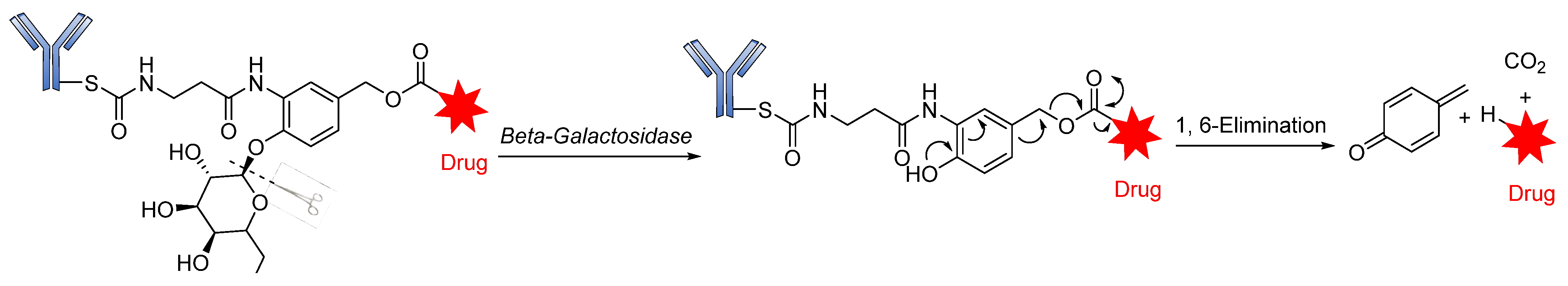
Glycosidase-Sensitive Linkers
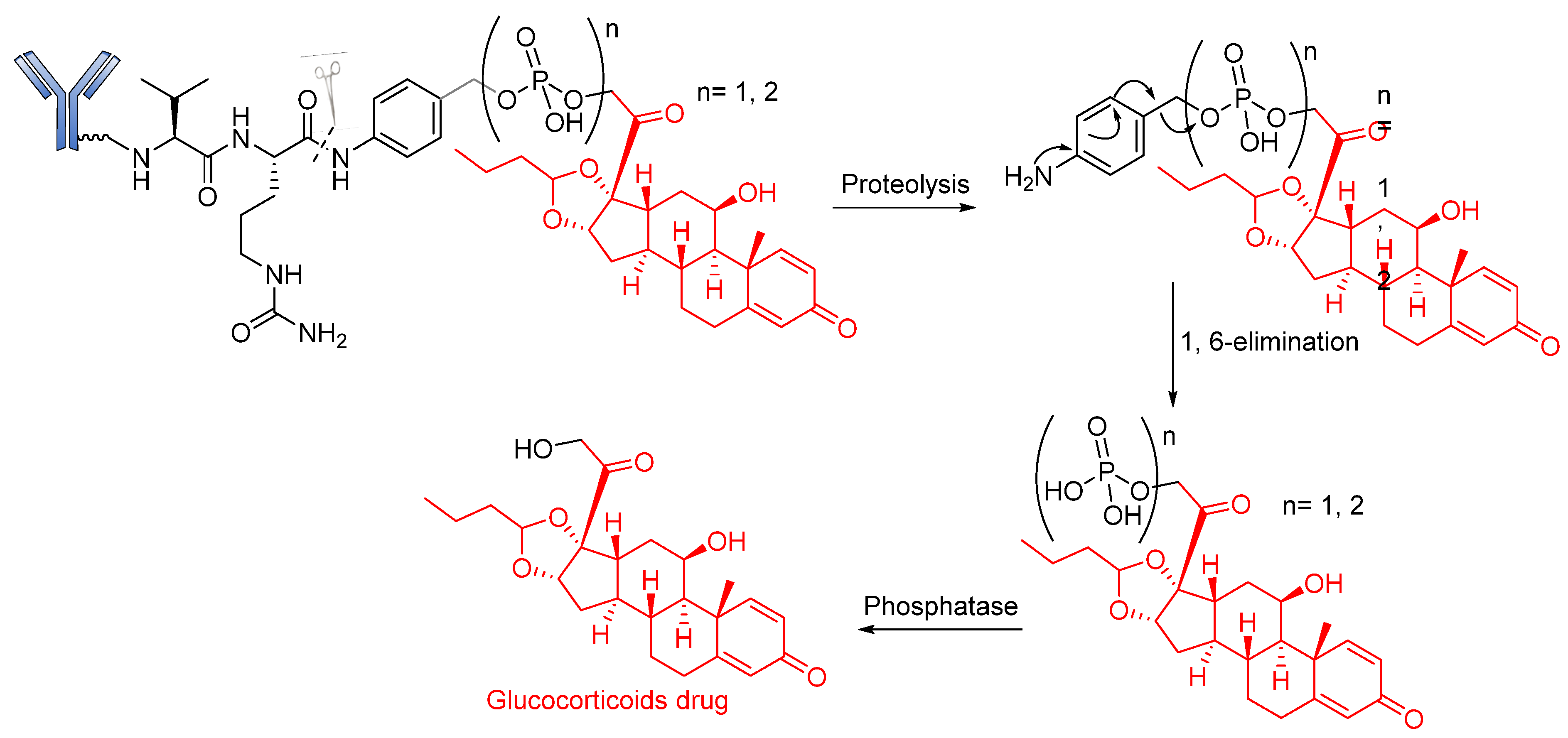
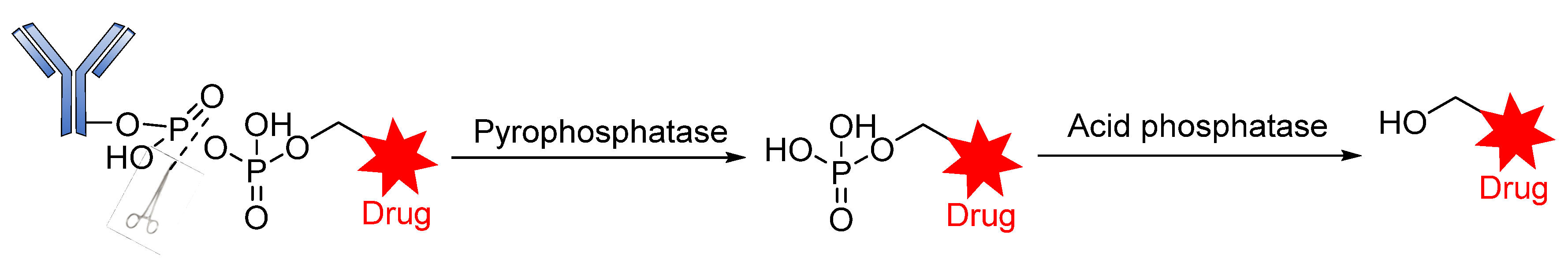
Phosphatase-Cleavable Linkers
3.2. Non-Cleavable Linkers
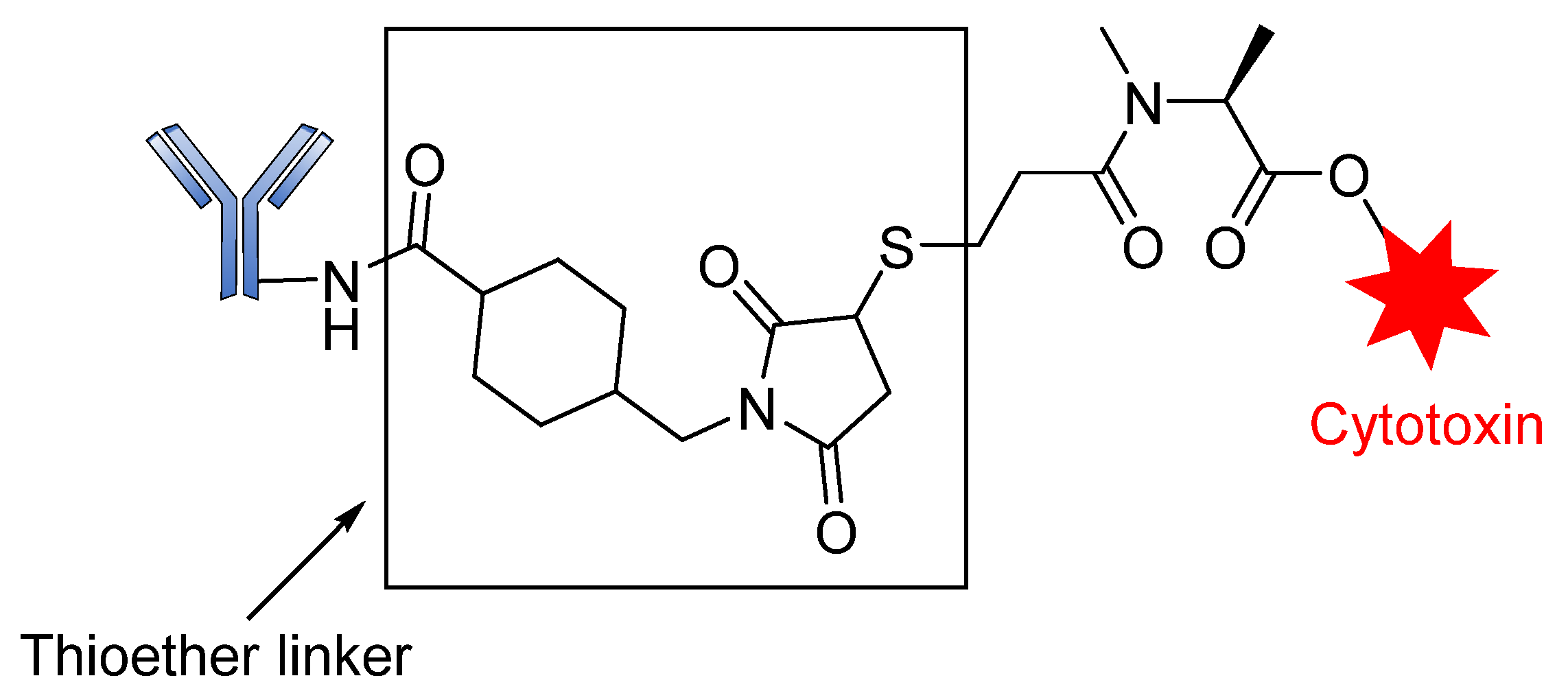
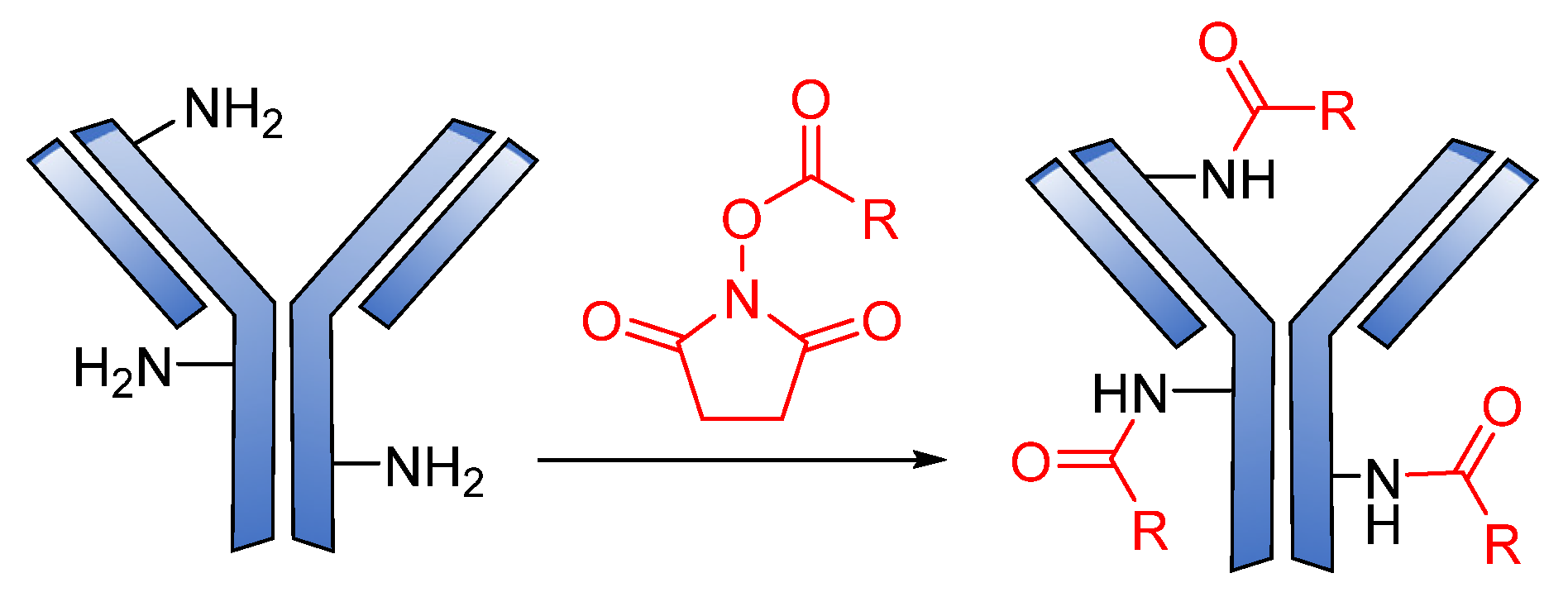
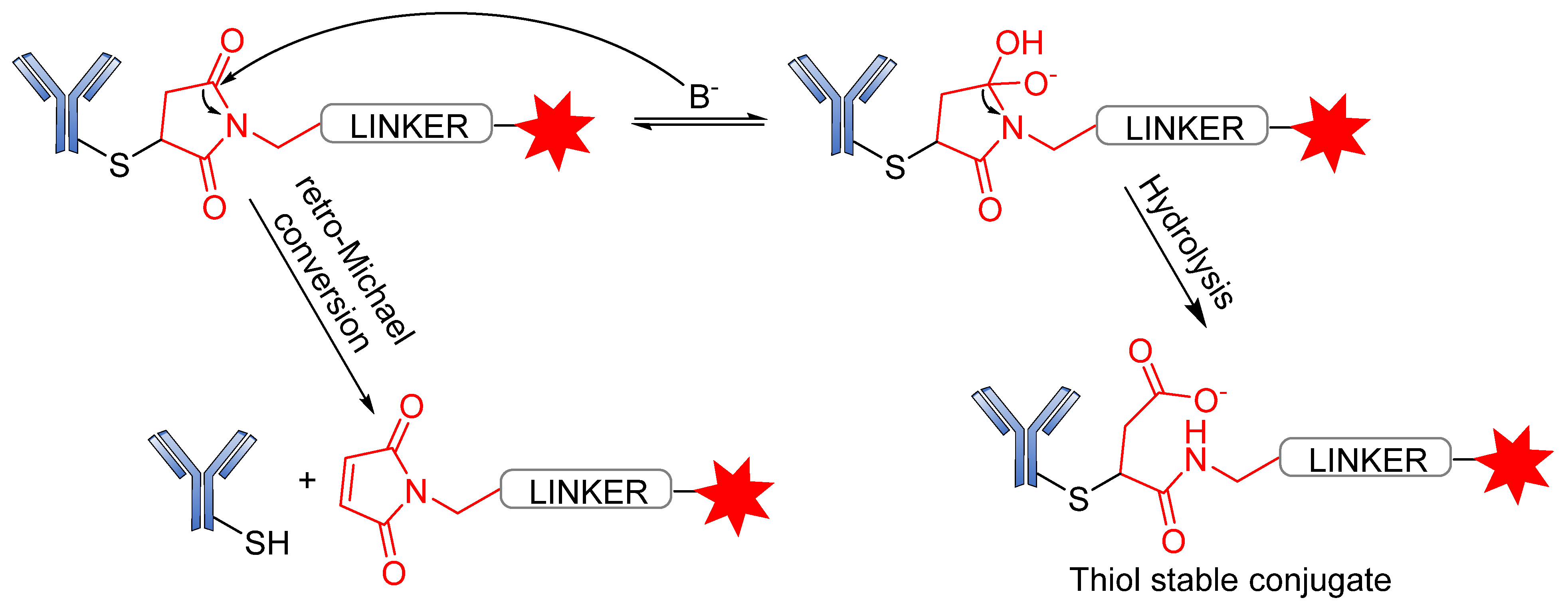
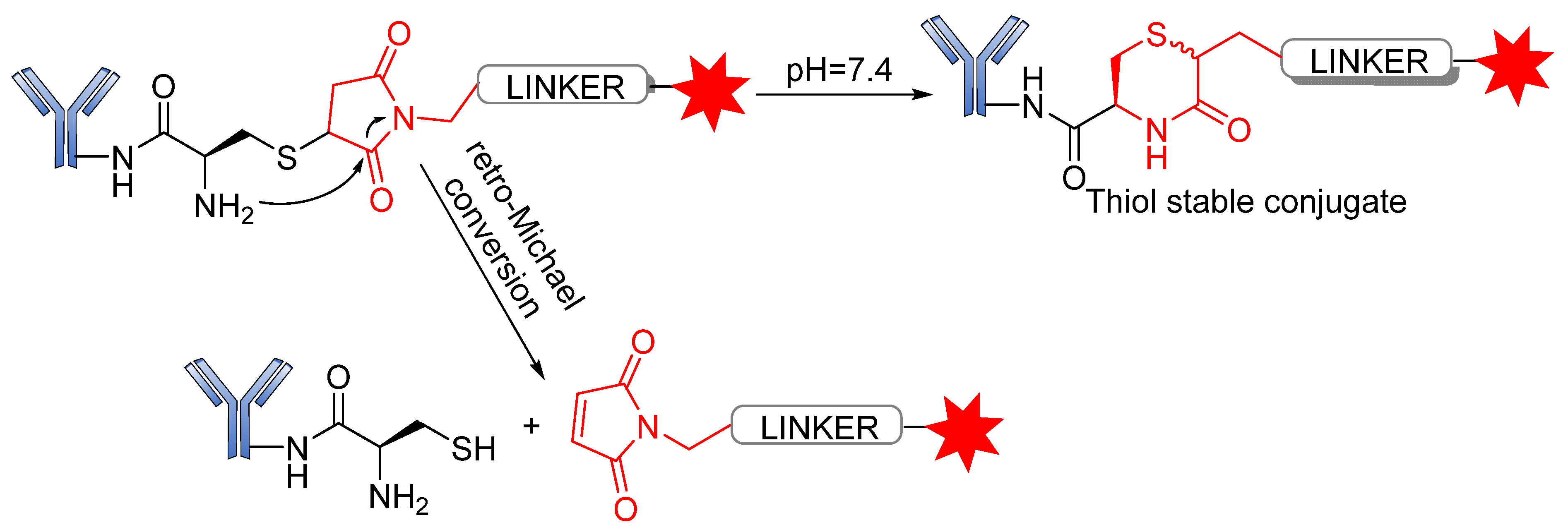
3.3. Conjugation Strategies
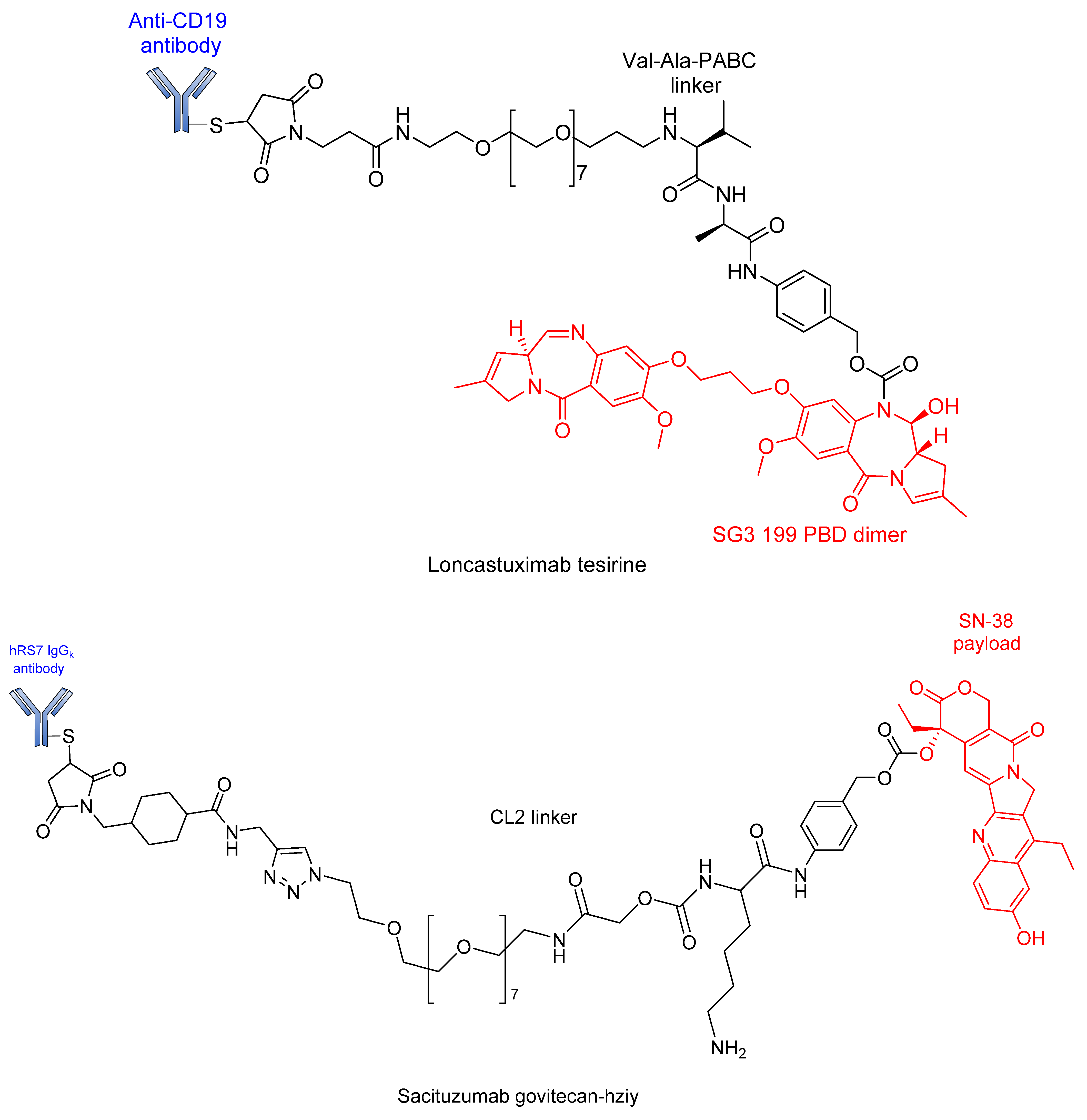
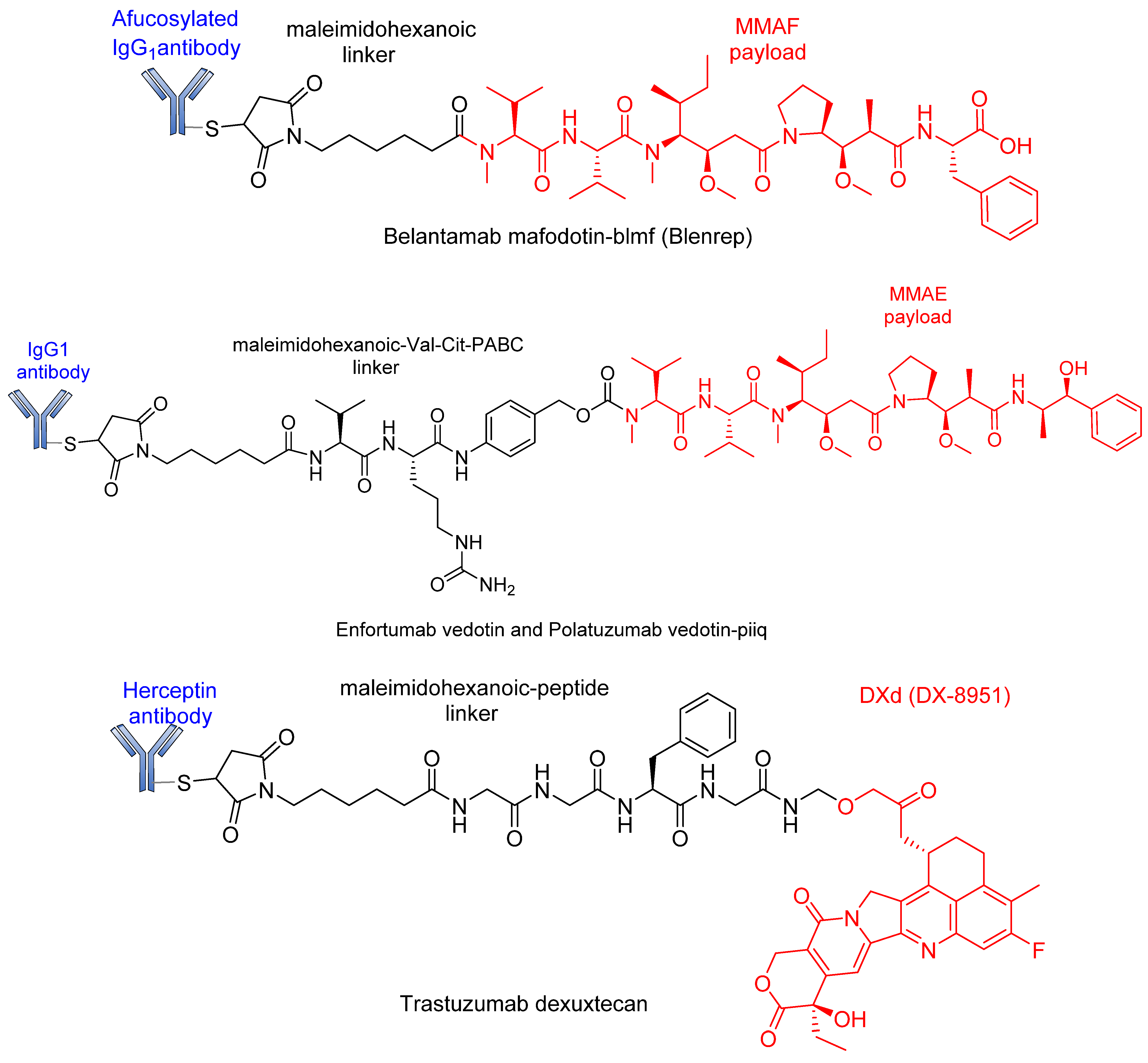
4. Approved ADCs and ADCs in Clinical Trials
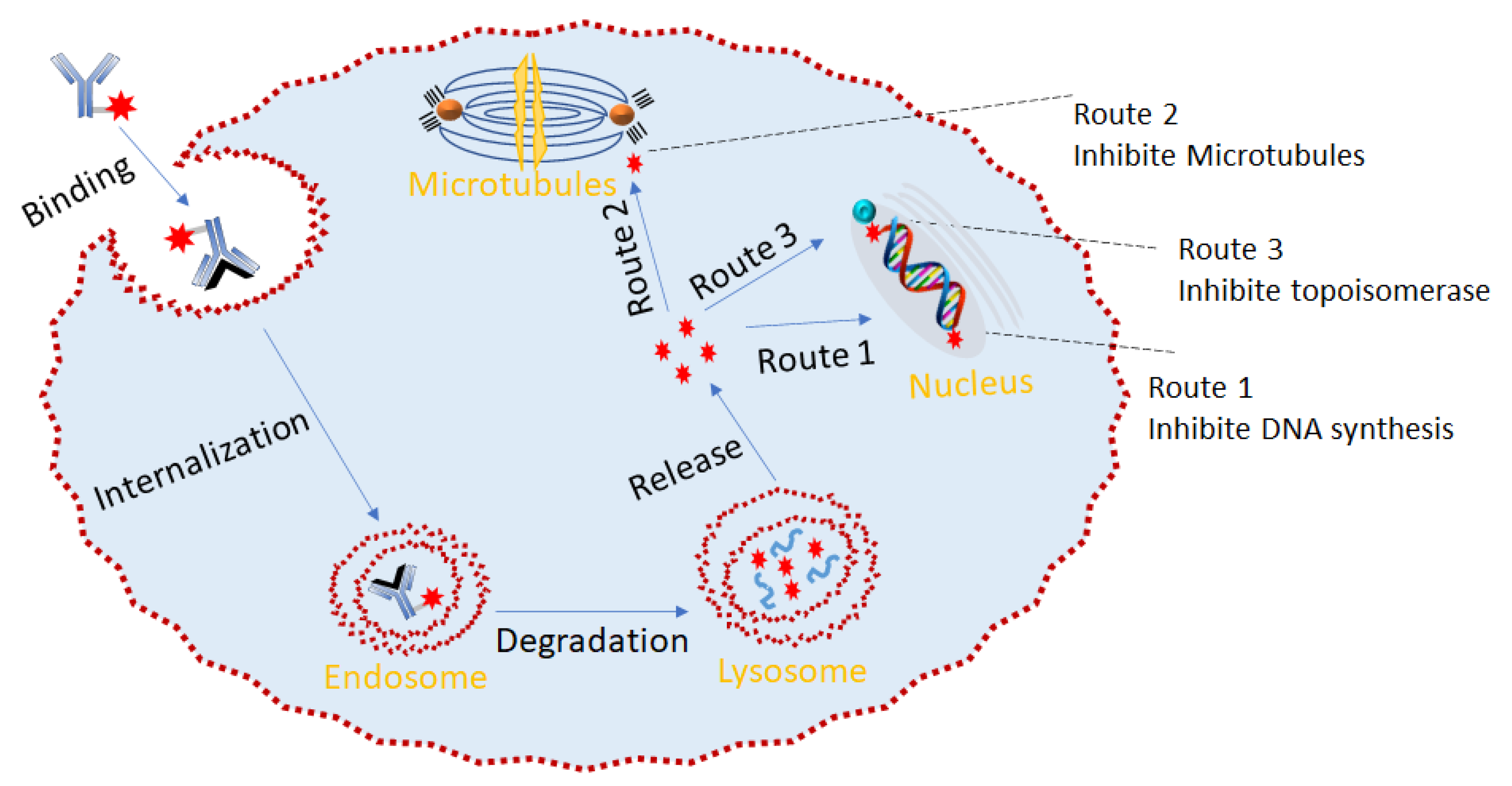
5. ADC Mechanism of Action
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, C.; Cheng, W.; Yuan, Q.; Müllen, K.; Yin, M. From Dyestuff chemistry to cancer theranostics: The rise of rylenecarboximides. Acc. Chem. Res. 2019, 52, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhou, Y. The role of surgery in the treatment of gastric cancer. J. Surg. Oncol. 2010, 101, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Hurwitz, M.D.; Krishnan, S.; Asea, A. Combined hyperthermia and radiotherapy for the treatment of cancer. Cancers 2011, 3, 3799–3823. [Google Scholar] [CrossRef]
- Garaci, E.; Pica, F.; Sinibaldi-Vallebona, P.; Pierimarchi, P.; Mastino, A.; Matteucci, C.; Rasi, G. Thymosin α1 in combination with cytokines and chemotherapy for the treatment of cancer. Int. Immu. Pharm. 2003, 3, 1145–1150. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-drug conjugates for cancer therapy: Chemistry to clinical implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Rehman, K.; Tariq, M.; Akash, M.S.; Gillani, Z.; Qazi, M.H. Effect of HA 14-1 on Apoptosis-Regulating Proteins in HeLa Cells. Chem. Biol. Drug Des. 2014, 83, 317–323. [Google Scholar] [CrossRef]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody–drug conjugates: An emerging concept in cancer therapy. Angew. Chem. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Kopp, A.; Thurber, G.M. Severing Ties: Quantifying the Payload Release from Antibody Drug Conjugates. Cell Chem. Biol. 2019, 26, 1631–1633. [Google Scholar] [CrossRef]
- Loadman, P. Anticancer drug development. Br. J. Cancer 2002, 86, 1665–1666. [Google Scholar] [CrossRef] [Green Version]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, J.; Wang, Y.; Cheng, J.; Ji, S.; Zhuang, X.; Chen, X. Sequentially responsive shell-stacked nanoparticles for deep penetration into solid tumors. Adv. Mater. 2017, 29, 1701170. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Blanco-Fernandez, B.; Puga, A.M.; Concheiro, A. Crosslinked ionic polysaccharides for stimuli-sensitive drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 1148–1171. [Google Scholar] [CrossRef]
- Zhao, H.; Li, L.; Zheng, C.; Hao, Y.; Niu, M.; Hu, Y.; Chang, J.; Zhang, Z.; Wang, L. An intelligent dual stimuli-responsive photosensitizer delivery system with O2-supplying for efficient photodynamic therapy. Colloids Surf. B Biointerfaces 2018, 167, 299–309. [Google Scholar] [CrossRef]
- Xu, F.; Zhong, H.; Chang, Y.; Li, D.; Jin, H.; Zhang, M.; Wang, H.; Jiang, C.; Shen, Y.; Huang, Y. Targeting death receptors for drug-resistant cancer therapy: Codelivery of pTRAIL and monensin using dual-targeting and stimuli-responsive self-assembling nanocomposites. Biomaterials 2018, 158, 56–73. [Google Scholar] [CrossRef]
- Aghebati-Maleki, L.; Shabani, M.; Baradaran, B.; Motallebnezhad, M.; Majidi, J.; Yousefi, M. Receptor tyrosine kinase-like orphan receptor 1 (ROR-1): An emerging target for diagnosis and therapy of chronic lymphocytic leukemia. Biomed. Pharmacother. 2017, 88, 814–822. [Google Scholar] [CrossRef]
- Amani, N.; Dorkoosh, F.A.; Mobedi, H. ADCs, as novel revolutionary weapons for providing a step forward in targeted therapy of malignancies. Curr. Drug Deliv. 2020, 17, 23–51. [Google Scholar] [CrossRef]
