
Mechanistically Coupled PK (MCPK) Model to Describe Enzyme Induction and Occupancy Dependent DDI of Dabrafenib Metabolism



Abstract
:1. Introduction
2. Materials and Methods
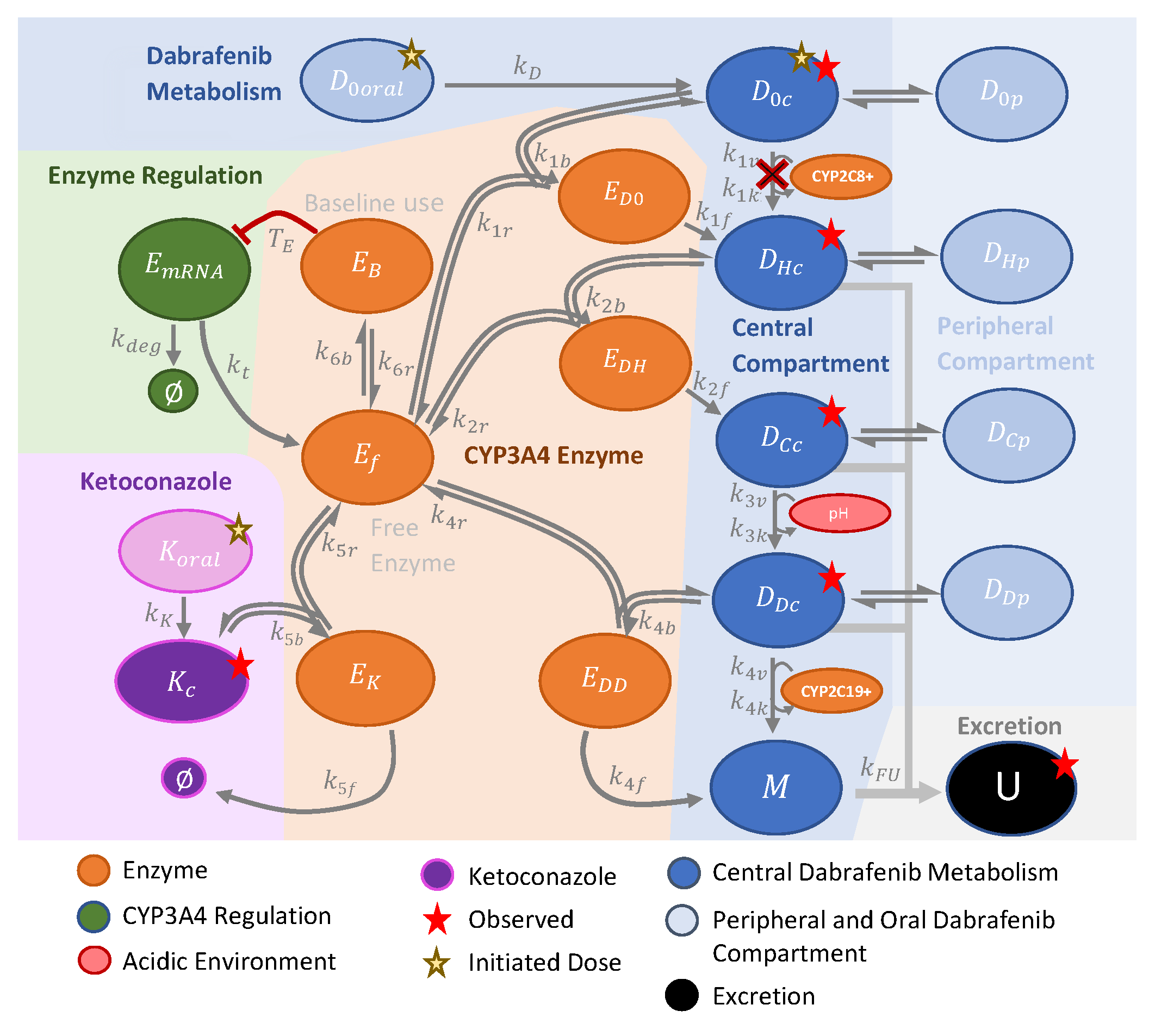
2.1. Modeling Methods

2.2. Cells and Reagents
2.3. Hydrogel
2.4. 3D Melanoma Spheroids
3. Results
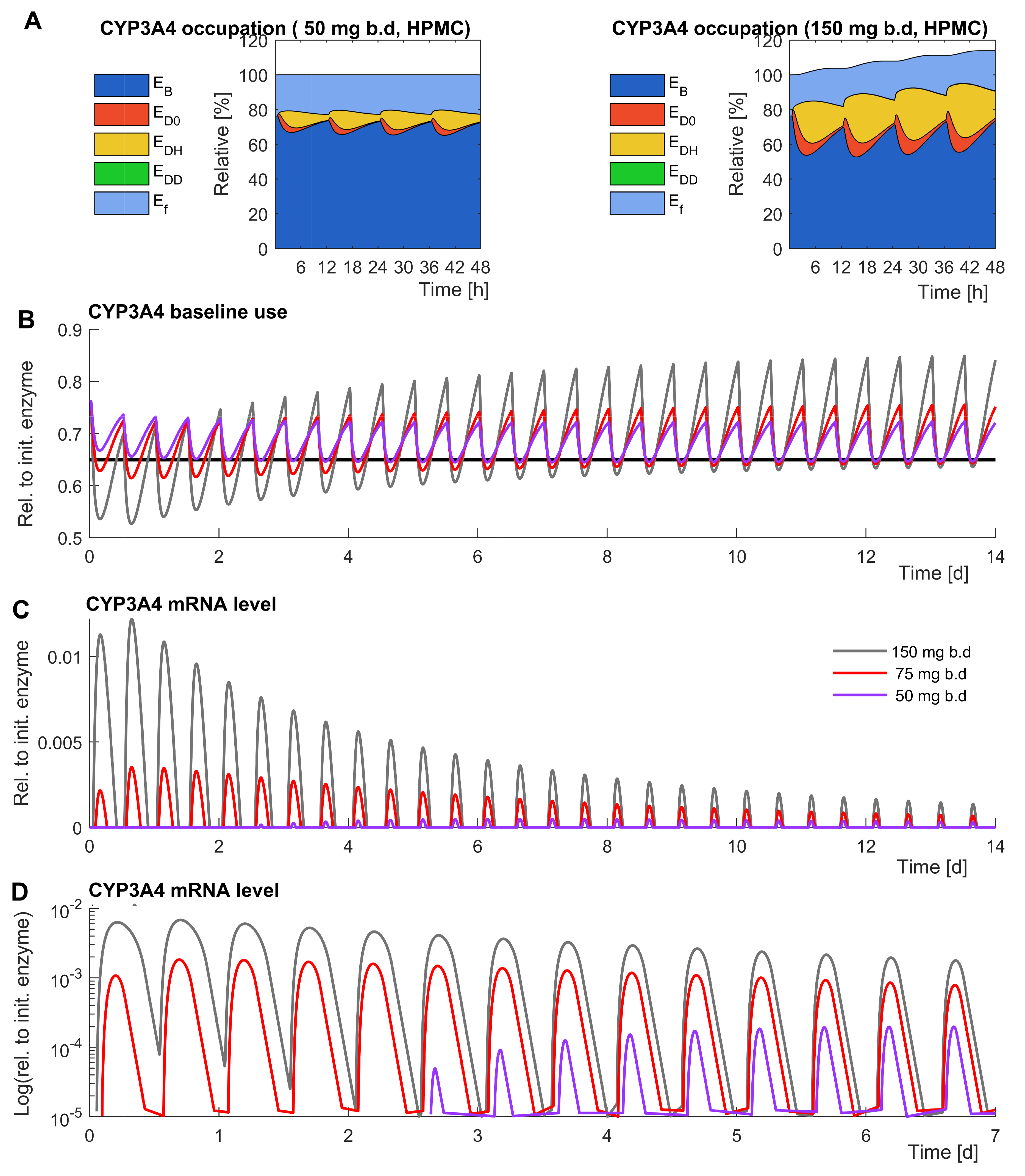
3.1. The Model Reproduces Data

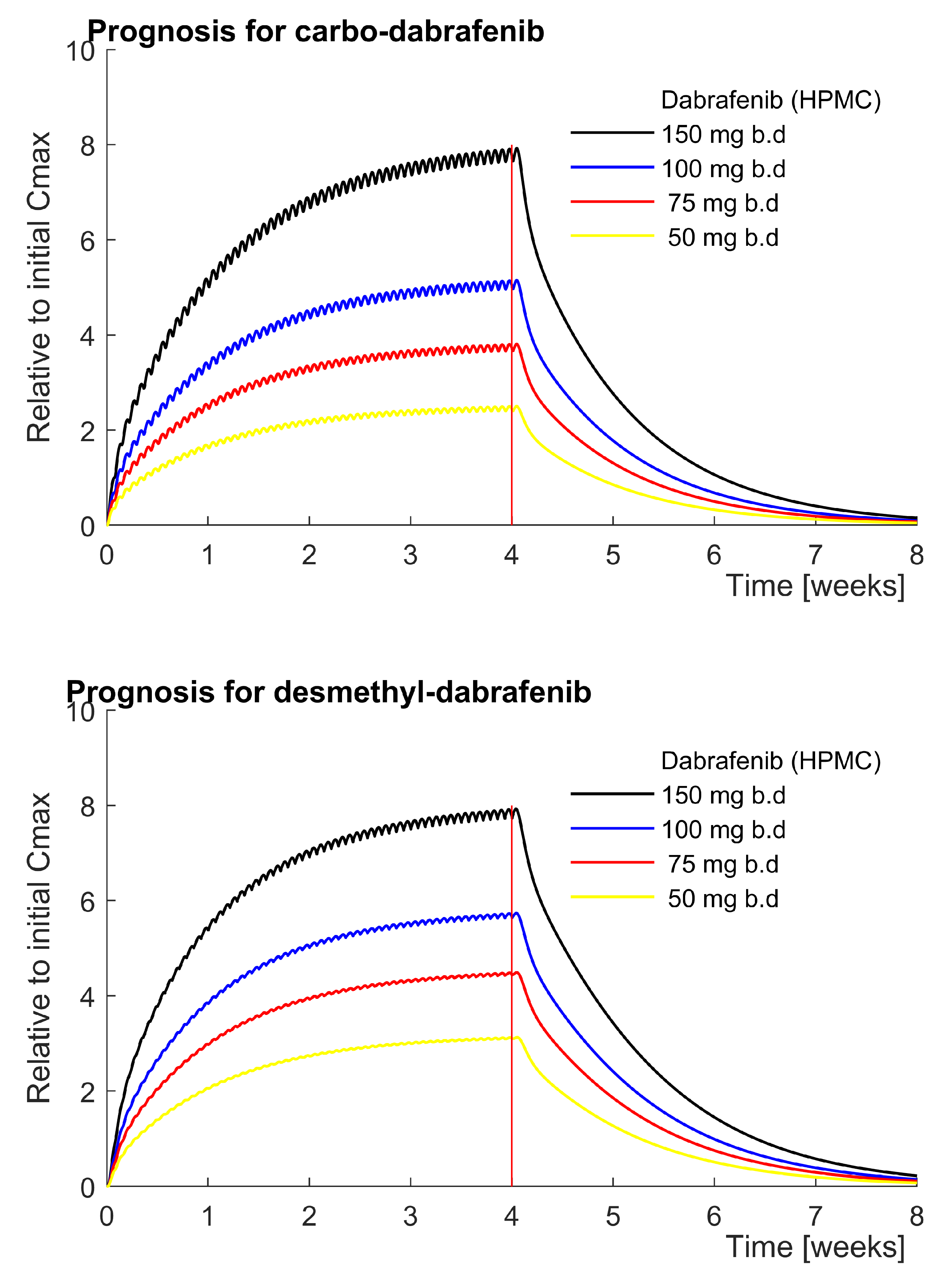
3.1.1. Carbo- and Desmethyl-Dabrafenib Accumulate According to the Model
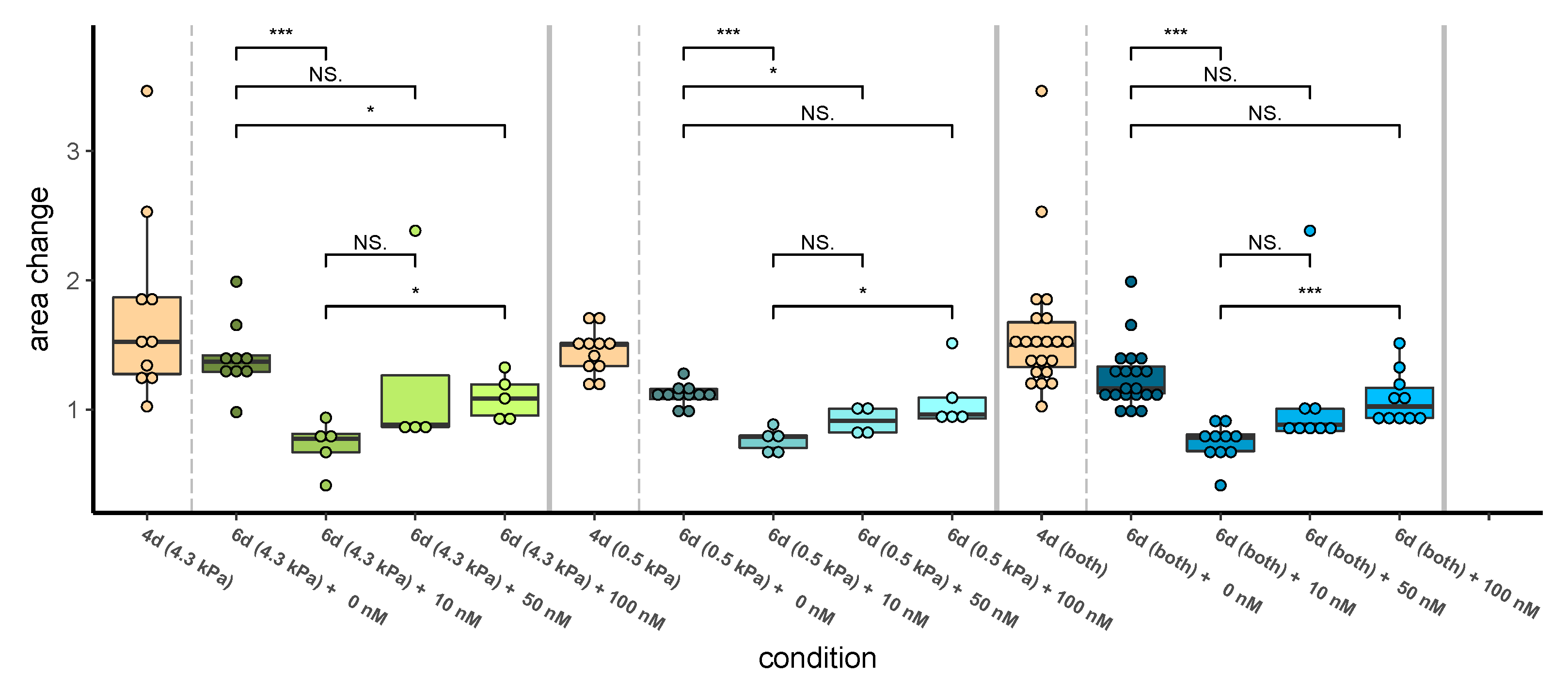
3.1.2. Higher Dabrafenib Levels Do Not Significantly Reduce Growth of 451LU Spheroids
4. Discussion
4.1. Model Extensions Are Well Supported by Data
4.2. More Experimental Evidence Might Allow the Consideration of PXR
4.3. Accumulating Carbo-Dabrafenib and Desmethyl-Dabrafenib Concentrations Are Plausible
4.4. Acidity Might Shift the Local Balance towards Active Desmethyl-Dabrafenib
4.5. Dabrafenib Is Ineffective If Highly Dosed in a Fibronectin-Supplemented Environment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Model Equations and Parameters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Value | Unit | Name | Value | Unit | Name | Value | Unit |
|---|---|---|---|---|---|---|---|---|
| (sol) | 4.55 | 8.63 | 2.26 | |||||
| 40.1 | 13.5 | 1.18 | ||||||
| 240.2 | 2.72 | 8.8 | ||||||
| 0.98 | 39.4 | 24.0 | ||||||
| 1.83 | 11.1 | 5.05 | ||||||
| 51.05 | 0.62 | 1.03 | ||||||
| 0.02 | 0.14 | 5.76 | ||||||
| 0.65 | 17.28 | 1.79 | ||||||
| 0.61 | 2.16 | |||||||
| 10.107 | 20.2 |
References
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF inhibitors amplify the proapoptotic activity of MEK inhibitors by inducing ER stress in NRAS-mutant melanoma. Clin. