
Alpinetin Suppresses Zika Virus-Induced Interleukin-1β Production and Secretion in Human Macrophages
 , ,
, ,  , , and
, , and 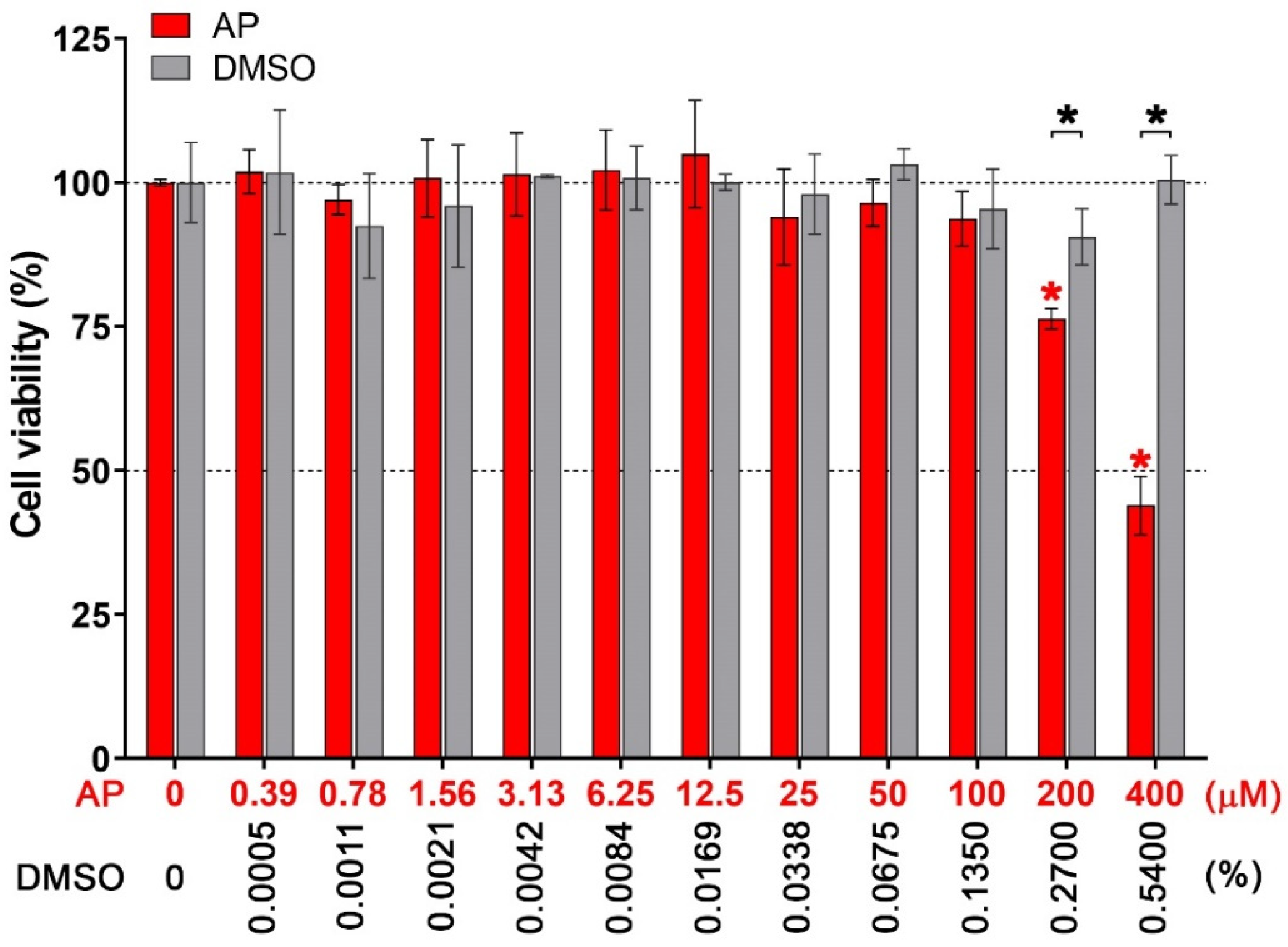{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. Stimulation