- Liu, H.; Bolleddula, J.; Nichols, A.; Tang, L.; Zhao, Z.; Prakash, C. Metabolism of bioconjugate therapeutics: Why, when, and how? Drug Metab. Rev. 2020, 52, 66–124. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Dhimolea, E. The future of antibodies as cancer drugs. Drug Discov. Today 2012, 17, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A. Antibody drug conjugates as cancer therapeutics. Antibodies 2013, 2, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M. Drug-conjugated antibodies for the treatment of cancer. Br. J. Clin. Pharmacol. 2013, 76, 248–262. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody–drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent developments in ADC technology: Preclinical studies signal future clinical trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–drug conjugates: The last decade. Pharm. J. 2020, 13, 245. [Google Scholar] [CrossRef]
- Lambert, J.M. Antibody–drug conjugates (ADCs): Magic bullets at last! Mol. Pharm. 2015, 12, 1701–1702. [Google Scholar] [CrossRef]
- Drachman, J.G.; Senter, P.D. Antibody-drug conjugates: The chemistry behind empowering antibodies to fight cancer. Hematol. Am. Soc. Hematol. Educ. Program Book 2013, 2013, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Gerber, H.-P.; Sapra, P. Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol. Immunol. 2015, 67, 107–116. [Google Scholar] [CrossRef]
- Hughes, B. Antibody–drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Antibody–drug conjugates: Basic concepts, examples and future perspectives. J. Control Release 2012, 161, 422–428. [Google Scholar] [CrossRef]
- Iyer, U.; Kadambi, V. Antibody drug conjugates—Trojan horses in the war on cancer. Toxicol. Appl. Pharmacol. 2011, 64, 207–212. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Donaghy, H. In Effects of Antibody, Drug and Linker on the Preclinical and Clinical Toxicities of Antibody-Drug Conjugates, mAbs; Taylor & Francis: Abingdon, UK, 2016; pp. 659–671. [Google Scholar]
- Lambert, J.M.; van Delft, F. Introduction to Antibody–Drug Conjugates. Drug Discov. 2021, 1–31. [Google Scholar] [CrossRef]
- Lee, P.; Kim, C.-U.; Seo, S.H.; Kim, D.-J. Current status of COVID-19 vaccine development: Focusing on antigen design and clinical trials on later stages. Immune Netw. 2021, 21, e4. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, G.; Tang, T.; Liang, T. Identification of tumor antigens and immune subtypes of pancreatic adenocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Davey, A.S.; Call, M.E.; Call, M.J. The influence of chimeric antigen receptor structural domains on clinical outcomes and associated toxicities. Cancers 2021, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody–drug conjugates—A new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapra, P.; Shor, B. Monoclonal antibody-based therapies in cancer: Advances and challenges. Pharmacol. Ther. 2013, 138, 452–469. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody–drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjugate Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Nolting, B. Linker technologies for antibody–drug conjugates. In Antibody-Drug Conjugates; Springer: Berlin/Heidelberg, Germany, 2013; pp. 71–100. [Google Scholar]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C. Synthesis and evaluation of hydrophilic linkers for antibody–maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Grandjean, C.; Rousselot-Pailley, P.; Joly, P.; Bourel-Bonnet, L.; Santraine, V.; Gras-Masse, H.; Melnyk, O. Solid-phase functionalization of peptides by an α-hydrazinoacetyl group. J. Org. Chem. 2003, 68, 7033–7040. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody−calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjugate Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef]