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessner, H.; Schmitz, J.; Tabatabai, G.; Schmid, A.M.; Calaminus, C.; Sinnberg, T.; Weide, B.; Eigentler, T.K.; Garbe, C.; Schittek, B.; et al. PI3K pathway inhibition achieves potent antitumor activity in melanoma brain metastases in vitro and in vivo. Clin. Cancer Res. 2016, 22, 5818–5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.H.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 770–784. [Google Scholar] [CrossRef]
- Larkin, J.; Minor, D.; D’Angelo, S.; Neyns, B.; Smylie, M.; Miller, W.H., Jr.; Gutzmer, R.; Linette, G.; Chmielowski, B.; Lao, C.D.; et al. Overall survival in patients with advanced melanoma who received nivolumab versus investigator’s choice chemotherapy in CheckMate 037: A randomized, controlled, open-label phase III trial. J. Clin. Oncol. 2018, 36, 383. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, H.T.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.; et al. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF-inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2014, 20, 4449–4458. [Google Scholar] [CrossRef] [Green Version]
- Ouellet, D.; Gibiansky, E.; Leonowens, C.; O’Hagan, A.; Haney, P.; Switzky, J.; Goodman, V.L. Population pharmacokinetics of dabrafenib, a BRAF inhibitor: Effect of dose, time, covariates, and relationship with its metabolites. J. Clin. Pharmacol. 2014, 54, 696–706. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Chu, X.; Gibson, C.; Cui, D.; Yee, K.L.; Ballard, J.; Cabalu, T.; Hochman, J. Drug–drug interaction studies: Regulatory guidance and an industry perspective. AAPS J. 2013, 15, 629–645. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C.; Talaly, P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis–Menten kinetic systems. J. Biol. Chem. 1977, 252, 6438–6442. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; Wiley-Blackwell: Weinheim, Germany, 2012. [Google Scholar]
