
An Overview of Nanotechnologies for Drug Delivery to the Brain


Abstract
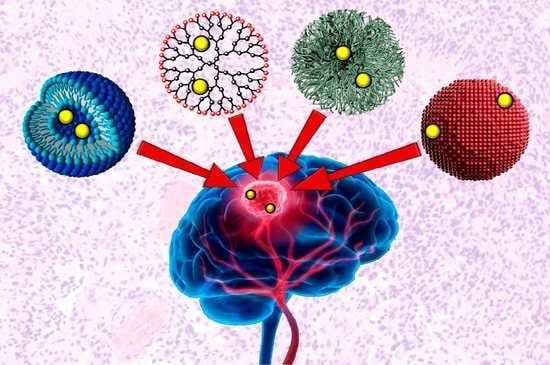
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction and Background
2. Delivery of Therapeutics to the Brain
2.1. The Blood–Brain Barrier (BBB)
Transport of Molecules across the BBB
- Paracellular transport: The transport of molecules through the intercellular spaces between the endothelial cells is the most restrictive form of transport through the BBB due to the tight junctions (Figure 1i) [21]. Paracellular transport through the BBB is mostly dictated by the environmental concentration gradient; i.e., a greater concentration gradient leads to more transport between the endothelial cells, with minor contributions from the molecule’s surface charge, its size and its lipophilicity [19,21]. The tight junctions contain tiny aqueous pores, which means only small hydrophilic compounds are able to make it across the BBB [21,22].
- Diffusion: Passive transcellular diffusion across the BBB through the endothelial cells (Figure 1ii) mainly depends on the lipophilicity of the molecule; higher lipophilicity means higher diffusion. Size also plays an important role here, with smaller compounds being relatively easily permeated. As with paracellular diffusion of molecules between the endothelial cells, the environmental concentration gradient also affects this pathway [19,22]. Compounds and particles that cross the BBB through the lipid-mediated transcellular diffusion must have a molecular weight of less than 500 Da [23,24]. A 100-fold decrease in BBB permeability has been reported for molecules of molecular weight of 450 Da when compared to smaller, 200 Da molecules [24,25].
- Influx transporters: For molecules that are unable to cross the BBB via diffusion due to size or molecular weight restrictions, a very efficient protein-transporter system is in place to help them across. Transport across the BBB through influx transporter proteins, also referred to as carrier-mediated transport (CMT), involves transporters binding to and carrying specific molecules through the BBB (Figure 1iii) [26]. These transporters assist slightly larger lipophilic molecules to permeate through the BBB that show an affinity towards specific endogenous BBB transporters. Molecules are bound to the transporter and carried across the endothelial cell lining of the BBB into the CNS [18,26].
- Receptor-mediated transcytosis (RMT): One of the important ways macromolecules pass through the BBB is via RMT. Receptors selective towards specific ligands enable a number of large molecules such as sugars, proteins, hormones, etc., to pass through the BBB [27]. RMT takes place in the following three distinct steps: Refs. [27,28,29]
- (a)
- Receptor-mediated endocytosis of molecule by the receptor;
- (b)
- Transport of vesicle across the membrane;
- (c)
- Release of molecule into the extracellular space of endothelial cells.
RMT differs from CMT mechanistically. Protein carriers that are able to freely move across the endothelial cells of the BBB aid a substrate by binding to and carrying it across the BBB themselves. In contrast, molecules crossing the BBB using RMT only bind to specific receptor-proteins on the membrane of the endothelial cells to transcytose across the BBB (Figure 1iv) [18,30]. - Absorptive-mediated transcytosis (AMT): Cationic molecules are not readily transported across the BBB via the previously discussed pathways; however, they have the ability to bind to and absorb into the luminal surface of the endothelial cells that make up the BBB [31]. Following the bond between the endothelial cells and the molecule, AMT follows the same path as RMT, in which endocytosis is followed by transport across the endothelial cell lining, ending with exocytosis of the molecule (Figure 1v) [28,31].
- Efflux transporters: This transport system is in charge of discarding unwanted molecules back into the systemic bloodstream using efflux proteins via CMT (Figure 1vi). The efflux transporter system employed by the BBB is very effective and is one of the major hurdles for drugs to treat brain or CNS diseases [28,32]. One of the most important proteins in the efflux pump is the P-glycoprotein (P-gp), which is responsible for the elimination of a number of drugs and molecules from the BBB [33].

2.2. Drug-Delivery Routes to the Brain
- Viral vectors: As part of their replication cycle, viruses attack their host to introduce their own genetic material into the host cell. The inserted genetic material is composed of basic instructions on how to produce more viruses. The viruses end up effectively taking over the host cell completely to fulfill their own needs [39]. The infected host cells carry out these instructions, with more and more viruses being produced, eventually cascading into taking over more host cells [39,40]. The genetic material that guides the host cells to assist the replication of viruses can be substituted with genetic instructions that would be beneficial for the host, for example, instructions to invade, infect and kill cancerous cells. Essentially, viral vectors can be used to deliver specific genes to fight or prevent diseases (gene therapy) [41,42]. In addition to gene therapy, viral vectors have been generating interest as drug carriers in recent years. The drugs can be encapsulated or infused with the vector, which can be functionalized for targeted delivery. Overall, there are still several issues associated with using viral vectors as drug-delivery systems, mainly linked with their high cost of production and their safety, as administration of viruses always carries a certain level of risk [43,44,45].
- Exosomes: Exosomes are naturally occurring vesicles that are being used as drug carriers to penetrate the BBB for smaller drugs, proteins and nucleic acids; they are of increasing interest due to their high biocompatibility. Functionalized exosomes have been seen to be able to cross the BBB through RMT [46]. The limitations associated with using exosomes as drug-delivery systems have to do with the variety of drugs that can be loaded into them. Whether a drug is a suitable contender to be used in an exosome DDS is mainly dependant on the drug’s physical and chemical characteristics [43,47].
- Drug delivery via active transporters: Essential amino acids, such as phenylalanine, leucine, tyrosine, isoleucine, valine, tryptophan, methionine and histidine, travel across the BBB into the brain and CNS via carrier-mediated influx. Smaller drugs with appropriate physico-chemical properties have been linked with these amino acids that work as active transporters and carry the molecule through the BBB endothelial cells. Similar to exosomes, the properties of the drug dictate whether using amino acids as active transporters for the delivery of the drug to the brain is going to be successful [48].
- Enhancing paracellular transport: The BBB has been shown to be disruptable, enabling certain drugs to pass through via paracellular diffusion which otherwise would not be able to [28,49]. There are two main ways whereby the BBB can be disrupted, i.e., by using osmotic agents that draw the cellular water out of the endothelial cells, resulting in them shrinking, enabling the drug passage through the tight junctions; and by using chemical agents that generate a temporary inflammatory reaction in the endothelial cells, which results in the tight junctions loosening temporarily, allowing the drug to pass through [38,49]. Paracellular transport is restrictive, which means only small hydrophilic drugs can cross through a disrupted BBB [38,50].
- Modification of drugs for transcytosis: Drugs can be modified by changing their physico-chemical properties (lipophilicity, size, charge, shape, etc.) to enhance AMT, or by conjugating them with specific ligands or antibodies to trigger RMT [38]. This is a very promising strategy for drug delivery to the brain owing to its range of versatility and cost-effectiveness relatively to other delivery systems [34,38,50].
