
Altered Blood–Brain Barrier Dynamics in the C9orf72 Hexanucleotide Repeat Expansion Mouse Model of Amyotrophic Lateral Sclerosis
 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. In Situ Transcardiac Brain Perfusion to Assess BBB Transport
2.4. Brain Microvessel Enrichment for Glut1 Quantification
2.5. BMEC Isolation for Proteomic Analysis
2.6. Proteomic Analysis of BMECs
2.7. Statistical Analysis of Data
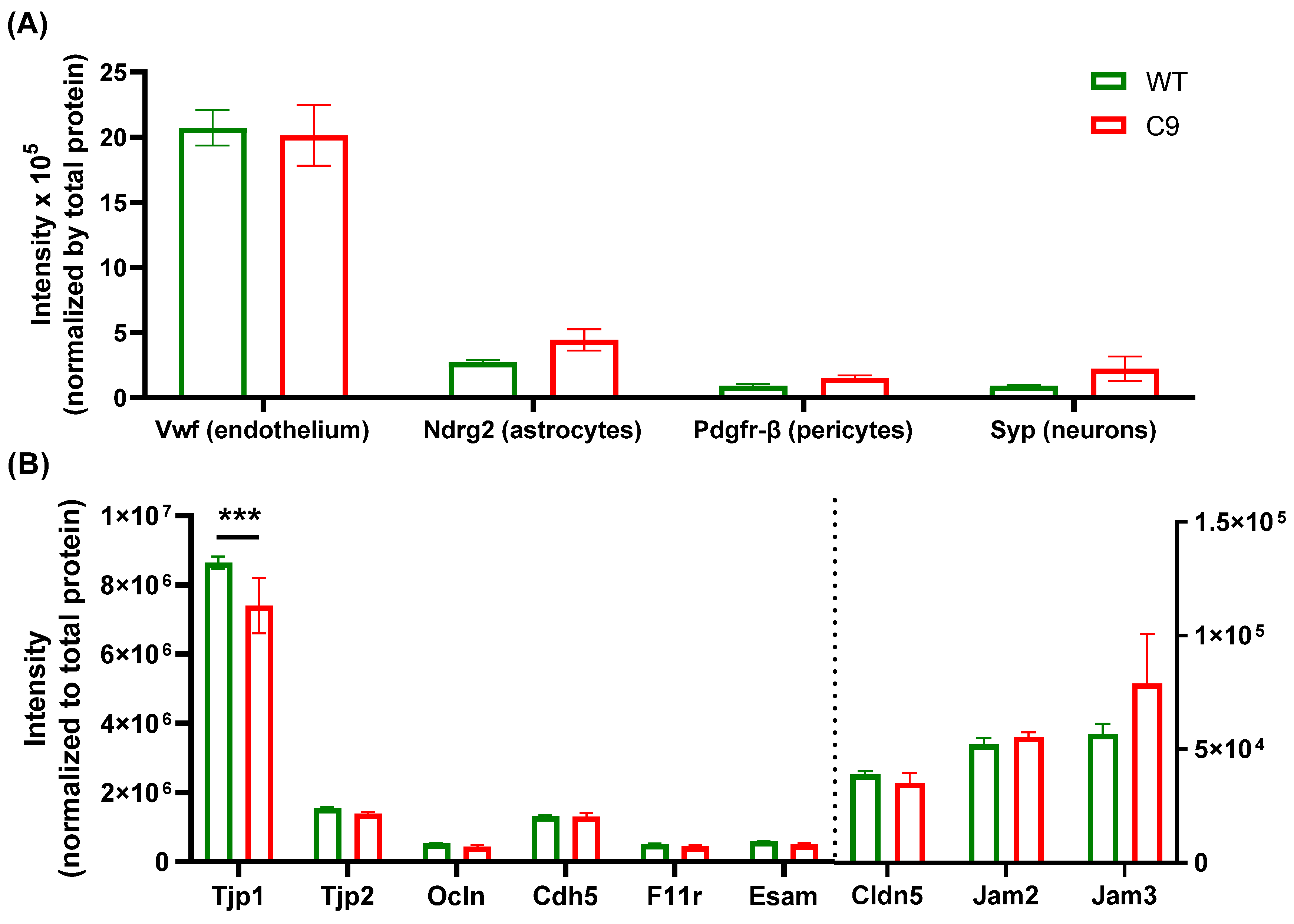
3. Results
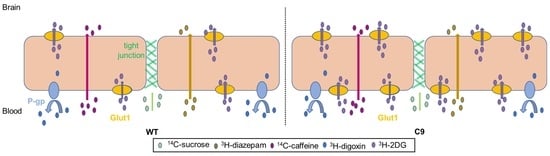
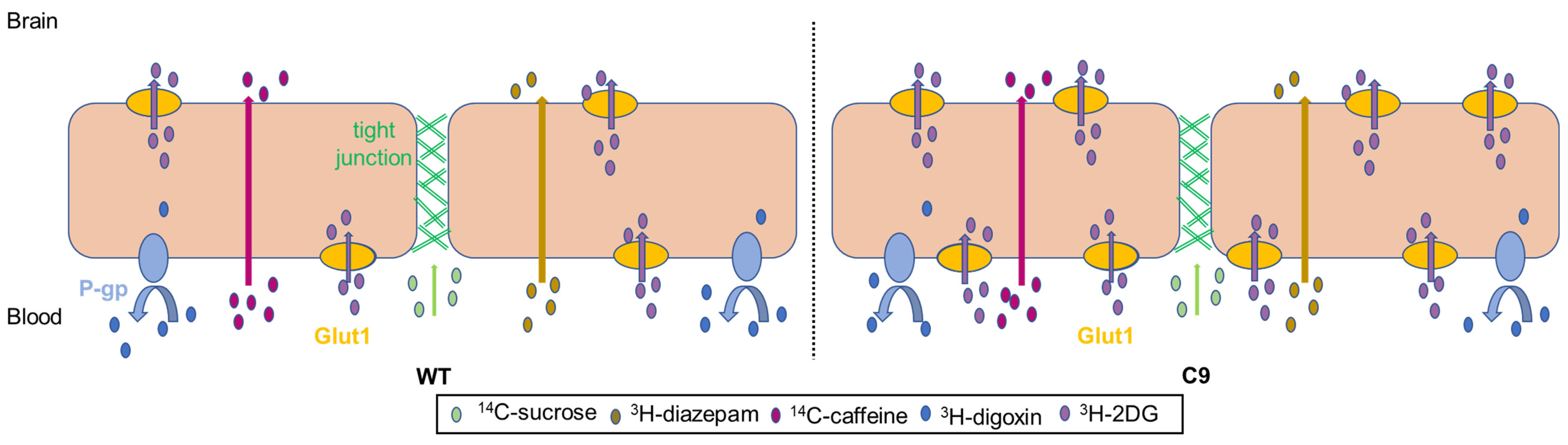
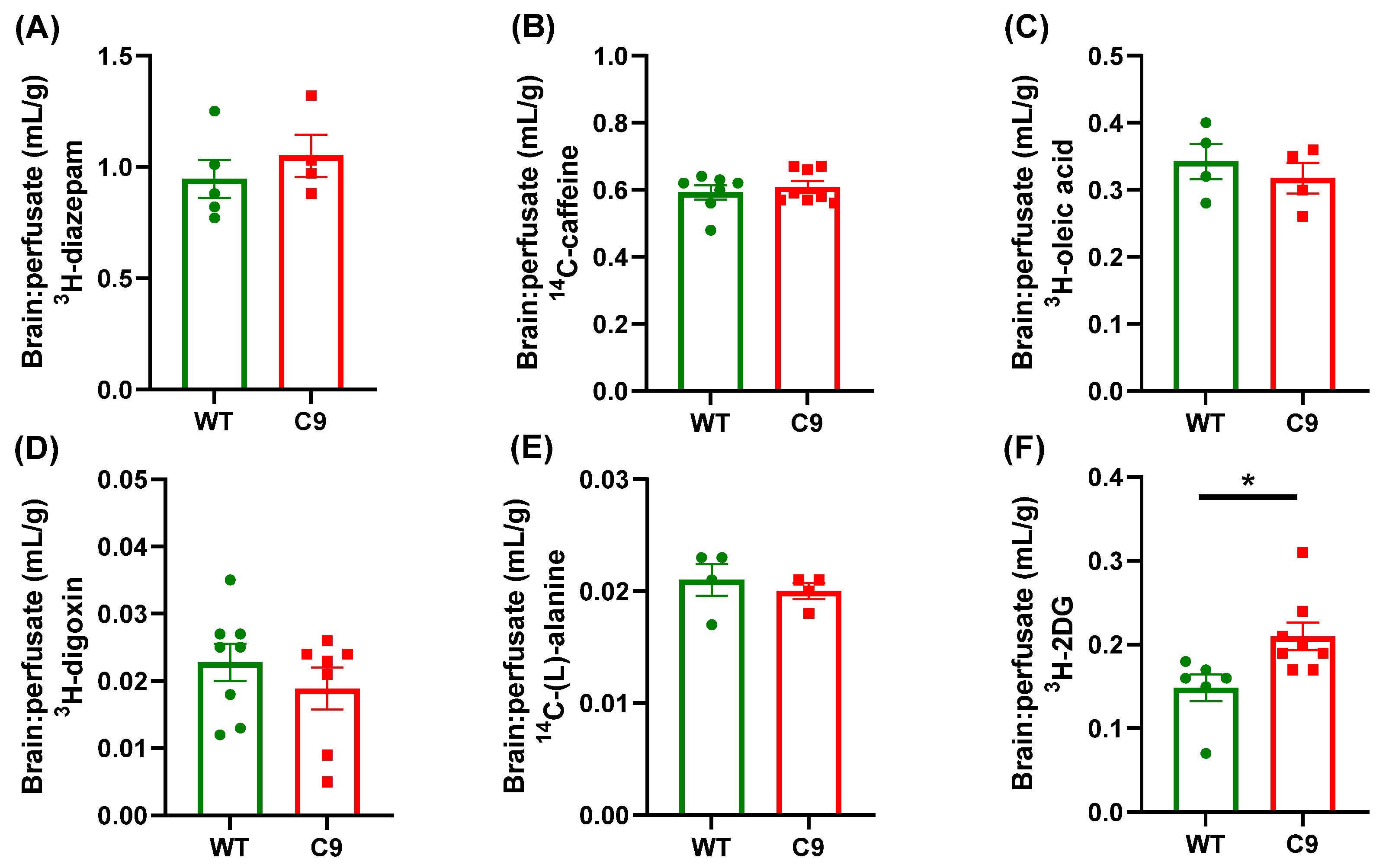
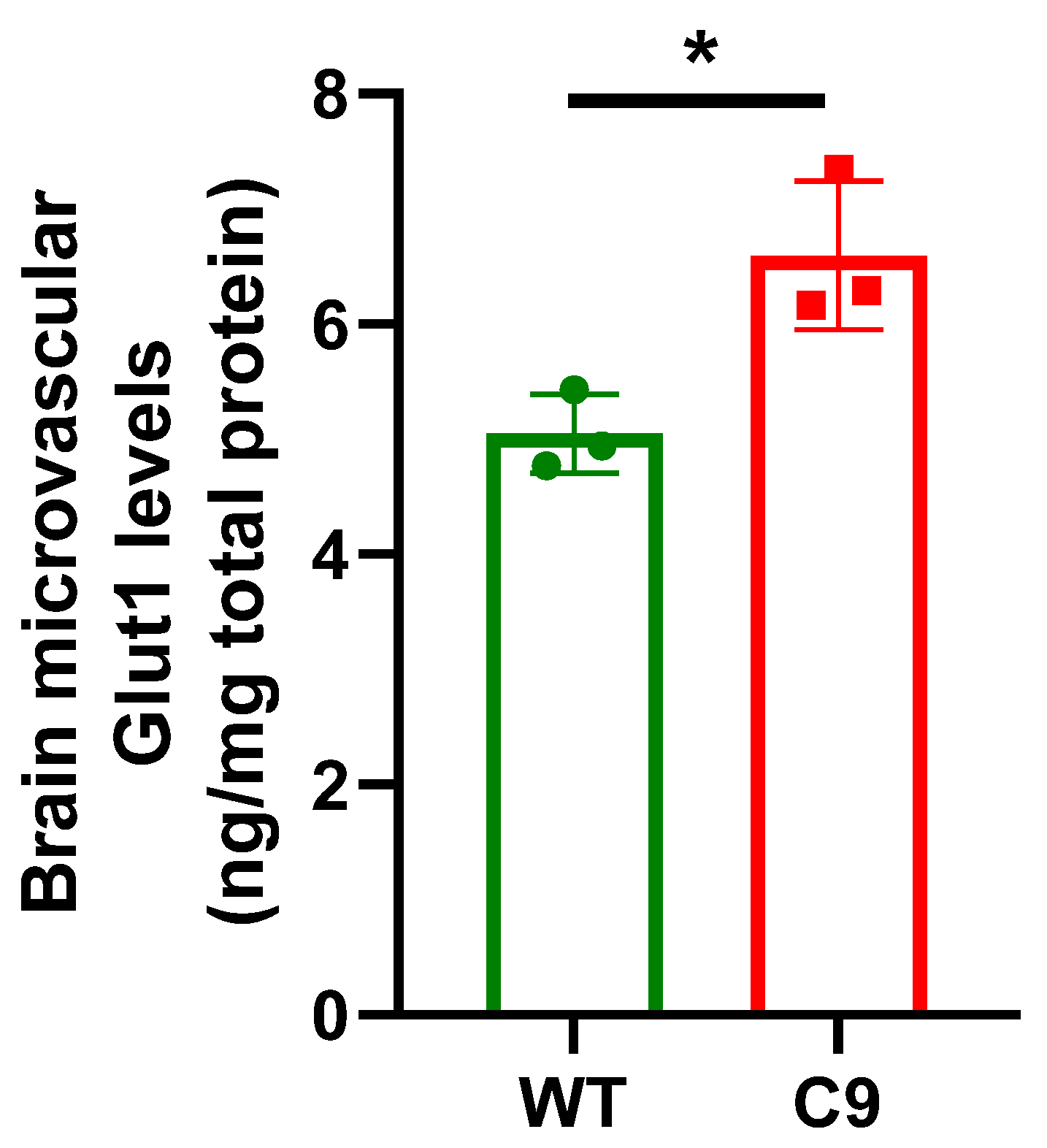