- Kalia, J.; Raines, R.T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef] [Green Version]
- Melnyk, O.; Fehrentz, J.A.; Martinez, J.; Gras-Masse, H. Functionalization of peptides and proteins by aldehyde or keto groups. Pept. Sci. 2000, 55, 165–186. [Google Scholar] [CrossRef]
- Yang, J.; Chen, H.; Vlahov, I.R.; Cheng, J.-X.; Low, P.S. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. USA 2006, 103, 13872–13877. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, S.; Luo, T.; Wang, C.; Zhao, J. Disulfide linkage: A potent strategy in tumor-targeting drug discovery. Curr. Med. Chem. 2012, 19, 2976–2983. [Google Scholar] [CrossRef]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Evans, D.Y.; Zhou, B.-B.; Damle, N.K. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother. Pharmacol. 2011, 67, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Mosure, K.; Knipe, J.O.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 2. Models of anticancer drugs paclitaxel (Taxol®), mitomycin C and doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3347–3352. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3341–3346. [Google Scholar] [CrossRef]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Albin, N.; Massaad, L.; Toussaint, C.; Mathieu, M.-C.; Morizet, J.; Parise, O.; Gouyette, A.; Chabot, G.G. Main drug-metabolizing enzyme systems in human breast tumors and peritumoral tissues. Cancer Res. 1993, 53, 3541–3546. [Google Scholar] [PubMed]
- Tranoy-Opalinski, I.; Legigan, T.; Barat, R.; Clarhaut, J.; Thomas, M.; Renoux, B.; Papot, S. β-Glucuronidase-responsive prodrugs for selective cancer chemotherapy: An update. Eur. J. Med. Chem. 2014, 74, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Fuselier, J.A.; Sun, L.; Woltering, S.N.; Murphy, W.A.; Vasilevich, N.; Coy, D.H. An adjustable release rate linking strategy for cytotoxin–peptide conjugates. Bioorg. Med. Chem. Lett. 2003, 13, 799–803. [Google Scholar] [CrossRef]
- Patterson, L.H.; McKeown, S.R.; Robson, T.; Gallagher, R.; Raleigh, S.M.; Orr, S. Antitumour prodrug development using cytochrome P450 (CYP) mediated activation. Anti-Cancer Drug Des. 1999, 14, 473–486. [Google Scholar]
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Firestone, R.; Willner, D.; Hofstead, S.; King, H.; Kaneko, T.; Braslawsky, G.; Greenfield, R.; Trail, P.; Lasch, S.; Henderson, A. Synthesis and antitumor activity of the immunoconjugate BR96-Dox. J. Control Release 1996, 39, 251–259. [Google Scholar] [CrossRef]
- Saleh, M.; Abbott, S.; Perron, V.; Lauzon, C.; Penney, C.; Zacharie, B. Synthesis and antimicrobial activity of 2-fluorophenyl-4, 6-disubstituted [1,3,5] triazines. Bioorg. Med. Chem. Lett. 2010, 20, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Nie, Y.; Huse, W.D.; Watkins, J.D. Humanization of a murine monoclonal antibody by simultaneous optimization of framework and CDR residues. J. Mol. Biol. 1999, 294, 151–162. [Google Scholar] [CrossRef]
- Ducry, L.; Stump, B. Antibody−drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Van der Velden, V.H.; te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef]
- Ravandi, F.; Estey, E.H.; Appelbaum, F.R.; Lo-Coco, F.; Schiffer, C.A.; Larson, R.A.; Burnett, A.K.; Kantarjian, H.M. Gemtuzumab ozogamicin: Time to resurrect? J. Clin. Oncol. 2012, 30, 3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, A.; Shinjo, K.; Yamakage, N.; Ono, T.; Hirano, I.; Matsui, H.; Shigeno, K.; Nakamura, S.; Tobita, T.; Maekawa, M. CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: Analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma. Br. J. Haematol. 2009, 146, 34–43. [Google Scholar] [CrossRef]