2.3. Glioblastomas
2.3.1. Treatment of GBMs
2.3.2. GBMs and Transferrin
3. Characteristics of an Effective Nanoparticle DDS
3.1. Biocompatibility
3.2. Size, Surface Morphology and Functionalizability
3.3. Surface Charge
3.4. Drug Loading Capacity
3.5. Functionalizability
3.5.1. End-Group Modification
3.5.2. Crosslinkage
3.5.3. Surface Modification through Physical Adsorption Using Electrostatic Forces
4. Nanotechnologies for Brain Drug Delivery
4.1. Inorganic Nanoparticles
4.1.1. Gold Nanoparticles (AuNPs)
4.1.2. Magnetic Nanoparticles (MNPs)
4.2. Organic Nanoparticles
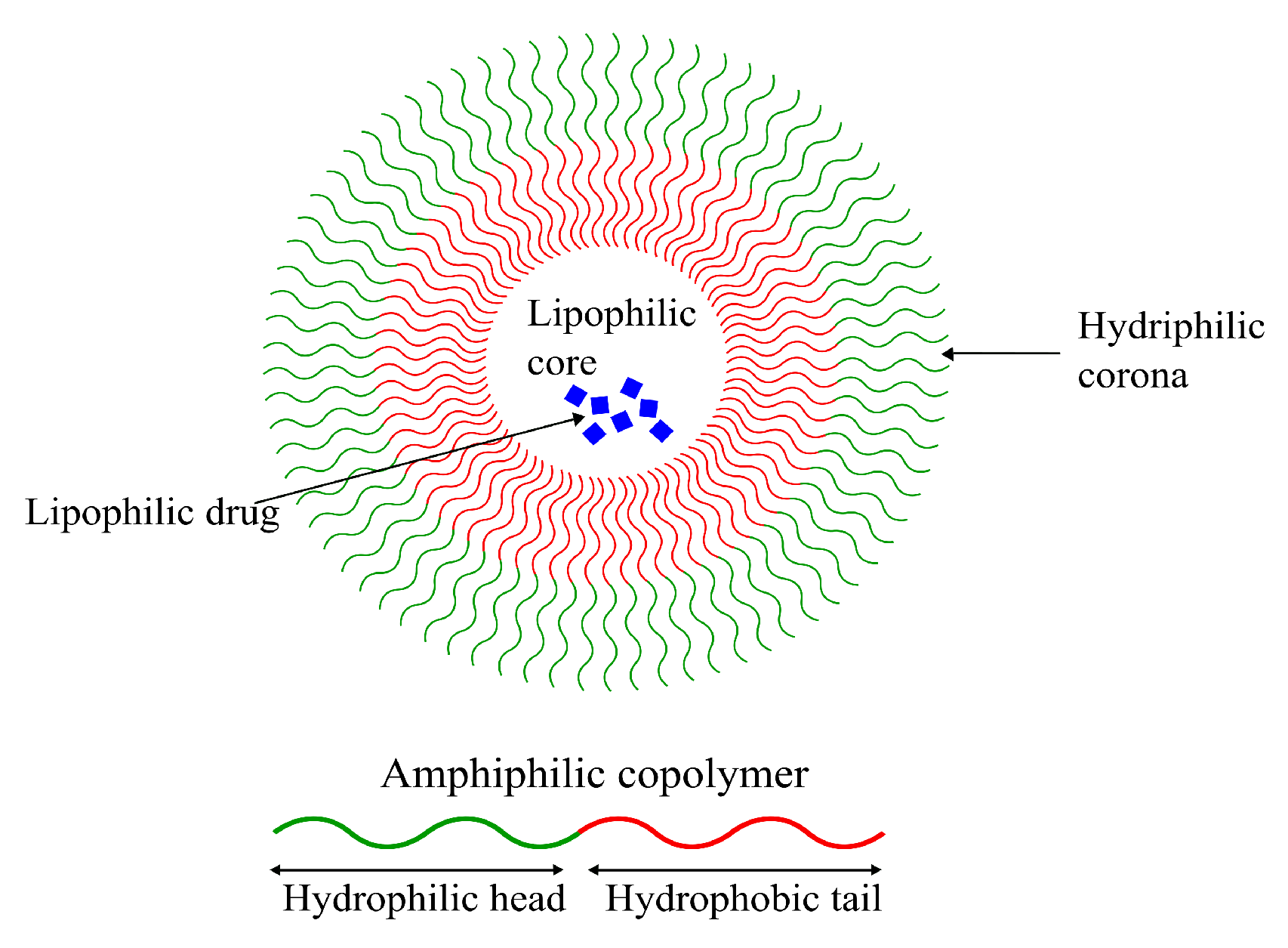
4.2.1. Polymeric Micelles (PM)
4.2.2. Dendrimers
4.2.3. Liposomes
4.2.4. Niosomes
4.2.5. Microemulsions
4.2.6. Polymeric Solid Nanoparticles (SNPs)
5. SNPs in Drug-Delivery Applications
5.1. Functionalization of SNPs
SNPs Made Using Layer-by-Layer (LbL) Assembly Method
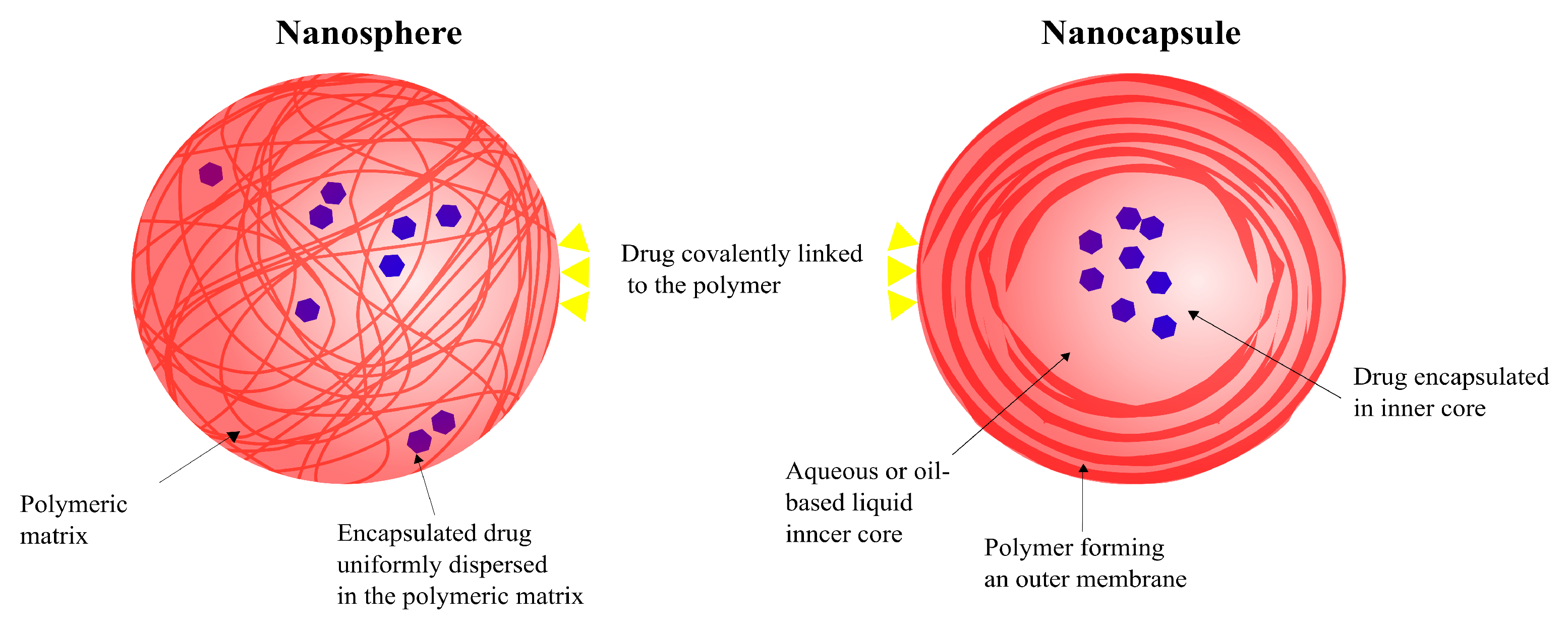
5.2. Types of SNPs: Nanocapsules and Nanospheres
5.3. Commonly Used Polymers for SNPs in Brain Drug Delivery
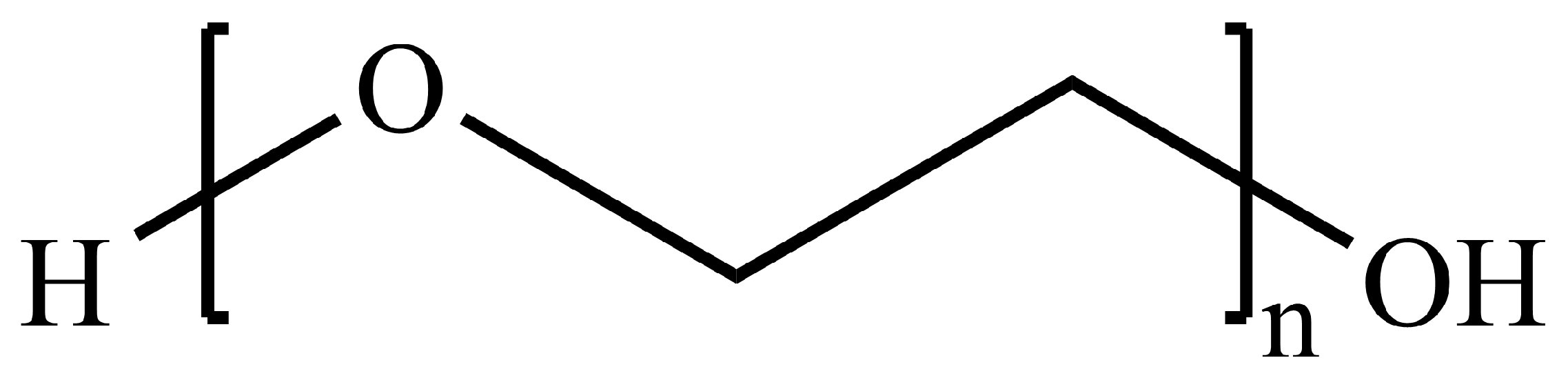
5.3.1. PLGA and PEG
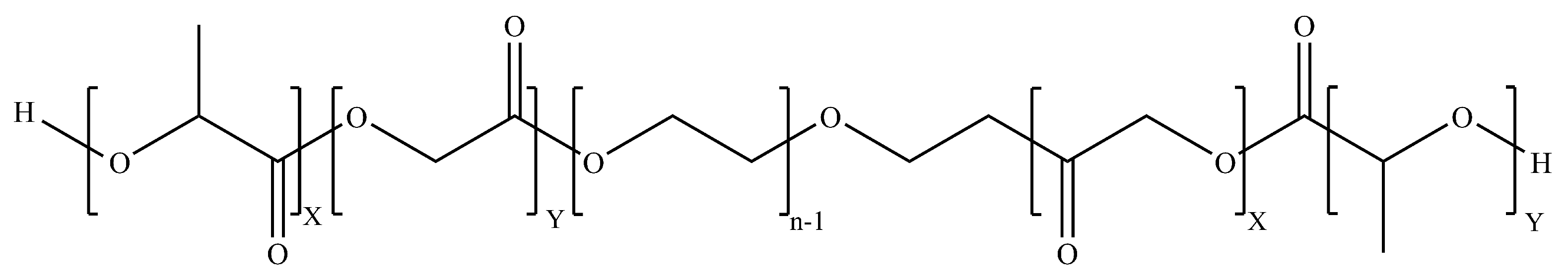
5.3.2. PLGA–PEG–PLGA (PEP)
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DDS(s) | Drug-delivery system(s) |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| CMT, RMT and AMT | Carrier-mediated, receptor-mediated and absorptive-mediated transcytosis |
| P-gp | P-glycoprotein |
| WHO | World Health Organization |
| GBM | Glioblastoma multiforme |
| Tf, oTf and Lf | serum-Transferrin, ovo-Transferrin and Lactoferrin |
| TfR | Transferrin receptor |
| EPR | Enhanced permeability and retention |
| NP(s) | Nanoparticle(s) |
| AuNP(s) | Gold nanoparticle(s) |
| MNP(s) | Magnetic nanoparticle(s) |
| MRI | Magnetic resonance imaging |
| PM(s) | Polymeric micelle(s) |
| SNP(s) | Polymeric solid nanoparticle(s) |
| Apo-A1 and Apo-E | Apolipoprotein A1 and Apolipoprotein E |
| LbL assembly | Layer-by-layer assembly |
| LA and GA | Lactic acid and Glycolic acid |
| Abbreviated Chemicals: | |
| FTIC | Fluorescein isothiocyanate |
| IO | Iron oxide |
| PS-PAA | Poly(styrene)-poly(acrylic acid) |
| PEG-PLA | Poly(ethylene)glycol-poly(lactic acid) |
| PEG-DSPE | Distearyl-sn-glycero-3-phosphoethanolamine-N-methoxy poly |
| (ethylene glycol) | |
| PEG-PLA | Poly(ethylene glycol)-poly(lactic acid) |
| PEG-PLGA | Poly(ethylene glycol)-poly(lactic-co-glycolic acid) |
| DPX | Dapoxetine |
| PAMAM | Polyamidoamine |
| IL-13R2 | Interleukin-13-receptor-2 |
| EDC | N-Ethyl-N-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| NHS | N-hydroxysuccinimide |
| PLGA | Poly(lactic-co-glycolic acid) |
| PEG | Poly(ethylene(glycol |
| BSA | Bovine serum albumin |
| FR | Folate receptor alpha |
| Cur | Curcumin |
| g7 | Gly-l-Phe-D-Thr-Gly-l-Phe-l-Leu-LSer(O--D-Glucose)-CONH |
| TMC | Trimethylated chitosan |
| PEP | PLGA-PEG-PLGA |
References
- Dias, D.A.; Urban, S.; Roessner, U. A Historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- Bongaarts, J. Human population growth and the demographic transition. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2985–2990. [Google Scholar] [CrossRef] [Green Version]
- Perrott, G.S.J.; Holland, D.F. Population trends and problems of public health. Milbank Q. 2005, 83, 569. [Google Scholar] [CrossRef] [Green Version]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.W. Early drug discovery and the rise of pharmaceutical chemistry. Drug Test. Anal. 2011, 3, 337–344. [Google Scholar] [CrossRef]
- Wein, W. Drug development: Successes, problems and pitfalls-the industry perspective. ESMO Open 2016, 1, e000033. [Google Scholar] [CrossRef] [Green Version]
- Scannell, J.W.; Bosley, J. When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis. PLoS ONE 2016, 11, e0147215. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.K. Drug delivery systems—An overview. Methods Mol. Biol. 2008, 437, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Škalko-Basnet, N. Biologics: The role of delivery systems in improved therapy. Biol. Targets Ther. 2014, 8, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, R.; Saini, S.; Sharma, S. Nanotechnology: The Future Medicine. J. Cutan. Aesthetic Surg. 2010, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Nanomedicine Revolution: Part 1: Emerging Concepts. Pharm. Ther. 2012, 37, 512. [Google Scholar]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J. Nanomater. 2019, 2019, 3702518. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2009, 37, 13–25. [Google Scholar] [CrossRef]
- Kusuhara, H.; Sugiyama, Y. Efflux transport systems for drugs at the blood–brain barrier and blood–cerebrospinal fluid barrier (Part 1). Drug Discov. Today 2001, 6, 150–156. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef] [Green Version]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx J. Am. Soc. Exp. NeuroTherapeutics 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Itallie, C.M.; Anderson, J.M. The Molecular Physiology of Tight Junction Pores. Physiology 2004, 19, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Nam, L.; Coll, C.; Erthal, L.; de la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779. [Google Scholar] [CrossRef] [Green Version]
- Mikitsh, J.L.; Chacko, A.M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Med. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood–brain barrier endogenous transporters as therapeutic targets: A new model for small molecule CNS drug discovery. Expert Opin. Ther. Targets 2015, 19, 1059–1072. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier and neurotherapeutics. NeuroRx J. Am. Soc. Exp. NeuroTherapeutics 2005, 2, 1–2. [Google Scholar] [CrossRef]