- Albrecht, M. Mathematical Histopathology and Systems Pharmacology of Melanoma. Ph.D. Thesis, University of Luxembourg, Esch-sur-Alzette, Luxemburg, 2019. [Google Scholar]
- Lawrence, S.K.; Nguyen, D.; Bowen, C.; Richards-Peterson, L.; Skordos, K.W. The metabolic drug-drug interaction profile of dabrafenib: In vitro investigations and quantitative extrapolation of the P450-mediated DDI risk. Drug Metab. Dispos. 2014, 42, 1180–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bershas, D.A.; Ouellet, D.; Mamaril-Fishman, D.B.; Nebot, N.; Carson, S.W.; Blackman, S.C.; Morrison, R.A.; Adams, J.L.; Jurusik, K.E.; Knecht, D.M.; et al. Metabolism and disposition of oral dabrafenib in cancer patients: Proposed participation of aryl nitrogen in carbon-carbon bond cleavage via decarboxylation following enzymatic oxidation. Drug Metab. Dispos. 2013, 41, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Heel, R.C.; Brogden, R.N.; Carmine, A.; Morley, P.A.; Speight, T.M.; Avery, G.S. Ketoconazole: A review of its therapeutic efficacy in superficial and systemic fungal infections. Drugs 1982, 23, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Suttle, A.B.; Grossmann, K.F.; Ouellet, D.; Richards-Peterson, L.E.; Aktan, G.; Gordon, M.S.; LoRusso, P.M.; Infante, J.R.; Sharma, S.; Kendra, K.; et al. Assessment of the drug interaction potential and single-and repeat-dose pharmacokinetics of the BRAF inhibitor dabrafenib. J. Clin. Pharmacol. 2015, 55, 392–400. [Google Scholar] [CrossRef]
- Knight, C.G.; Willenbrock, F.; Murphy, G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992, 296, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Vörsmann, H.; Groeber, F.; Walles, H.; Busch, S.; Beissert, S.; Walczak, H.; Kulms, D. Development of a human three-dimensional organotypic skin-melanoma spheroid model for in vitro drug testing. Cell Death Dis. 2013, 4, 719. [Google Scholar] [CrossRef]