- Mthembu, S.N.; Sharma, A.; Albericio, F.; de la Torre, B.G. Breaking a Couple: Disulfide Reducing Agents. ChemBioChem 2020, 21, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellogg, B.A.; Garrett, L.; Kovtun, Y.; Lai, K.C.; Leece, B.; Miller, M.; Payne, G.; Steeves, R.; Whiteman, K.R.; Widdison, W. Disulfide-linked antibody−maytansinoid conjugates: Optimization of in vivo activity by varying the steric hindrance at carbon atoms adjacent to the disulfide linkage. Bioconjugate Chem. 2011, 22, 717–727. [Google Scholar] [CrossRef]
- Socinski, M.A.; Kaye, F.J.; Spigel, D.R.; Kudrik, F.J.; Ponce, S.; Ellis, P.M.; Majem, M.; Lorigan, P.; Gandhi, L.; Gutierrez, M.E. Phase 1/2 study of the CD56-targeting antibody-drug conjugate lorvotuzumab mertansine (IMGN901) in combination with carboplatin/etoposide in small-cell lung cancer patients with extensive-stage disease. Clin. Lung Cancer 2017, 18, 68–76.e2. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjugate Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Stephan, J.P.; Kozak, K.R.; Wong, W.L.T. Challenges in developing bioanalytical assays for characterization of antibody–drug conjugates. Bioanalysis 2011, 3, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Isse, K.; Fujihira, T.; Takenoyama, M.; Saunders, L.; Bheddah, S.; Nakanishi, Y.; Okamoto, I. Prevalence of Delta-like protein 3 expression in patients with small cell lung cancer. Lung Cancer 2018, 115, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Caculitan, N.G.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; Haley, B. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody–drug conjugates. Cancer Res. 2017, 77, 7027–7037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J. Development and properties of β-glucuronide linkers for monoclonal antibody−drug conjugates. Bioconjugate Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; De Brabander, J.; Miyamoto, J.; Senter, P.D. Expanded utility of the β-glucuronide linker: ADCs that deliver phenolic cytotoxic agents. ACS Med. Chem. Lett. 2010, 1, 277–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodych, S.; Michel, C.; Delacroix, S.; Koniev, O.; Ehkirch, A.; Eberova, J.; Cianférani, S.; Renoux, B.; Krezel, W.; Poinot, P. Development and evaluation of β-galactosidase-sensitive antibody-drug conjugates. Eur. J. Med. Chem. 2017, 142, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Dooney, D.; Zhang, R.; Liang, L.; Brandish, P.E.; Cheng, M.; Feng, G.; Beck, A.; Bresson, D.; Firdos, J. Novel phosphate modified cathepsin B linkers: Improving aqueous solubility and enhancing payload scope of ADCs. Bioconjugate Chem. 2016, 27, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paula Costa Monteiro, I.; Madureira, P.; De Vasconscelos, A.; Humberto Pozza, D.; Andrade de Mello, R. Targeting HER family in HER2-positive metastatic breast cancer: Potential biomarkers and novel targeted therapies. Pharmacogenomics 2015, 16, 257–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antibody–drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Chen, Y.; Wang, J.; Zheng, L.; Hutchinson, M.; Persson, J.; Ji, J. Characterization of ring-opening reaction of succinimide linkers in ADCs. J. Pharm. Sci. 2019, 108, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumey, L.N.; Charati, M.; He, T.; Sousa, E.; Ma, D.; Han, X.; Clark, T.; Casavant, J.; Loganzo, F.; Barletta, F. Mild method for succinimide hydrolysis on ADCs: Impact on ADC potency, stability, exposure, and efficacy. Bioconjugate Chem. 2014, 25, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Lahnsteiner, M.; Kastner, A.; Mayr, J.; Roller, A.; Keppler, B.K.; Kowol, C.R. Improving the Stability of Maleimide–Thiol Conjugation for Drug Targeting. Chem. Eur. J. 2020, 26, 15867. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-drug conjugate-based therapeutics: State of the science. JNCI J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D.A.; Soria, J.-C. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: Gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A.; Dubowchik, G.M.; Lowinger, T.B. Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. Pharmacol. Ther. 2018, 181, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.A.; Flagella, K.; Beyer, J.; Tibbitts, J.; Kaur, S.; Saad, O.; Yi, J.-H.; Girish, S.; Dybdal, N.; Reynolds, T. Preclinical safety profile of trastuzumab emtansine (T-DM1): Mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol. Appl. Pharmacol. 2013, 273, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Sangha, R.; Davies, A.; Dang, N.H.; Ogura, M.; MacDonald, D.A.; Ananthakrishnan, R.; Paccagnella, M.L.; Vandendries, E.; Boni, J.; Goh, Y.T. Phase 1 study of inotuzumab ozogamicin combined with R-GDP for the treatment of patients with relapsed/refractory CD22+ B-cell non-Hodgkin lymphoma. J. Drug Assess. 2017, 6, 10–17. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, G.B.; Albericio, F. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2018, 23, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Moxetumomab pasudotox: First global approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreitman, R.J.; Dearden, C.E.; Zinzani, P.L.L.; Delgado, J.; Robak, T.; le Coutre, P.D.; Gjertsen, B.T.; Troussard, X.; Roboz, G.J.; Karlin, L. Moxetumomab Pasudotox-Tdfk in heavily pretreated patients with relapsed/refractory hairy cell leukemia (HCL): Long-term follow-up from the pivotal Phase 3 Trial. Blood 2019, 134, 2808. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-drug conjugates: Possibilities and challenges. Avi. J. Med. Bio. 2019, 11, 3. [Google Scholar]
- De la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2019. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.; Weinstock, C.; Zhang, L.; Charlab, R.; Dorff, S.E.; Gong, Y.; Hsu, V.; Li, F.; Ricks, T.K.; Song, P. FDA approval summary: Enfortumab vedotin for locally advanced or metastatic urothelial carcinoma. Clin. Cancer Res. 2021, 27, 922–927. [Google Scholar] [CrossRef]
- Jain, R.K.; Skelton IV, W.P.; Zhang, J. Emerging treatment options for the treatment of metastatic urothelial cancer: Therapeutic potential of enfortumab vedotin. Cancer Manag. Res. 2020, 12, 8379. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab deruxtecan: First approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody—Drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A. First-in-human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef] [Green Version]
- Zeng, P.; Chen, M.-B.; Zhou, L.-N.; Tang, M.; Liu, C.-Y.; Lu, P.-H. Impact of TROP2 expression on prognosis in solid tumors: A Systematic Review and Meta-analysis. Sci. Rep. 2016, 6, 33658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M. Efficacy and safety of anti-trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2017, 35, 2141. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Guarino, M.J.; Masters, G.; Purcell, W.T.; Starodub, A.N.; Horn, L.; Scheff, R.J.; Bardia, A.; Messersmith, W.A.; Berlin, J. Therapy of advanced non-small-cell lung cancer with an SN-38-anti-Trop-2 drug conjugate, sacituzumab govitecan. J. Clin. Oncol. 2017, 35, 2790–2797. [Google Scholar] [CrossRef] [PubMed]
- Torre, B.G.; Albericio, F. The pharmaceutical industry in 2020. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2021, 26, 627. [Google Scholar] [CrossRef]