- Lawther, B.K.; Kumar, S.; Krovvidi, H. Blood-brain barrier. Contin. Educ. Anaesth. Crit. Care Pain 2011, 11, 128–132. [Google Scholar] [CrossRef]
- de Lange, E.C. The Physiological Characteristics and Transcytosis Mechanisms of the Blood-Brain Barrier (BBB). Curr. Pharm. Biotechnol. 2012, 13, 2319–2327. [Google Scholar] [CrossRef]
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert Opin. Drug Deliv. 2019, 16, 271–285. [Google Scholar] [CrossRef]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-Glycoprotein in Pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Scherrmann, J.M. Drug delivery to brain via the blood–brain barrier. Vasc. Pharmacol. 2002, 38, 349–354. [Google Scholar] [CrossRef]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and physiology of the blood-brain barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, T.; Zhou, J.; Piepmeier, J.M.; Saltzman, W.M. Polymeric nanoparticles for drug delivery to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 701–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahnke, K.; Doolittle, N.D.; Muldoon, L.L.; Neuwelt, E.A. Implications of the blood-brain barrier in primary central nervous system lymphoma. Neurosurg. Focus 2006, 21, E11. [Google Scholar] [CrossRef]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, L.S.; Matsudaira, P.; Baltimore, D.; Darnell, J. Viruses: Structure, Function, and Uses. In Molecular Cell Biology, 4th ed.; Freeman, W.H., Ed.; W. H. Freeman & Company: New York, NY, USA, 2000; Chapter 6.3. [Google Scholar]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, G.A.R.; Paiva, R.d.M.A. Gene therapy: Advances, challenges and perspectives. Einstein 2017, 15, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Ghivizzani, S.C. Viral Vectors for Gene Therapy. Pharmacol. Ther. 1998, 80, 35–47. [Google Scholar] [CrossRef]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Lockney, D.; Franzen, S.; Lommel, S. Viruses as nanomaterials for drug delivery. Methods Mol. Biol. 2011, 726, 207–221. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Lakhal, S.; Wood, M.J. Exosome nanotechnology: An emerging paradigm shift in drug delivery. BioEssays 2011, 33, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Peura, L.; Malmioja, K.; Huttunen, K.; Leppänen, J.; Hämäläinen, M.; Forsberg, M.M.; Gynther, M.; Rautio, J.; Laine, K. Design, synthesis and brain uptake of LAT1-targeted amino acid prodrugs of dopamine. Pharm. Res. 2013, 30, 2523–2537. [Google Scholar] [CrossRef]
- Rodriguez, A.; Tatter, S.; Debinski, W. Neurosurgical Techniques for Disruption of the Blood–Brain Barrier for Glioblastoma Treatment. Pharmaceutics 2015, 7, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Guerin, J.B.; Giannini, C.; Morris, J.M.; Eckel, L.J.; Kaufmann, T.J. 2016 Updates to the WHO Brain Tumor Classification System: What the Radiologist Needs to Know. RadioGraphics 2017, 37, 2164–2180. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma, 1st ed.; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 8; pp. 143–155. [Google Scholar] [CrossRef]
- Voth, B.; Nagasawa, D.T.; Pelargos, P.E.; Chung, L.K.; Ung, N.; Gopen, Q.; Tenn, S.; Kamei, D.T.; Yang, I. Transferrin receptors and glioblastoma multiforme: Current findings and potential for treatment. J. Clin. Neurosci. 2015, 22, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.D.M.; Bonavia, R.; Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.N.; Baker, H.M.; Kidd, R.D. Lactoferrin and transferrin: Functional variations on a common structural framework. Biochem. Cell Biol. 2002, 80, 27–34. [Google Scholar] [CrossRef]
- Kell, D.B.; Heyden, E.L.; Pretorius, E. The Biology of Lactoferrin, an Iron-Binding Protein That Can Help Defend Against Viruses and Bacteria. Front. Immunol. 2020, 11, 1221. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Acero-Lopez, A. Ovotransferrin: Structure, bioactivities, and preparation. Food Res. Int. 2012, 46, 480–487. [Google Scholar] [CrossRef]
- Wally, J.; Buchanan, S.K. A structural comparison of human serum transferrin and human lactoferrin. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2007, 20, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Gatter, K.C.; Brownt, G.; Trowbridget, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Fishman, J.B.; Rubin, J.B.; Handrahan, J.V.; Connor, J.R.; Fine, R.E. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res. 1987, 18, 299–304. [Google Scholar] [CrossRef]
- Sun, T.; Wu, H.; Li, Y.; Huang, Y.; Yao, L.; Chen, X.; Han, X.; Zhou, Y.; Du, Z. Targeting transferrin receptor delivery of temozolomide for a potential glioma stem cell-mediated therapy. Oncotarget 2017, 8, 74451. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.; Bortot, B.; Ruozi, B.; Dolcetta, D.; Vandelli, M.A.; Forni, F.; Severini, G.M. Potential use of polymeric nanoparticles for drug delivery across the blood-brain barrier. Curr. Med. Chem. 2013, 20, 2212–2225. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Fan, Y.; Feng, Q.; Cui, F.Z. Biocompatibility and Toxicity of Nanoparticles and Nanotubes. J. Nanomater. 2012, 2012, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of engineered nanoparticles for drug delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Calzoni, E.; Cesaretti, A.; Polchi, A.; Di Michele, A.; Tancini, B.; Emiliani, C. Biocompatible Polymer Nanoparticles for Drug Delivery Applications in Cancer and Neurodegenerative Disorder Therapies. J. Funct. Biomater. 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Sadat, S.M.A.; Jahan, S.T.; Haddadi, A. Effects of Size and Surface Charge of Polymeric Nanoparticles on in Vitro and in Vivo Applications. J. Biomater. Nanobiotechnol. 2016, 07, 91–108. [Google Scholar] [CrossRef] [Green Version]