- van der Graaf, P.H.; Benson, N. Systems pharmacology: Bridging systems biology and pharmacokinetics-pharmacodynamics (PKPD) in drug discovery and development. Pharm. Res. 2011, 28, 1460–1464. [Google Scholar] [CrossRef]
- Wolkenhauer, O. Why model? Front. Physiol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Tang, C.; Sant, V.; Li, S.; Poloyac, S.M.; Xie, W. A molecular aspect in the regulation of drug metabolism: Does PXR-induced enzyme expression always lead to functional changes in drug metabolism? Curr. Pharmacol. Rep. 2016, 2, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Novotna, A.; Krasulová, K.; Bartoňková, I.; Korhoňová, M.; Bachleda, P.; Anzenbacher, P.; Dvořák, Z. Dual effects of ketoconazole cis-enantiomers on CYP3A4 in human hepatocytes and HepG2 cells. PLoS ONE 2014, 9, 111286. [Google Scholar] [CrossRef]
- Huang, H.; Wang, H.; Sinz, M.; Zoeckler, M.; Staudinger, J.; Redinbo, M.R.; Teotico, D.G.; Locker, J.; Kalpana, G.V.; Mani, S. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 2007, 26, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, I.; Hafner-Blumenstiel, V.; Markert, C.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Effect of the CYP3A inhibitor ketoconazole on the PXR-mediated induction of CYP3A activity. Eur. J. Clin. Pharmacol. 2013, 69, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.; McSheehy, P.M.J.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the V600EBRAF oncogene. Oncotarget 2013, 4, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouton, J.W.; Theuretzbacher, U.; Craig, W.A.; Tulkens, P.M.; Derendorf, H.; Cars, O. Tissue concentrations: Do we ever learn? J. Antimicrob. Chemother. 