3.1. BBB Transport of 3H-2DG Was Increased in C9 Mice Relative to WT Mice, in Association with Increased Brain Microvascular Glut1 Abundance
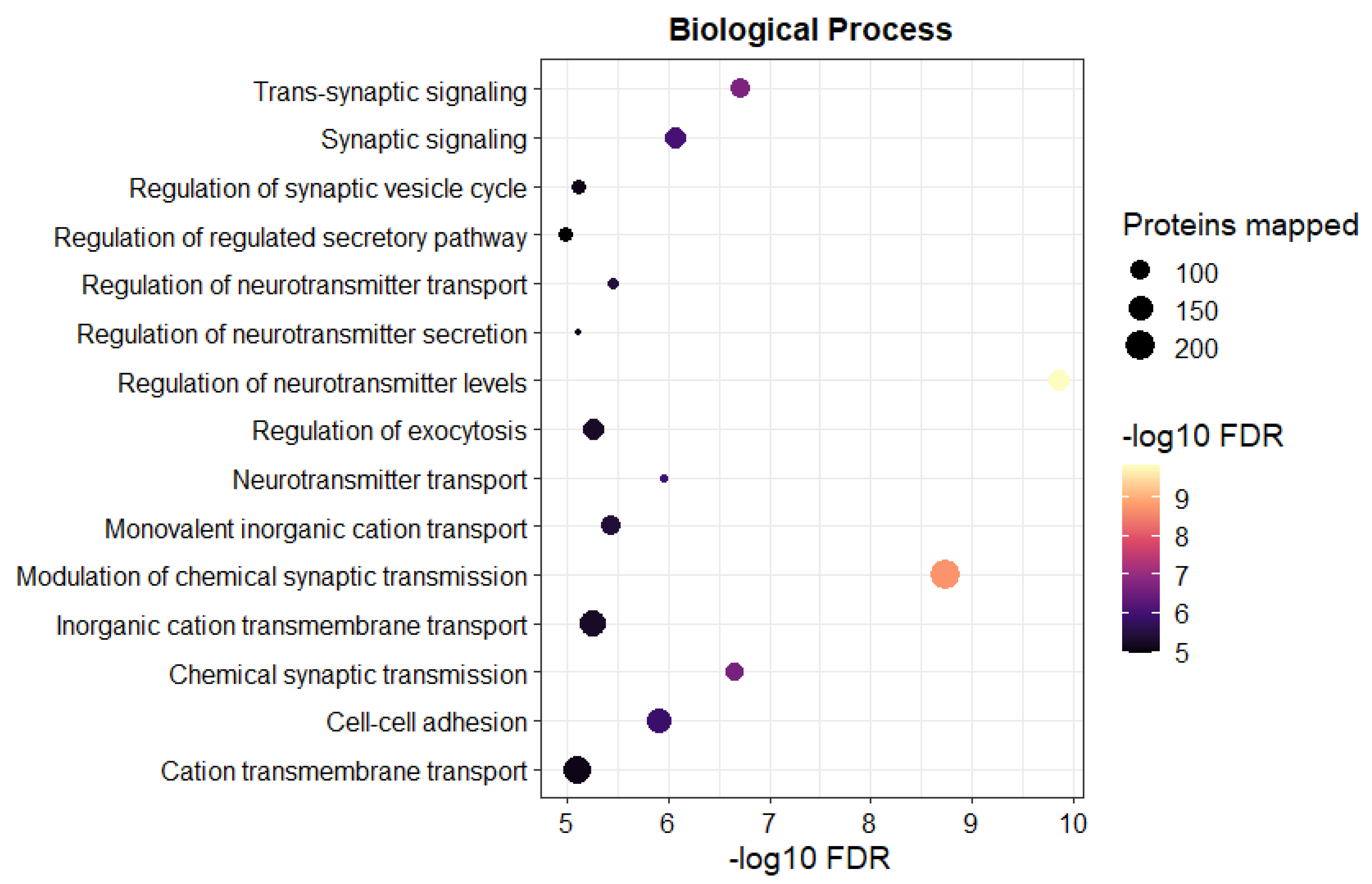
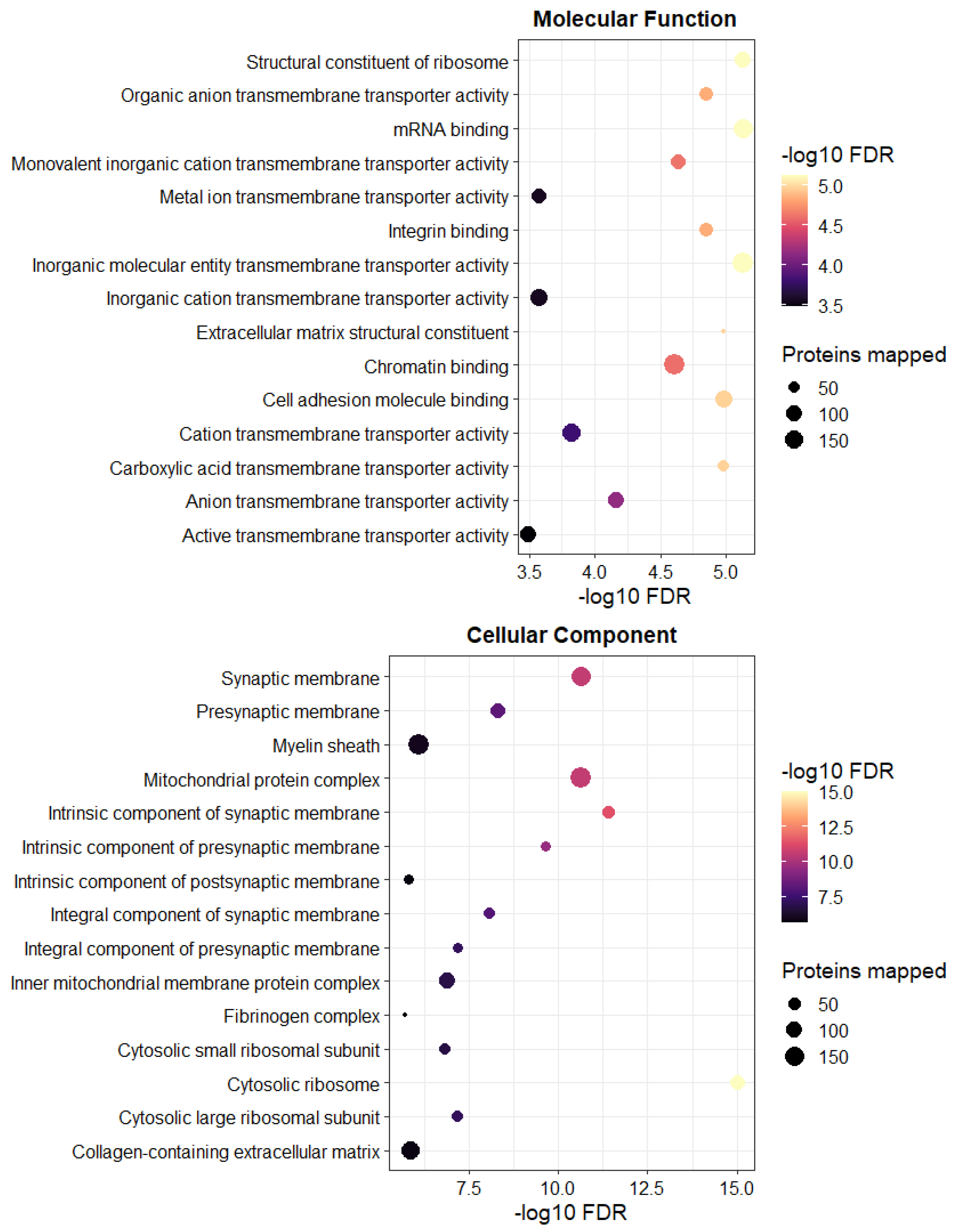
3.2. The Tandem Mass Tag (TMT)-Proteomics Studies Revealed an Altered Proteome Profile of C9 BMECs Compared to WT BMECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2013, 17, 17–23. [Google Scholar] [CrossRef]
- Byrne, S.; Elamin, M.; Bede, P.; Shatunov, A.; Walsh, C.; Corr, B.; Heverin, M.; Jordan, N.; Kenna, K.; Lynch, C.; et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: A population-based cohort study. Lancet Neurol. 