- Nandhakumar, S.; Dhanaraju, M.D.; Sundar, V.D.; Heera, B. Influence of surface charge on the in vitro protein adsorption and cell cytotoxicity of paclitaxel loaded poly(ϵ-caprolactone) nanoparticles. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 249–258. [Google Scholar] [CrossRef]
- Honary, S.; Zahir, F. Effect of zeta potential on the properties of nano-drug delivery systems—A review (Part 2). Trop. J. Pharm. Res. 2013, 12, 265–273. [Google Scholar] [CrossRef]
- Jawahar, N.; Meyyanathan, S. Polymeric nanoparticles for drug delivery and targeting: A comprehensive review. Int. J. Health Allied Sci. 2012, 1, 217. [Google Scholar] [CrossRef]
- Mout, R.; Moyano, D.F.; Rana, S.; Rotello, V.M. Surface functionalization of nanoparticles for nanomedicine. Chem. Soc. Rev. 2012, 41, 2539–2544. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Surface Functionalization and Bioconjugation of Nanoparticles for Biomedical Applications. Ph.D. Thesis, The University of Western Ontario, London, ON, Canada, 2014. [Google Scholar]
- Arora, B.; Tandon, R.; Attri, P.; Bhatia, R. Chemical Crosslinking: Role in Protein and Peptide Science. Curr. Protein Pept. Sci. 2017, 18, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Agarwal, A.; Jain, A.; Agrawal, R.K.; Jain, S.K. Bioconjugation of polymers: A novel platform for targeted drug delivery. Curr. Pharm. Des. 2011, 17, 1108–1125. [Google Scholar] [CrossRef]
- Mattson, G.; Conklin, E.; Desai, S.; Nielander, G.; Savage, M.D.; Morgensen, S. A practical approach to crosslinking. Mol. Biol. Rep. 1993, 17, 167–183. [Google Scholar] [CrossRef]
- Li, S.; Mouradov, D.; King, G.; Liu, T.; Ross, I.; Kobe, B.; Woods, V.L., Jr.; Huber, T. Study of Protein Three-Dimensional Structure and Dynamics Using Peptide Amide Hydrogen/Deuterium Exchange Spectrometry (DXMS) and Chemical Cross-Linking with Mass Spectrometry to Constrain Molecular Modelling. In Structural Bioinformatics, 2nd ed.; Gu, J., Bourne, P.E., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; Chapter 7; pp. 171–207. [Google Scholar]
- Hermanson, G.T. Zero-Length Crosslinkers. In Bioconjugate Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 4; pp. 259–275. [Google Scholar]
- Hermanson, G.T. Heterobifunctional Crosslinkers. In Bioconjugate Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 6; pp. 299–341. [Google Scholar]
- Wong, S.S. Zero-Length Cross-Linking Reagents. In Chemistry of Protein Conjugation and Cross-Linking, 1st ed.; CRC Press: Boca Raton, FL, USA, 1991; Chapter 6; pp. 195–203. [Google Scholar]
- Hermanson, G.T. Homobifunctional Crosslinkers. In Bioconjugate Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 5; pp. 275–299. [Google Scholar]
- Montalbetti, C.A.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Canalle, L.A.; Löwik, D.W.P.M.; van Hest, J.C.M. Polypeptide–polymer bioconjugates. Chem. Soc. Rev. 2010, 39, 329–353. [Google Scholar] [CrossRef] [Green Version]
- Tavano, L.; Muzzalupo, R.; Mauro, L.; Pellegrino, M.; Andò, S.; Picci, N. Transferrin-Conjugated Pluronic Niosomes as a New Drug Delivery System for Anticancer Therapy. Langmuir 2013, 29, 12638–12646. [Google Scholar] [CrossRef]
- Rafael, D.; Martínez, F.; Andrade, F.; Seras-Franzoso, J.; Garcia-Aranda, N.; Gener, P.; Sayós, J.; Arango, D.; Abasolo, I.; Schwartz, S. Efficient EFGR mediated siRNA delivery to breast cancer cells by Cetuximab functionalized Pluronic® F127/Gelatin. Chem. Eng. J. 2018, 340, 81–93. [Google Scholar] [CrossRef]
- Zweifel, G.; Nantz, M. Modern Organic Synthesis: An Introduction, 1st ed.; W. H. Freeman and Company: New York, NY, USA, 2007. [Google Scholar]
- Tojo, G.; Fernández, M. (Eds.) Chromium-based Reagents. In Oxidation of Alcohols to Aldehydes and Ketones; Springer: Berlin/Heidelberg, Germany, 2006; Chapter 1; Volume 1, p. 95. [Google Scholar] [CrossRef]
- Ding, H.; Yong, K.T.; Law, W.C.; Roy, I.; Hu, R.; Wu, F.; Zhao, W.; Huang, K.; Erogbogbo, F.; Bergey, E.J.; et al. Non-invasive tumor detection in small animals using novel functional Pluronic nanomicelles conjugated with anti-mesothelin antibody. Nanoscale 2011, 3, 1813. [Google Scholar] [CrossRef]
- Tian, J.L.; Zhao, Y.Z.; Jin, Z.; Lu, C.T.; Tang, Q.Q.; Xiang, Q.; Sun, C.Z.; Zhang, L.; Xu, Y.Y.; Gao, H.S.; et al. Synthesis and characterization of Poloxamer 188-grafted heparin copolymer. Drug Dev. Ind. Pharm. 2010, 36, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Suthiwangcharoen, N.; Nagarajan, R. Nanoarmoring of Proteins by Conjugation to Block Copolymer Micelles. Methods Enzymol. 2017, 590, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Zenke, M.; Cotten, M.; Beug, H.; Birnstiel, M.L. Transferrin-polycation conjugates as carriers for DNA uptake into cells. Proc. Natl. Acad. Sci. USA 1990, 87, 3410–3414. [Google Scholar] [CrossRef] [Green Version]
- Bellocq, N.C.; Pun, S.H.; Jensen, G.S.; Davis, M.E. Transferrin-Containing, Cyclodextrin Polymer-Based Particles for Tumor-Targeted Gene Delivery. Bioconjug. Chem. 2003, 14, 1122–1132. [Google Scholar] [CrossRef]