2007, 61, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentilcore, G.; Madonna, G.; Mozzillo, N.; Ribas, A.; Cossu, A.; Palmieri, G.; Ascierto, P.A. Effect of dabrafenib on melanoma cell lines harbouring the BRAF V600D/R mutations. BMC Cancer 2013, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Adelmann, C.H.; Ching, G.; Du, L.; Saporito, R.C.; Bansal, V.; Pence, L.J.; Liang, R.; Lee, W.; Tsai, K.Y. Comparative profiles of BRAF inhibitors: The paradox index as a predictor of clinical toxicity. Oncotarget 2016, 7, 30453. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Abel, E.V.; Koomen, J.M.; Fang, B.; Wood, E.R.; Chen, Y.A.; Fisher, K.J.; Iyengar, S.; Dahlman, K.B.; Wargo, J.A.; et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene 2016, 35, 1225. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Aplin, A.E. Akt3-mediated resistance to apoptosis in B-RAF–targeted melanoma cells. Cancer Res. 2010, 70, 6670–6681. [Google Scholar] [CrossRef] [Green Version]
- Seip, K.; Fleten, K.G.; Barkovskaya, A.; Nygaard, V.; Haugen, M.H.; Engesæter, B.Ø; Mælandsmo, G.M.; Prasmickaite, L. Fibroblast-induced switching to the mesenchymal-like phenotype and PI3K/mTOR signaling protects melanoma cells from BRAF inhibitors. Oncotarget 2016, 7, 19997. [Google Scholar] [CrossRef]
- Wei, S.C.; Yang, J. Forcing through tumor metastasis: The interplay between tissue rigidity and epithelial–mesenchymal transition. Trends Cell Biol. 2016, 26, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weder, G.; Hendriks-Balk, M.C.; Smajda, R.; Rimoldi, D.; Liley, M.; Heinzelmann, H.; Meister, A.; Mariotti, A. Increased plasticity of the stiffness of melanoma cells correlates with their acquisition of metastatic properties. Nanomedicine 2014, 10, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue force programs cell fate and tumor aggression. Cancer Discov. 2017, 7, 1224–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179. [Google Scholar] [CrossRef]