- Markham, A. Belantamab mafodotin: First approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Minor, C. Belantamab Mafodotin-Blmf (Blenrep). Oncology Times 2021, 43, 15. [Google Scholar] [CrossRef]
- Snyder, J. Belantamab Mafodotin-blmf: Management and Prevention of Ocular Toxicities. HOPA 2021, 14. [Google Scholar]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Peng, H. Perspectives on the development of antibody-drug conjugates targeting ROR1 for hematological and solid cancers. Antib. Ther. 2021, 4, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef]
- Tong, J.T.; Harris, P.W.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., III; Robert, F. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet 2017, 18, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Moek, K.; de Groot, D.; de Vries, E.; Fehrmann, R. The antibody–drug conjugate target landscape across a broad range of tumour types. Ann. Oncol. 2017, 28, 3083–3091. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Camidge, D.R. Antibody drug conjugates in thoracic malignancies. Lung Cancer 2018, 124, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Maric, G.; Annis, M.; Dong, Z.; Rose, A.; Ng, S.; Perkins, D.; MacDonald, P.; Ouellet, V.; Russo, C.; Siegel, P. GPNMB cooperates with neuropilin-1 to promote mammary tumor growth and engages integrin α 5 β 1 for efficient breast cancer metastasis. Oncogene 2015, 34, 5494–5504. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Barris, D.M.; Piperdi, S.; Kuo, V.; Everts, S.; Geller, D.; Houghton, P.; Kolb, E.A.; Hawthorne, T.; Gill, J. Targeting glycoprotein NMB with antibody-drug conjugate, glembatumumab vedotin, for the treatment of Osteosarcoma. Pediatr. Blood Cancer 2016, 63, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Spitaleri, G.; Rodon, J.; Dotti, K.; Isambert, N.; Salvagni, S.; Moreno, V.; Assadourian, S.; Gomez, C.; Harnois, M. A first-in-human phase I study of SAR125844, a selective MET tyrosine kinase inhibitor, in patients with advanced solid tumours with MET amplification. Eur. J. Cancer 2017, 87, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickler, J.H.; Weekes, C.D.; Nemunaitis, J.; Ramanathan, R.K.; Heist, R.S.; Morgensztern, D.; Angevin, E.; Bauer, T.M.; Yue, H.; Motwani, M. First-in-Human Phase I, Dose-Escalation and-Expansion Study of Telisotuzumab Vedotin, an Antibody-Drug Conjugate Targeting c-Met, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 78, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Morgan, H.E.; Smart, K.; Zahari, N.M.; Pumford, S.; Ellis, I.O.; Robertson, J.F.; Nicholson, R.I. The emerging role of the LIV-1 Subfamily of Zinc Transporters in Breast Cancer. Mol. Med. 2007, 13, 396–406. [Google Scholar] [CrossRef]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.P.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alpha–targeting antibody–drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A phase i expansion study. J. Clin. Oncol. 2017, 35, 1112. [Google Scholar] [CrossRef]
- Carol, H.; Szymanska, B.; Evans, K.; Boehm, I.; Houghton, P.J.; Smith, M.A.; Lock, R.B. The anti-CD19 antibody–drug conjugate SAR3419 prevents hematolymphoid relapse postinduction therapy in preclinical models of pediatric acute lymphoblastic leukemia. Clin. Cancer Res. 2013, 19, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Ribrag, V.; Dupuis, J.; Tilly, H.; Morschhauser, F.; Laine, F.; Houot, R.; Haioun, C.; Copie, C.; Varga, A.; Lambert, J. A dose-escalation study of SAR3419, an anti-CD19 antibody maytansinoid conjugate, administered by intravenous infusion once weekly in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2014, 20, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Siegel, D.S.; Chanan-Khan, A.A.; Somlo, G.; Heffner, L.T.; Jagannath, S.; Zimmerman, T.; Munshi, N.C.; Madan, S.; Mohrbacher, A. Indatuximab ravtansine (BT062) in combination with low-dose dexamethasone and lenalidomide or pomalidomide: Clinical activity in patients with relapsed/refractory multiple myeloma. Blood 2016, 128, 4486. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Hassan, R.; Moore, K.N.; Santin, A.; Kindler, H.L.; Nemunaitis, J.J.; Seward, S.M.; Rajagopalan, P.; Walter, A.; Sarapa, N. Phase I study of anti-mesothelin antibody drug conjugate anetumab ravtansine (AR). J. Clin. Oncol. 2016, 34, 2509. [Google Scholar] [CrossRef]
- Boni, V.; Rixe, O.; Rasco, D.; Gomez-Roca, C.; Calvo, E.; Morris, J.C.; Tolcher, A.W.; Assadourian, S.; Guillemin, H.; Delord, J.-P. Abstract A73: A Phase I first-in-human (FIH) study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs)(NCT01156870). Clin. Trials 2013, 12, A73. [Google Scholar]
- Gomez-Roca, C.A.; Boni, V.; Moreno, V.; Morris, J.C.; Delord, J.-P.; Calvo, E.; Papadopoulos, K.P.; Rixe, O.; Cohen, P.; Tellier, A. A phase I study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs)(NCT01156870). J. Clin. Oncol. 2016, 34, 2511. [Google Scholar] [CrossRef]
- Thompson, J.A.; Motzer, R.J.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; Kim, S.K. Phase I Trials of Anti-ENPP3 Antibody–Drug Conjugates in Advanced Refractory Renal Cell Carcinomas. Clin. Cancer Res. 2018, 24, 4399–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parslow, A.C.; Parakh, S.; Lee, F.-T.; Gan, H.K.; Scott, A.M. Antibody–drug conjugates for cancer therapy. Biomedicines 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC Structure | Linkage Type | Cleavage Mechanism | Products Formed | Cleavage Site |
|---|---|---|---|---|
 | pH sensitive | Hydrolysis |  | Lysosome/Endosome [57,58,59,60] |
 | Reduction sensitive | Reduction |  | Cytoplasm [56,61,62,63] |
 | Peptide based linkage | Proteolysis |  | Lysosome [64,65,66,67] |
 | β-glucoronide | Glyosidase 1, 6-Elimination  |  | Lysosome [68,69] |
 | Phosphatase cleavage | Phosphatase |  | Lysosome [70] |
 | Carbamate | Esterase |  | Lysosome [71,72] |
 | Ester | Esterase |  | Lysosome [72,73] |
| API | Trade Name | Developer | mAb | Linker | Cytotoxin | Target Antigen | Indication(s) | Phase |
|---|---|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | Mylotarg | Pfizer | Hp67.6 (Hz IgG4) | Hydrazone | Calicheamicin | CD33 | Acute myeloid leukemia | Approved 2000 [112], withdrawn 2010 [113]; reapproved 2017 [19]. |
| Brentuximab vedotin (SGN-35) | Adcetris | Millennim/Takeda/Seattle Genetics | cAC10 (SGN-30, Ch-IgG1) | Dipeptide (VC) | MMAE | CD30 | Hodgkin lymphoma, systemic anaplastic large cell lymphoma | Accelerated approval 2011 [87]; full approval 2015 [114]. |
| Trastuzumab emtansine | Kadcyla | Roche/Genetech | Trastuzumab (Hz IgG1) | Non-cleavable (SMCC) | DM1 | HER2 | HER2-positive breast cancer | Approved 2013 [115,116] |
| Inotuzumab ozogamicin | Besponsa | Pfizer | G5/44 (Hz IgG4) | Hydrazone | Calicheamicin | CD22 | Acute lymphoblastic leukemia | Approved 2017 [117,118] |
| Moxetumomab pasudotox-tdfk | Lumoxiti | AstraZeneca | Anti-CD22 | Hydrazone | Pasudotox-tdfk | CD22 | Relapsed hairy cell leukemia | Approved 2018 [119,120] |
| Polatuzumab vedotin (RG7596, TAB-897, DCDS4501A | Polivy | Genentech/Roche | Anti-CD79b (Hz IgG1) | Dipeptide (VC) | MMAE | CD79b | Relapsed or refractory diffuse large B-cell lymphoma | Approved 2019 [19,121,122] |
| Enfortumab vedotin | Padcev | Agensys/Astellas | Enfortumab | Dipeptide (VC) | MMAE | Nectin4 | Solid and urothelial tumors | Approved 2019 [122,123,124] |