- Huang, R.Q.; Qu, Y.H.; Ke, W.L.; Zhu, J.H.; Pei, Y.Y.; Jiang, C. Efficient gene delivery targeted to the brain using a transferrin-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. FASEB J. 2007, 21, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogris, M.; Walker, G.; Blessing, T.; Kircheis, R.; Wolschek, M.; Wagner, E. Tumor-targeted gene therapy: Strategies for the preparation of ligand-polyethylene glycol-polyethylenimine/DNA complexes. J. Control. Release 2003, 91, 173–181. [Google Scholar] [CrossRef]
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-loaded polymeric nanoparticles for cancer stem cell targeting. Front. Pharmacol. 2017, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Wiley, D.T.; Webster, P.; Gale, A.; Davis, M.E. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 8662–8667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, R.; Reifenberger, G.; Wechsler, W. Transferrin receptor expression in tumours of the human nervous system: Relation to tumour type, grading and tumour growth fraction. Virchows Arch. A Pathol. Anat. Histopathol. 1990, 416, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Jallouli, Y.; Kroubi, M.; Yuan, X.B.; Feng, W.; Kang, C.S.; Pu, P.Y.; Betbeder, D. Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood-brain barrier. Int. J. Pharm. 2009, 379, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Pramanik, C.; Heinz, O.; Ding, Y.; Mishra, R.K.; Marchon, D.; Flatt, R.J.; Estrela-Lopis, I.; Llop, J.; Moya, S.; et al. Nanoparticle decoration with surfactants: Molecular interactions, assembly, and applications. Surf. Sci. Rep. 2017, 72, 1–58. [Google Scholar] [CrossRef]
- Ambruosi, A.; Gelperina, S.; Khalansky, A.; Tanski, S.; Theisen, A.; Kreuter, J. Influence of surfactants, polymer and doxorubicin loading on the anti-tumour effect of poly(butyl cyanoacrylate) nanoparticles in a rat glioma model. J. Microencapsul. 2006, 23, 582–592. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Cheng, J.; Yuan, Z. Recent advances on inorganic nanoparticle-based cancer therapeutic agents. Int. J. Environ. Res. Public Health 2016, 13, 1182. [Google Scholar] [CrossRef]
- Agarwal, V.; Chatterjee, K. Recent advances in the field of transition metal dichalcogenides for biomedical applications. Nanoscale 2018, 10, 16365–16397. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- Hirsjarvi, S.; Passirani, C.; Benoit, J.P. Passive and Active Tumour Targeting with Nanocarriers. Curr. Drug Discov. Technol. 2011, 8, 188–196. [Google Scholar] [CrossRef]
- Chauhan, M.K.; KChauhan, M.; Manchanda, A.; Khurana, J.K.; Jain, P.; Sharma, D.; Jain, S.; Bansal, S. Nanotechnology: The nano soldiers in the war against cancer. J. Pharm. Res. 2011, 4, 4420–4423. [Google Scholar]
- Bazak, R.; Houri, M.; Achy, S.E.; Hussein, W.; Refaat, T. Passive targeting of nanoparticles to cancer: A comprehensive review of the literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Stirland, D.L.; Nichols, J.W.; Miura, S.; Bae, Y.H. Mind the gap: A survey of how cancer drug carriers are susceptible to the gap between research and practice. J. Control. Release 2013, 172, 1045–1064. [Google Scholar] [CrossRef] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Clemons, T.D.; Singh, R.; Sorolla, A.; Chaudhari, N.; Hubbard, A.; Iyer, K.S. Distinction Between Active and Passive Targeting of Nanoparticles Dictate Their Overall Therapeutic Efficacy. Langmuir 2018, 34, 15343–15349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer active targeting by nanoparticles: A comprehensive review of literature. J. Cancer Res. Clin. Oncol. 2015, 141, 769–784. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.H. An introduction to the clinical practice of theranostics in oncology. Br. J. Radiol. 2018, 91, 20180440. [Google Scholar] [CrossRef]
- Bhujwalla, Z.M.; Kakkad, S.; Chen, Z.; Jin, J.; Hapuarachchige, S.; Artemov, D.; Penet, M.F. Theranostics and metabolotheranostics for precision medicine in oncology. J. Magn. Reson. 2018, 291, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Ehlerding, E.B.; Chen, F.; Cai, W. Biodegradable and Renal Clearable Inorganic Nanoparticles. Adv. Sci. 2016, 3, 1500223. [Google Scholar] [CrossRef]
- Dreaden, E.C.; Austin, L.A.; MacKey, M.A.; El-Sayed, M.A. Size matters: Gold nanoparticles in targeted cancer drug delivery. Ther. Deliv. 2012, 3, 457–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Pandit, S.; Mokkapati, V.R.; Garg, A.; Ravikumar, V.; Mijakovic, I. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int. J. Mol. Sci. 2018, 19, 1979. [Google Scholar] [CrossRef]
- Dhar, S.; Daniel, W.L.; Giljohann, D.A.; Mirkin, C.A.; Lippard, S.J. Polyvalent oligonucleotide gold nanoparticle conjugates as delivery vehicles for platinum(IV) warheads. J. Am. Chem. Soc. 2009, 131, 14652–14653. [Google Scholar] [CrossRef]
- Dreaden, E.C.; Mwakwari, S.C.; Sodji, Q.H.; Oyelere, A.K.; El-Sayed, M.A. Tamoxifen-poly(ethylene glycol)-thiol gold nanoparticle conjugates: Enhanced potency and selective delivery for breast cancer treatment. Bioconjug. Chem. 2009, 20, 2247–2253. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.Y.; Zhang, J.W.; Li, R.F.; Wang, Z.X.; Wang, W.J.; Wang, W. Unique roles of gold nanoparticles in drug delivery, targeting and imaging applications. Molecules 2017, 22, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, J.D.; Khanal, B.P.; Zubarev, E.R. Paclitaxel-functionalized gold nanoparticles. J. Am. Chem. Soc. 2007, 129, 11653–11661. [Google Scholar] [CrossRef]