- Levesque, M.P.; Cheng, P.F.; Raaijmakers, M.I.G.; Saltari, A.; Dummer, R. Metastatic melanoma moves on: Translational science in the era of personalized medicine. Cancer Metast. Rev. 2017, 36, 7–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchenreuther, J.; Leask, A. Why target the tumor stroma in melanoma? J. Cell Commun. Signal 2018, 474, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astashkina, A.; Mann, B.; Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol. Ther. 2012, 134, 82–106. [Google Scholar] [CrossRef]
- Yeo, K.R.; Jamei, M.; Yang, J.; Tucker, G.T.; Rostami-Hodjegan, A. Physiologically based mechanistic modelling to predict complex drug–drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut-the effect of diltiazem on the time-course of exposure to triazolam. Eur. J. Pharm. Sci. 2010, 39, 298–309. [Google Scholar]
- Huynh, H.H.; Pressiat, C.; Sauvageon, H.; Madelaine, I.; Maslanka, P.; Lebbé, C.; Thieblemont, C.; Goldwirt, L.; Mourah, S. Development and validation of a simultaneous quantification method of 14 tyrosine kinase inhibitors in human plasma using LC-MS/MS. Ther. Drug. Monit. 2017, 39, 43–54. [Google Scholar] [CrossRef]
- Schäuble, S.; Stavrum, A.K.; Puntervoll, P.; Schuster, S.; Heiland, I. Effect of substrate competition in kinetic models of metabolic networks. FEBS Lett. 2013, 587, 2818–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruyere, A.; Decleves, X.; Bouzom, F.; Ball, K.; Marques, C.; Treton, X.; Pocard, M.; Valleur, P.; Bouhnik, Y.; Panis, Y.; et al. Effect of variations in the amounts of P-glycoprotein (ABCB1), BCRP (ABCG2) and CYP3A4 along the human small intestine on PBPK models for predicting intestinal first pass. Mol. Pharm. 2010, 7, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Choi, W.