2012, 11, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Pan, Y.; Nicolazzo, J.A. Altered blood–brain barrier and blood–spinal cord barrier dynamics in amyotrophic lateral sclerosis: Impact on medication efficacy and safety. J. Cereb. Blood Flow Metab. 2022, 179, 2577–2588. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef] [Green Version]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.; Sanberg, P.R. Impaired blood–brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Jablonski, M.R.; Jacob, D.A.; Campos, C.; Miller, D.S.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Selective increase of two ABC drug efflux transporters at the blood–spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol. Dis. 2012, 47, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Milane, A.; Fernandez, C.; Dupuis, L.; Buyse, M.; Loeffler, J.-P.; Farinotti, R.; Meininger, V.; Bensimon, G. P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 472, 166–170. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Saánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Kumar, V.; Hasan, G.M.; Hassan, I. Unraveling the role of RNA mediated toxicity of C9orf72 repeats in C9-FTD/ALS. Front. Neurosci. 2017, 11, 711. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, C. TDP-43—The key to understanding amyotrophic lateral sclerosis. Rare Dis. 2014, 2, e944443. [Google Scholar] [CrossRef] [Green Version]
- Ormeño, F.; Hormazabal, J.; Moreno, J.; Riquelme, F.; Rios, J.; Criollo, A.; Albornoz, A.; Alfaro, I.E.; Budini, M. Chaperone mediated autophagy degrades TDP-43 protein and is affected by TDP-43 aggregation. Front. Mol. Neurosci. 2020, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in amyotrophic lateral sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef]
- Perry, T.L.; Krieger, C.; Ba, S.H.; Eisen, A. Amyotrophic lateral sclerosis: Amino acid levels in plasma and cerebrospinal fluid. Ann. Neurol. 1990, 28, 12–17. [Google Scholar] [CrossRef]
- Pan, Y.; Scanlon, M.J.; Owada, Y.; Yamamoto, Y.; Porter, C.J.H.; Nicolazzo, J.A. Fatty acid-binding protein 5 facilitates the blood–brain barrier transport of docosahexaenoic acid. Mol. Pharm. 2015, 12, 4375–4385. [Google Scholar] [CrossRef]
- Pan, Y.; Choy, K.H.C.; Marriott, P.J.; Chai, S.Y.; Scanlon, M.J.; Porter, C.J.H.; Short, J.L.; Nicolazzo, J.A. Reduced blood-brain barrier expression of fatty acid-binding protein 5 is associated with increased vulnerability of APP/PS1 mice to cognitive deficits from low omega-3 fatty acid diets. J. Neurochem. 2017, 144, 81–92. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 19 January 2022).