- Oliveira, J.P.; Prado, A.R.; Keijok, W.J.; Antunes, P.W.P.; Yapuchura, E.R.; Guimarães, M.C.C. Impact of conjugation strategies for targeting of antibodies in gold nanoparticles for ultrasensitive detection of 17β-estradiol. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jazayeri, M.H.; Amani, H.; Pourfatollah, A.A.; Pazoki-Toroudi, H.; Sedighimoghaddam, B. Various methods of gold nanoparticles (GNPs) conjugation to antibodies. Sens. Bio-Sens. Res. 2016, 9, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Meyers, J.D.; Broome, A.M.; Kenney, M.E.; Basilion, J.P.; Burda, C. Deep penetration of a PDT drug into tumors by noncovalent drug-gold nanoparticle conjugates. J. Am. Chem. Soc. 2011, 133, 2583–2591. [Google Scholar] [CrossRef] [Green Version]
- Doane, T.; Burda, C. Nanoparticle mediated non-covalent drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef]
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of Nanoparticles. Small 2008, 4, 26–49. [Google Scholar] [CrossRef]
- Murphy, C.J.; Gole, A.M.; Stone, J.W.; Sisco, P.N.; Alkilany, A.M.; Goldsmith, E.C.; Baxter, S.C. Gold nanoparticles in biology: Beyond toxicity to cellular imaging. Acc. Chem. Res. 2008, 41, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xu, K.; Ji, L.; Tang, B. Effect of gold nanoparticles on glutathione depletion-induced hydrogen peroxide generation and apoptosis in HL7702 cells. Toxicol. Lett. 2011, 205, 86–95. [Google Scholar] [CrossRef]
- Rosli, N.S.B.; Rahman, A.A.; Aziz, A.A.; Shamsuddin, S. Determining the size and concentration dependence of gold nanoparticles in vitro cytotoxicity (IC 50 ) test using WST-1 assay. AIP Conf. Proc. 2015, 1657, 060001. [Google Scholar] [CrossRef]
- Prades, R.; Guerrero, S.; Araya, E.; Molina, C.; Salas, E.; Zurita, E.; Selva, J.; Egea, G.; López-Iglesias, C.; Teixidó, M.; et al. Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials 2012, 33, 7194–7205. [Google Scholar] [CrossRef] [PubMed]
- Sela, H.; Cohen, H.; Elia, P.; Zach, R.; Karpas, Z.; Zeiri, Y. Spontaneous penetration of gold nanoparticles through the blood brain barrier (BBB). J. Nanobiotechnol. 2015, 13, 71. [Google Scholar] [CrossRef] [Green Version]
- Ruff, J.; Hüwel, S.; Kogan, M.J.; Simon, U.; Galla, H.J. The effects of gold nanoparticles functionalized with ß-amyloid specific peptides on an in vitro model of blood–brain barrier. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1645–1652. [Google Scholar] [CrossRef]
- Gonzalez-Carter, D.A.; Ong, Z.Y.; McGilvery, C.M.; Dunlop, I.E.; Dexter, D.T.; Porter, A.E. L-DOPA functionalized, multi-branched gold nanoparticles as brain-targeted nano-vehicles. Nanomed. Nanotechnol. Biol. Med. 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Senyei, A.; Widder, K.; Czerlinski, G. Magnetic guidance of drug-carrying microspheres. J. Appl. Phys. 1978, 49, 3578–3583. [Google Scholar] [CrossRef]
- Qiao, R.; Jia, Q.; Hüwel, S.; Xia, R.; Liu, T.; Gao, F.; Galla, H.J.; Gao, M. Receptor-mediated delivery of magnetic nanoparticles across the blood-brain barrier. ACS Nano 2012, 6, 3304–3310. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Mahmoud, W.E.; Al-Ghamdi, A.A.; Bronstein, L.M. Magnetic Drug Delivery: Where the Field Is Going. Front. Chem. 2018, 6, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBain, S.C.; Yiu, H.H.; Dobson, J. Magnetic nanoparticles for gene and drug delivery. Int. J. Nanomed. 2008, 3, 169. [Google Scholar] [CrossRef] [Green Version]
- D’Agata, F.; Ruffinatti, F.A.; Boschi, S.; Stura, I.; Rainero, I.; Abollino, O.; Cavalli, R.; Guiot, C. Magnetic nanoparticles in the central nervous system: Targeting principles, applications and safety issues. Molecules 2018, 23, 9. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, L.B.; Thomsen, M.S.; Moos, T. Targeted drug delivery to the brain using magnetic nanoparticles. Ther. Deliv. 2015, 6, 1145–1155. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Sagar, V.; Agudelo, M.; Pilakka-Kanthikeel, S.; Subba, V.; Atluri, R.; Raymond, A.; Thangavel, S.; Nair, M.P. Enhanced blood-brain barrier transmigration using a novel Transferrin-embedded fluorescent magnetoliposome nanoformulation HHS Public Access. Nanotechnology 2014, 25, 55101. [Google Scholar] [CrossRef]
- Chertok, B.; Moffat, B.A.; David, A.E.; Yu, F.; Bergemann, C.; Ross, B.D.; Yang, V.C. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials 2008, 29, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Chertok, B.; David, A.E.; Yang, V.C. Polyethyleneimine-modified iron oxide nanoparticles for brain tumor drug delivery using magnetic targeting and intra-carotid administration. Biomaterials 2010, 31, 6317–6324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chertok, B.; David, A.E.; Yang, V.C. Brain tumor targeting of magnetic nanoparticles for potential drug delivery: Effect of administration route and magnetic field topography. J. Control. Release 2011, 155, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Wang, Y.; He, S.; Ku, S.; Gu, W.; Ye, L. Transferrin-conjugated, fluorescein-loaded magnetic nanoparticles for targeted delivery across the blood–brain barrier. J. Mater. Sci. Mater. Med. 2013, 24, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Takemoto, H. Polymeric Micelles. In Encyclopedia of Polymeric Nanomaterials; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–7. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Y.; Li, S. Polymeric micelles: Nanocarriers for cancer-targeted drug delivery. AAPS PharmSciTech 2014, 15, 862–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, S.C.; Chan, D.P.Y.; Shoichet, M.S. Polymeric micelle stability. Nano Today 2012, 7, 53–65. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov. Today 2003, 8, 1112–1120. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. [Google Scholar] [CrossRef]