-G.; Lee, S.; Lee, H.S. Revisiting the metabolism and bioactivation of ketoconazole in human and mouse using liquid chromatography–mass spectrometry-based metabolomics. Int. J. Mol. Sci. 2017, 18, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneshmend, T.K.; Warnock, D.W.; Ene, M.D.; Johnson, E.M.; Potten, M.R.; Richardson, M.D.; Williamson, P.J. Influence of food on the pharmacokinetics of ketoconazole. Antimicrob. Agents Chemother. 1984, 25, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Bucher, J. Dynamische Modellierung und Simulation des Arzneimittelmetabolismus in Humanen Leberzellen: Identifizierbarkeit, Robustheit und Inter-Individuelle Variabilität. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2011. [Google Scholar]
- Wei, Z.; Jiang, S.; Zhang, Y.; Wang, X.; Peng, X.; Meng, C.; Liu, Y.; Wang, H.; Guo, L.; Qin, S.; et al. The effect of microRNAs in the regulation of human CYP3A4: A systematic study using a mathematical model. Sci. Rep. 2014, 4, 4283. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albrecht, M.; Kogan, Y.; Kulms, D.; Sauter, T. Mechanistically Coupled PK (MCPK) Model to Describe Enzyme Induction and Occupancy Dependent DDI of Dabrafenib Metabolism. Pharmaceutics 2022, 14, 310. https://doi.org/10.3390/pharmaceutics14020310
Albrecht M, Kogan Y, Kulms D, Sauter T. Mechanistically Coupled PK (MCPK) Model to Describe Enzyme Induction and Occupancy Dependent DDI of Dabrafenib Metabolism. Pharmaceutics. 2022; 14(2):310. https://doi.org/10.3390/pharmaceutics14020310
Chicago/Turabian StyleAlbrecht, Marco, Yuri Kogan, Dagmar Kulms, and Thomas Sauter. 2022. "Mechanistically Coupled PK (MCPK) Model to Describe Enzyme Induction and Occupancy Dependent DDI of Dabrafenib Metabolism" Pharmaceutics 14, no. 2: 310. https://doi.org/10.3390/pharmaceutics14020310