- Al-Majdoub, Z.M.; Al Feteisi, H.; Achour, B.; Warwood, S.; Neuhoff, S.; Rostami-Hodjegan, A.; Barber, J. Proteomic quantification of human blood–brain barrier SLC and ABC transporters in healthy individuals and dementia patients. Mol. Pharm. 2019, 16, 1220–1233. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Smith, Q.R.; Allen, D.D. The Blood-Brain Barrier: Biology and Research Protocols; Sukriti, N., Ed.; Humana Press: Totowa, NJ, USA, 2003; pp. 209–218. [Google Scholar]
- Cattelotte, J.; André, P.; Ouellet, M.; Bourasset, F.; Scherrmann, J.-M.; Cisternino, S. In situ mouse carotid perfusion model: Glucose and cholesterol transport in the eye and brain. J. Cereb. Blood Flow Metab. 2008, 28, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Mizojiri, K. Drug-protein binding and blood-brain barrier permeability. J. Pharmacol. Exp. Ther. 1999, 288, 912–918. [Google Scholar]
- Low, Y.L.; Jin, L.; Morris, E.R.; Pan, Y.; Nicolazzo, J.A. Pioglitazone increases blood-brain barrier expression of fatty acid-binding protein 5 and docosahexaenoic acid trafficking into the brain. Mol. Pharm. 2020, 17, 873–884. [Google Scholar] [CrossRef]
- Jin, L.; Pan, Y.; Tran, N.L.L.; Polychronopoulos, L.N.; Warrier, A.; Brouwer, K.L.R.; Nicolazzo, J.A. Intestinal permeability and oral absorption of selected drugs are reduced in a mouse model of familial Alzheimer’s disease. Mol. Pharm. 2020, 17, 1527–1537. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Ayala, V.; Jové, M.; Dalfó, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Metabolic dysfunctions in amyotrophic lateral sclerosis pathogenesis and potential metabolic treatments. Front. Neurosci. 2017, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- Devraj, K.; Klinger, M.E.; Myers, R.L.; Mokashi, A.; Hawkins, R.A.; Simpson, I.A. GLUT-1 glucose transporters in the blood-brain barrier: Differential phosphorylation. J. Neurosci. Res. 2011, 89, 1913–1925. [Google Scholar] [CrossRef] [Green Version]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [Green Version]
- Patching, S.G. Glucose Transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2016, 54, 1046–1077. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Z.; Zou, W.; Guo, H.; Liu, M.; Ma, Y.; Zhang, L. The appropriate marker for astrocytes: Comparing the distribution and expression of three astrocytic markers in different mouse cerebral regions. BioMed Res. Int. 2019, 2019, 9605265. [Google Scholar] [CrossRef]
- Smyth, L.C.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.; Park, T.I.; Dragunow, M. Markers for human brain pericytes and smooth muscle cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef]
- Perry, A.; Brat, D.J. Practical Surgical Neuropathology: A Diagnostic Approach, 2nd ed.; Perry, A., Brat, D.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–17. [Google Scholar]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.-K.; Philips, M.-A.; et al. Cell type– and brain region–resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1831. [Google Scholar] [CrossRef]
- Pan, Y.; Nicolazzo, J.A. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv. Drug Deliv. Rev. 2018, 135, 62–74. [Google Scholar] [CrossRef]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Kelleher, N.L. Precision proteomics: The case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. USA 2008, 105, 18132–18138. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, L.T.; Trotti, D. EAAT2 and the molecular signature of amyotrophic lateral sclerosis. Adv. Neurobiol. 2017, 16, 117–136. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Martínez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na+-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef] [Green Version]
- Gliddon, C.M.; Shao, Z.; LeMaistre, J.L.; Anderson, C.M. Cellular distribution of the neutral amino acid transporter subtype ASCT2 in mouse brain. J. Neurochem. 2009, 108, 372–383. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The human SLC1A5 (ASCT2) amino acid transporter: From function to structure and role in cell biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.W.; Edmundson, C.L.; Miller, D.W.; Hatch, G.M. On the mechanism of oleate transport across human brain microvessel endothelial cells. J. Neurochem. 2009, 110, 1049–1057. [Google Scholar] [CrossRef]