- Sezgin-bayindir, Z.; Ergin, A.D.; Parmaksiz, M.; Elcin, A.E.; Elcin, Y.M.; Yuksel, N. Evaluation of various block copolymers for micelle formation and brain drug delivery: In vitro characterization and cellular uptake studies. J. Drug Deliv. Sci. Technol. 2016, 36, 120–129. [Google Scholar] [CrossRef]
- Abourehab, M.A.; Ahmed, O.A.; Balata, G.F.; Almalki, W.H. Self-assembled biodegradable polymeric micelles to improve dapoxetine delivery across the blood–brain barrier. Int. J. Nanomed. 2018, 13, 3679–3687. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Venkatraman, S.S.; Yang, Y.Y.; Guo, K.; Lu, J.; He, B.; Moochhala, S.; Kan, L. Polymeric micelles anchored with TAT for delivery of antibiotics across the blood-brain barrier. Biopolymers 2008, 90, 617–623. [Google Scholar] [CrossRef]
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, applications, and properties. Nanoscale Res. Lett. 2014, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Madaan, K.; Kumar, S.; Poonia, N.; Lather, V.; Pandita, D. Dendrimers in drug delivery and targeting: Drug-dendrimer interactions and toxicity issues. J. Pharm. Bioallied Sci. 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, M.H.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Mekuria, S.L.; Debele, T.A.; Tsai, H.C. PAMAM dendrimer based targeted nano-carrier for bio-imaging and therapeutic agents. RSC Adv. 2016, 6, 63761–63772. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Pang, Z. Dendrimer-based drug delivery systems for brain targeting. Biomolecules 2019, 9, 790. [Google Scholar] [CrossRef] [Green Version]
- Florendo, M.; Figacz, A.; Srinageshwar, B.; Sharma, A.; Swanson, D.; Dunbar, G.L.; Rossignol, J. Use of polyamidoamine dendrimers in brain diseases. Molecules 2018, 23, 2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Lv, L.; Shi, H.; Hua, Y.; Lv, W.; Wang, X.; Xin, H.; Xu, Q. PEGylated Polyamidoamine dendrimer conjugated with tumor homing peptide as a potential targeted delivery system for glioma. Colloids Surf. B Biointerfaces 2016, 147, 242–249. [Google Scholar] [CrossRef]
- Santos, S.D.; Xavier, M.; Leite, D.M.; Moreira, D.A.; Custódio, B.; Torrado, M.; Castro, R.; Leiro, V.; Rodrigues, J.; Tomás, H.; et al. PAMAM dendrimers: Blood-brain barrier transport and neuronal uptake after focal brain ischemia. J. Control. Release 2018, 291, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Ryman, B.E. Liposomes as carriers of enzymes or drugs: A new approach to the treatment of storage diseases. Biochem. J. 1971, 124, 58. [Google Scholar] [CrossRef] [Green Version]
- Weissig, V. Liposomes came first: The early history of liposomology. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1522. [Google Scholar] [CrossRef]
- Taira, M.C.; Chiaramoni, N.S.; Pecuch, K.M.; Alonso-Romanowski, S. Stability of Liposomal Formulations in Physiological Conditions for Oral Drug Delivery. Drug Deliv. 2004, 11, 123–128. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Szoka, F.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [Green Version]
- Zumbuehl, O.; Weder, H.G. Liposomes of controllable size in the range of 40 to 180 nm by defined dialysis of lipid/detergent mixed micelles. Biochim. Biophys. Acta-(BBA)-Biomembr. 1981, 640, 252–262. [Google Scholar] [CrossRef]
- Deamer, D.W. Preparation and Properties of Ether-injection Liposomes. Ann. N. Y. Acad. Sci. 1978, 308, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Li, X.; Zhang, T.; Song, Y.; She, Z.; Li, J.; Deng, Y. Progress involving new techniques for liposome preparation. Asian J. Pharm. Sci. 2014, 9, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Gurturk, Z.; Tezcaner, A.; Dalgic, A.D.; Korkmaz, S.; Keskin, D. Maltodextrin modified liposomes for drug delivery through the blood–brain barrier. MedChemComm 2017, 8, 1337–1345. [Google Scholar] [CrossRef]
- Iachetta, G.; Falanga, A.; Molino, Y.; Masse, M.; Jabès, F.; Mechioukhi, Y.; Laforgia, V.; Khrestchatisky, M.; Galdiero, S.; Valiante, S. gH625-liposomes as tool for pituitary adenylate cyclase-activating polypeptide brain delivery. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Valiante, S.; Falanga, A.; Cigliano, L.; Iachetta, G.; Busiello, R.A.; Marca, V.L.; Galdiero, M.; Lombardi, A.; Galdiero, S. Peptide gH625 enters into neuron and astrocyte cell lines and crosses the blood–brain barrier in rats. Int. J. Nanomed. 2015, 10, 1885. [Google Scholar] [CrossRef] [Green Version]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood–brain barrier. Int. J. Nanomed. 2016, 11, 5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani, P.; Penson, P.E.; Barreto, G.E.; Sahebkar, A. Recent Advancements in Liposome-Based Strategies for Effective Drug Delivery to the Brain. Curr. Med. Chem. 2020, 28, 4152–4171. [Google Scholar] [CrossRef]
- Karim, K.; Mandal, A.; Biswas, N.; Guha, A.; Chatterjee, S.; Behera, M.; Kuotsu, K. Niosome: A future of targeted drug delivery systems. J. Adv. Pharm. Technol. Res. 2010, 1, 374. [Google Scholar] [CrossRef]
- Uchegbu, I.F.; Vyas, S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 1998, 172, 33–70. [Google Scholar] [CrossRef]
- Baillie, A.J.; Coombs, G.H.; Dolan, T.F.; Laurie, J. Non-ionic surfactant vesicles, niosomes, as a delivery system for the anti-leishmanial drug, sodium stibogluconate. J. Pharm. Pharmacol. 1986, 38, 502–505. [Google Scholar] [CrossRef]