| Trastuzumab deruxtecan | Enhertu | AstraZeneca/Daiichi Sankyo | Trastuzumab (Herceptin) | Non-cleavable (mc) | Deruxtecan | HER2 | HER2-positive breast cancer | Approved 2019 [125,126] |
| Sacituzumab govitecan | Trodelvy | Immunomedics | hRS7 IgGk | Acid-labile ester | SN-38 | Trop-2 | Triple-negative breast cancer, urothelial and other cancers | Approved May 2020 [127,128,129,130,131,132] |
| Belantamab mafodotin-blmf | Blenrep | GlaxoSmithKline (GSK) | IgG1 | Non-cleavable (mc) | MMAE | BCMA | Multiple myeloma | Approved 2020 [132,133,134,135,136] |
| Loncastuximab tesirine-lpyl | Zynlonta | ADC Therapeutics | Anti-CD19 | Dipeptide (VA) | PBD | CD19 | Large B-cell lymphoma | Approved 2021 [137,138] |
| Tisotumab vedotin tftv | Tivdak | Seagen Inc | Tisotumab | Dipeptide (VC) | MMAE | Tissue factor | Metastatic cervical cancer | Approved 2021 [139,140] |
| ADC Name | Developer | mAb | Linker | Cytotoxin | Target Antigen | Indication(s) | Phase |
|---|---|---|---|---|---|---|---|
| Rovalpituzumab tesirine | Sanofi/ImmunoGen | Anti-DLL3 (Rovalpituzumab) | Dipeptide (VC) | PBD dimer | DLL3 | Small-cell lung cancer | III [141,142,143] |
| Glembatumumab vedotin | Seattle Genetics/Celldex/Progenics | CR-011 (Hu IgG2) | Cleavable dipeptide | MMAE | gpNMB | Metastatic breast cancer and melanoma | II/III [144,145] |
| PSMA ADC | Seattle Genetics/Progenics | Anti-PSMA (Hu IgG1) | Cleavable dipeptide | MMAE | PSMA | Prostate cancer | II [121] |
| Pinatuzumab vedotin | Roche/Genentech | Anti-CD22 (Hz IgG1) | Cleavable dipeptide | MMAE | CD22 | Diffuse large B-cell lymphoma, follicular non-Hodgkin lymphoma | II [121] |
| Telisotuzumab vedotin | AbbVie/Pierre Fabre | ABT-700 | Cleavable dipeptide | MMAE | ABT-700 | Advanced solid tumors cancer and non-small cell lung cancer | II [146,147] |
| Ladiratuzumab vedotin SGN-LIV1A | Seattle Genetics | Anti-LIV1 (Hz IgG1) | Cleavable dipeptide | MMAE | LIV-1 | Breast cancer, lung cancer | II [148] |
| Mirvetuximab soravtansine | ImmunoGen | M9346A | Cleavable disulfide | DM4 | FOLR1 | Ovarian, endometrial, non-small cell lung cancer | III [149,150] |
| Lorvotuzumab mertansine | ImmunoGen | huN901 (Hz IgG1) | Cleavable disulfide | DM1 | CD56 | Leukemia | II [88] |
| Coltuximab ravtansine | ImmunoGen | huB4 (Hz IgG1) | Cleavable disulfide | DM4 | CD19 | Diffuse large B cell lymphoma, acute lymphocytic leukaemia | II [93,151,152] |
| Indatuximab ravtansine | Biotest/ImmunoGen | Nbt062, Anti- CD138 (Ch IgG4) | Cleavable disulfide | DM4 | CD138 | Multiple myeloma | II [153] |
| Anetumab ravtansine | Bayer Health Care | Antimesothelin (Hz IgG1) | Cleavable disulfide | DM4 | Mesothelin | Mesothelioma and other solid tumors | II [115,154] |
| SAR566658 | Sanofi | DS6 (Hu IgG1) | Cleavable disulfide | DM4 | CA6 | Triple-negative breast cancer | II [155,156] |
| Depatuxizumab mafodotin | AbbVie | ABT-806 | Non-cleavable (mc) | MMAF | EGFR | Glioblastoma and other EGFR-positive tumors | III [129] |
| Naratuximab emtansine | ImmunoGen | K7153A humanized IgG1 | Non-cleavable (SMCC) | DM1 | CD37 | Diffuse large B cell lymphoma and follicular lymphoma | II [121] |
| AGS-16C3F | Agensys/Astellas | Anti-AGS16 (Hu IgG2a) | Non-cleavable (mc) | ENPP3 | ENPP3 | Renal cell carcinoma | II [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. https://doi.org/10.3390/pharmaceutics14020396
Sheyi R, de la Torre BG, Albericio F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics. 2022; 14(2):396. https://doi.org/10.3390/pharmaceutics14020396
Chicago/Turabian StyleSheyi, Rotimi, Beatriz G. de la Torre, and Fernando Albericio. 2022. "Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate" Pharmaceutics 14, no. 2: 396. https://doi.org/10.3390/pharmaceutics14020396