- Lee, G.S.; Pan, Y.; Scanlon, M.J.; Porter, C.J.; Nicolazzo, J.A. Fatty acid–binding protein 5 mediates the uptake of fatty acids, but not drugs, into human brain endothelial cells. J. Pharm. Sci. 2018, 107, 1185–1193. [Google Scholar] [CrossRef]
- E Umoh, M.; Dammer, E.; Dai, J.; Duong, D.; Lah, J.J.; I Levey, A.; Gearing, M.; Glass, J.D.; Seyfried, N.T. A proteomic network approach across the ALS - FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 2017, 10, 48–62. [Google Scholar] [CrossRef]
- González-Romero, R.; Eirín-López, J.M.; Ausió, J. Evolution of high mobility group nucleosome-binding proteins and its implications for vertebrate chromatin specialization. Mol. Biol. Evol. 2014, 32, 121–131. [Google Scholar] [CrossRef]
- Ueda, T.; Furusawa, T.; Kurahashi, T.; Tessarollo, L.; Bustin, M. The nucleosome binding protein HMGN3 modulates the transcription profile of pancreatic β cells and affects insulin secretion. Mol. Cell. Biol. 2009, 29, 5264–5276. [Google Scholar] [CrossRef] [Green Version]
- Shelkovnikova, T.; Robinson, H.K.; Troakes, C.; Ninkina, N.; Buchman, V.L. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 2013, 23, 2298–2312. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Name | WT/C9 Fold-Change | p Value |
|---|---|---|---|
| ABC transporters | |||
| Abca2 | Abc2 | 1.18 | 0.12 |
| Abca9 | Abc9 | 0.66 * | 0.03 |
| Abcb1a | P-gp | 1.23 | 0.14 |
| Abcb4 | Mdr3 | 1.09 | 0.20 |
| Abcb11 | Bsep | 1.35 * | 0.01 |
| Abcc1 | Mrp1 | 1.14 | 0.31 |
| Abcc9 | Mrp9 | 0.78 | 0.12 |
| Abcg2 | Bcrp | 1.21 | 0.07 |
| SLC transporters | |||
| Slc1a2 | Eaat2 | 0.42 * | 0.05 |
| Slc1a5 | Asct2 | 0.77 * | 0.04 |
| Slc6a6 | Taut | 1.21 * | 0.05 |
| Slc2a1 | Glut1 | 1.22 | 0.23 |
| Slc7a5 | Lat1 | 1.32 | 0.08 |
| Slc16a1 | Mct1 | 1.32 | 0.13 |
| Slc22a8 | Oat3 | 1.15 | 0.33 |
| Slc27a1 | Fatp1 | 0.70 * | 0.03 |
| Slc29a1 | Ent1 | 0.90 | 0.25 |
| Slc38a2 | Snat2 | 0.94 | 0.70 |
| Slco1a1 | Oatp1a1 | 3.51 | 0.17 |
| Slco1a4 | Oatp1a4 | 1.26 | 0.10 |
| Slco1a6 | Oatp1a6 | 1.33 | 0.08 |
| Slco1c1 | Oatp1c1 | 1.20 | 0.14 |
| Slco2b1 | Oatp2b1 | 0.94 | 0.59 |
| Gene | Protein Name | WT/C9 Fold-Change | Adjusted p Value |
|---|---|---|---|
| Atp5mc1 | ATP synthase F(0) complex subunit C1 | 1.63 | 0.0243 |
| Bcap29 | B-cell receptor-associated protein 29 | 1.51 | 0.0243 |
| Cdc42bpa | CDC42 binding protein kinase alpha | 1.27 | 0.0446 |
| Gulp1 | PTB domain-containing engulfment adapter protein 1 | 1.56 | 0.0258 |
| H2ac15 | histone H2A type 1-K | 1.61 | 0.0243 |
| Hmgn3 | high mobility group nucleosome-binding domain-containing protein 3 | 1.95 | 0.0073 |
| Isg15 | ubiquitin-like protein ISG15 | 1.42 | 0.0243 |
| Nckap1l | nck-associated protein 1-like | 0.64 | 0.0292 |
| Nono | non-POU domain-containing octamer-binding protein | 1.41 | 0.0243 |
| Rgs10 | regulator of G-protein signaling 10 | 0.44 | 0.0243 |
| Smco4 | single-pass membrane and coiled-coil domain-containing protein 4 | 1.79 | 0.0243 |
| Tmpo.1 | lamina-associated polypeptide 2 | 1.55 | 0.0355 |
| UPF0729 | UPF0729 protein C18orf32 homolog | 1.70 | 0.0355 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Kagawa, Y.; Sun, J.; Turner, B.J.; Huang, C.; Shah, A.D.; Schittenhelm, R.B.; Nicolazzo, J.A. Altered Blood–Brain Barrier Dynamics in the C9orf72 Hexanucleotide Repeat Expansion Mouse Model of Amyotrophic Lateral Sclerosis. Pharmaceutics 2022, 14, 2803. https://doi.org/10.3390/pharmaceutics14122803
Pan Y, Kagawa Y, Sun J, Turner BJ, Huang C, Shah AD, Schittenhelm RB, Nicolazzo JA. Altered Blood–Brain Barrier Dynamics in the C9orf72 Hexanucleotide Repeat Expansion Mouse Model of Amyotrophic Lateral Sclerosis. Pharmaceutics. 2022; 14(12):2803. https://doi.org/10.3390/pharmaceutics14122803
Chicago/Turabian StylePan, Yijun, Yoshiteru Kagawa, Jiaqi Sun, Bradley J. Turner, Cheng Huang, Anup D. Shah, Ralf B. Schittenhelm, and Joseph A. Nicolazzo. 2022. "Altered Blood–Brain Barrier Dynamics in the C9orf72 Hexanucleotide Repeat Expansion Mouse Model of Amyotrophic Lateral Sclerosis" Pharmaceutics 14, no. 12: 2803. https://doi.org/10.3390/pharmaceutics14122803