- Ge, X.; Wei, M.; He, S.; Yuan, W.E. Advances of non-ionic surfactant vesicles (niosomes) and their application in drug delivery. Pharmaceutics 2019, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, A.; Venkatesh, N.; Senthil, M.; Sanapalli, B.K.R.; Shanmugham, R.; Karri, V.V.S.R. Smart niosomes of temozolomide for enhancement of brain targeting. Nanobiomedicine 2018, 5, 1–11. [Google Scholar] [CrossRef]
- Danielsson, I.; Lindman, B. The definition of microemulsion. Colloids Surf. 1981, 3, 391–392. [Google Scholar] [CrossRef]
- Kale, S.N.; Deore, S.L. Emulsion micro emulsion and nano emulsion: A review. Syst. Rev. Pharm. 2016, 8, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Callender, S.P.; Mathews, J.A.; Kobernyk, K.; Wettig, S.D. Microemulsion utility in pharmaceuticals: Implications for multi-drug delivery. Int. J. Pharm. 2017, 526, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Patel, M.R.; Bhatt, K.K.; Patel, B.G.; Gaikwad, R.V. Microemulsion-based drug delivery system for transnasal delivery of Carbamazepine: Preliminary brain-targeting study. Drug Deliv. 2014, 23, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göppert, T.M.; Müller, R.H. Plasma Protein Adsorption of Tween 80- and Poloxamer 188-stabilized Solid Lipid Nanoparticles. J. Drug Target. 2003, 11, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Göppert, T.M.; Müller, R.H. Protein adsorption patterns on poloxamer- and poloxamine-stabilized solid lipid nanoparticles (SLN). Eur. J. Pharm. Biopharm. 2005, 60, 361–372. [Google Scholar] [CrossRef]
- Ritz, S.; Schöttler, S.; Kotman, N.; Baier, G.; Musyanovych, A.; Kuharev, J.; Landfester, K.; Schild, H.; Jahn, O.; Tenzer, S.; et al. Protein Corona of Nanoparticles: Distinct Proteins Regulate the Cellular Uptake. Biomacromolecules 2015, 16, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, R.; Gatto, F.; Bardi, G. Protein adsorption: A feasible method for nanoparticle functionalization? Materials 2019, 12, 1991. [Google Scholar] [CrossRef] [Green Version]
- Decher, G. Fuzzy nanoassemblies: Toward layered polymeric multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Ariga, K.; McShane, M.; Lvov, Y.M.; Ji, Q.; Hill, J.P. Layer-by-layer assembly for drug delivery and related applications. Expert Opin. Drug Deliv. 2011, 8, 633–644. [Google Scholar] [CrossRef]
- Matsusaki, M.; Akashi, M. Functional multilayered capsules for targeting and local drug delivery. Expert Opin. Drug Deliv. 2009, 6, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Garg, G.; Sharma, P. Nanospheres: A Novel Approach for Targeted Drug Delivery System. Int. J. Pharm. Sci. Rev. Res. 2010, 5, 84–88. [Google Scholar]
- Ibrahim, N.; Krishnakumar, K.; Dineshkumar, B.; Nair, S.K. An Overview on Nanosphere Drug Delivery. Eur. J. Pharm. Med. Res. 2018, 5, 192–198. [Google Scholar]
- Kothamasu, P.; Kanumur, H.; Ravur, N.; Maddu, C.; Parasuramrajam, R.; Thangavel, S. Nanocapsules: The weapons for novel drug delivery systems. BioImpacts BI 2012, 2, 71–81. [Google Scholar] [CrossRef]
- Teixeira, M.; Alonso, M.J.; Pinto, M.M.M.; Barbosa, C.M. Development and characterization of PLGA nanospheres and nanocapsules containing xanthone and 3-methoxyxanthone. Eur. J. Pharm. Biopharm. 2005, 59, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yeo, Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepmann, J.; Siepmann, F. Modeling of diffusion controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J.; Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Barbara, R.; Belletti, D.; Pederzoli, F.; Masoni, M.; Keller, J.; Ballestrazzi, A.; Vandelli, M.A.; Tosi, G.; Grabrucker, A.M. Novel Curcumin loaded nanoparticles engineered for Blood-Brain Barrier crossing and able to disrupt Abeta aggregates. Int. J. Pharm. 2017, 526, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.H.; Wang, Z.Y.; Sun, C.S.; Wang, C.Y.; Jiang, T.Y.; Wang, S.L. Trimethylated chitosan-conjugated PLGA nanoparticles for the delivery of drugs to the brain. Biomaterials 2010, 31, 908–915. [Google Scholar] [CrossRef]
- Green, A.E.; Rose, P.G. Pegylated liposomal doxorubicin in ovarian cancer. Int. J. Nanomed. 2006, 1, 229. [Google Scholar]
- Gabizon, A.A. Pegylated Liposomal Doxorubicin: Metamorphosis of an Old Drug into a New Form of Chemotherapy. Cancer Investig. 2001, 19, 424–436. [Google Scholar] [CrossRef]
- Hu, K.; Shi, Y.; Jiang, W.; Han, J.; Huang, S.; Jiang, X. Lactoferrin conjugated PEG-PLGA nanoparticles for brain delivery: Preparation, characterization and efficacy in Parkinson’s disease. Int. J. Pharm. 2011, 415, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Hsieh, W.Y.; Lee, W.F.; Zeng, D.T. Effects of surface modification of PLGA-PEG-PLGA nanoparticles on loperamide delivery efficiency across the blood-brain barrier. J. Biomater. Appl. 2011, 27, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Thermoreversible Gelation of PEG−PLGA−PEG Triblock Copolymer Aqueous Solutions. Macromolecules 1999, 32, 7064–7069. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayub, A.; Wettig, S. An Overview of Nanotechnologies for Drug Delivery to the Brain. Pharmaceutics 2022, 14, 224. https://doi.org/10.3390/pharmaceutics14020224
Ayub A, Wettig S. An Overview of Nanotechnologies for Drug Delivery to the Brain. Pharmaceutics. 2022; 14(2):224. https://doi.org/10.3390/pharmaceutics14020224
Chicago/Turabian StyleAyub, Ahsan, and Shawn Wettig. 2022. "An Overview of Nanotechnologies for Drug Delivery to the Brain" Pharmaceutics 14, no. 2: 224. https://doi.org/10.3390/pharmaceutics14020224