of THP-1 Cell Differentiation
2.3. ZIKV Infection and AP Treatment
2.4. MTT Assay
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Western Blot Analysis
2.7. Immunofluorescence Study
2.8. Plaque Assay
2.9. Statistical Analysis
3. Results
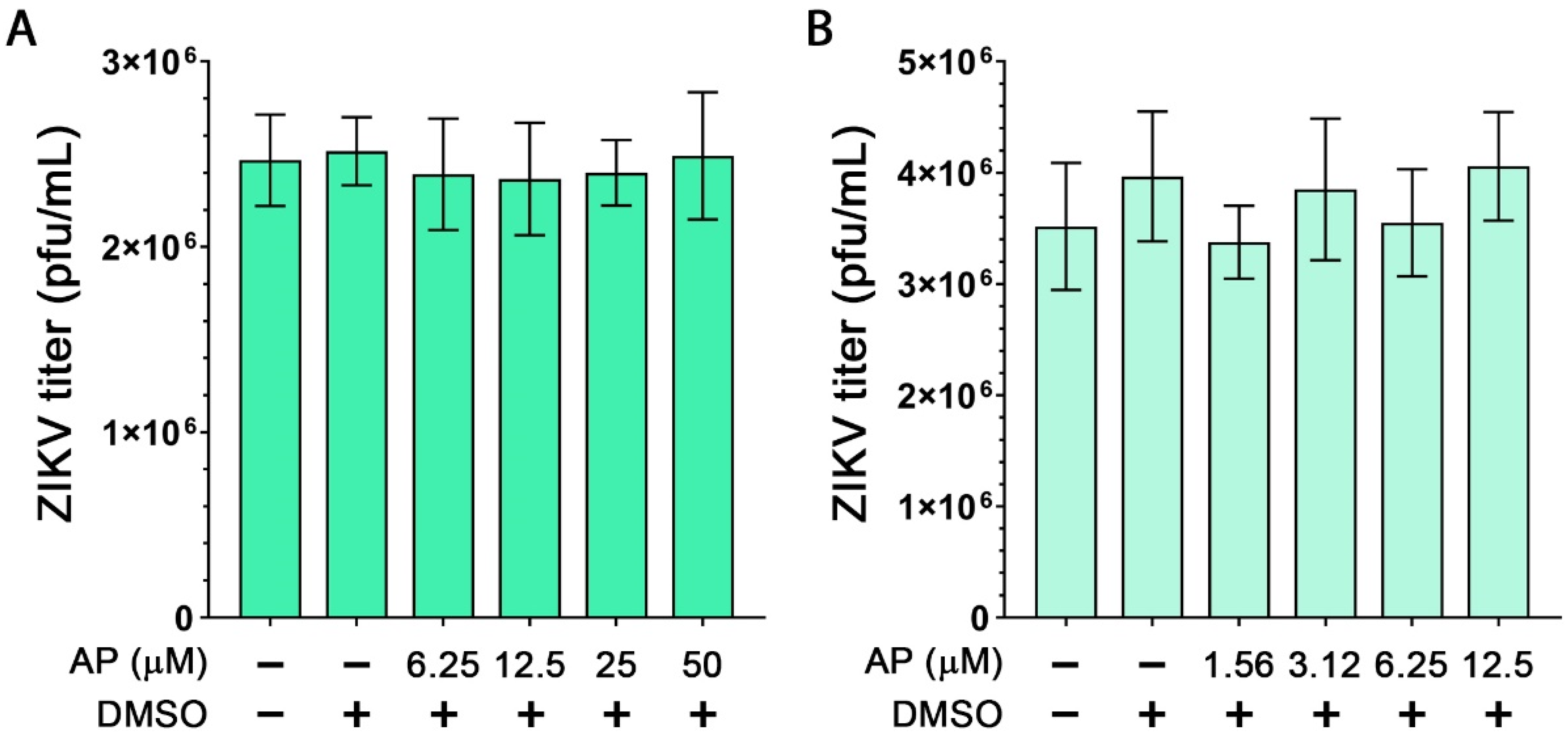
3.1. Effects of AP on the Viability of Human Macrophages
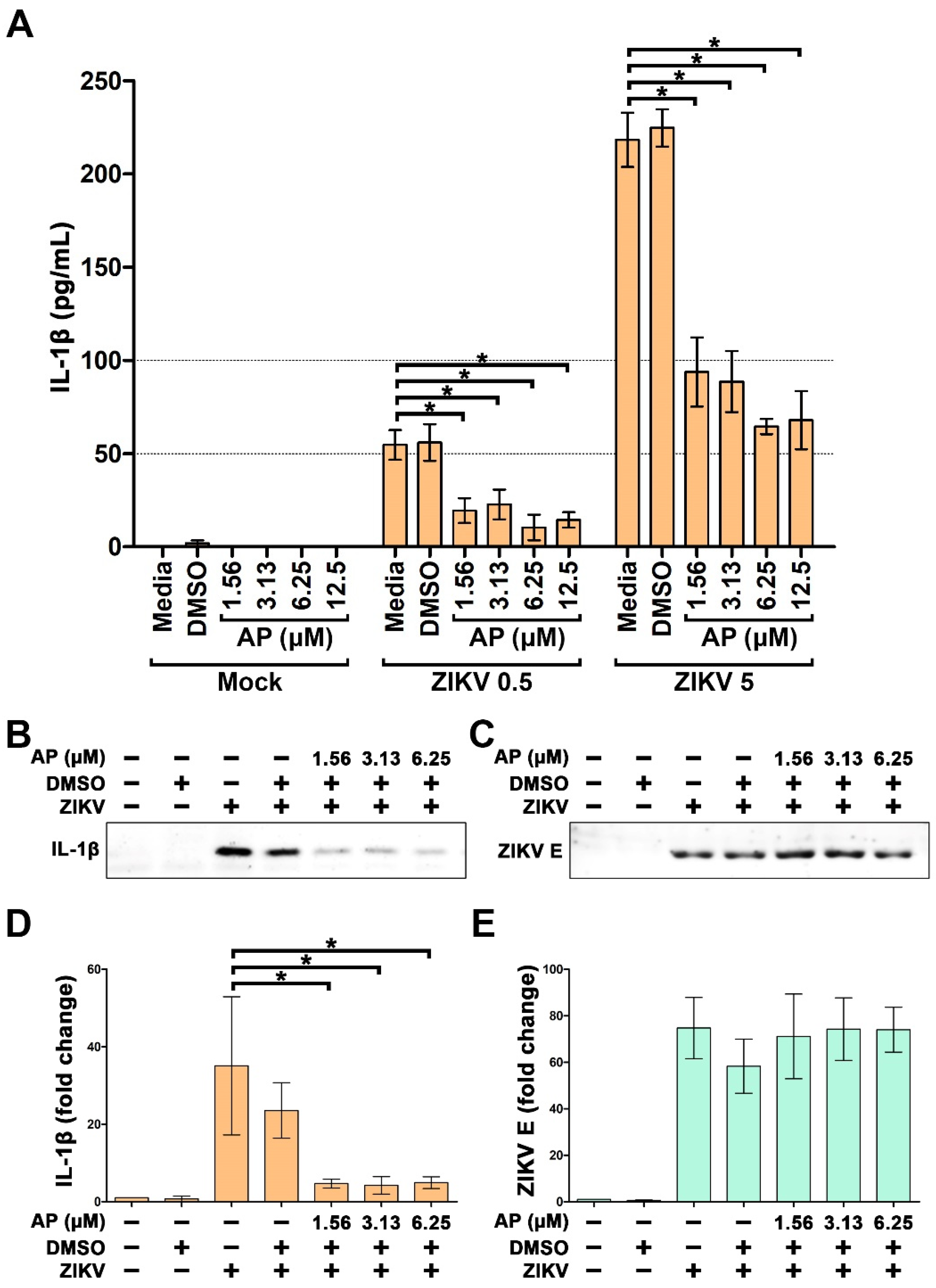
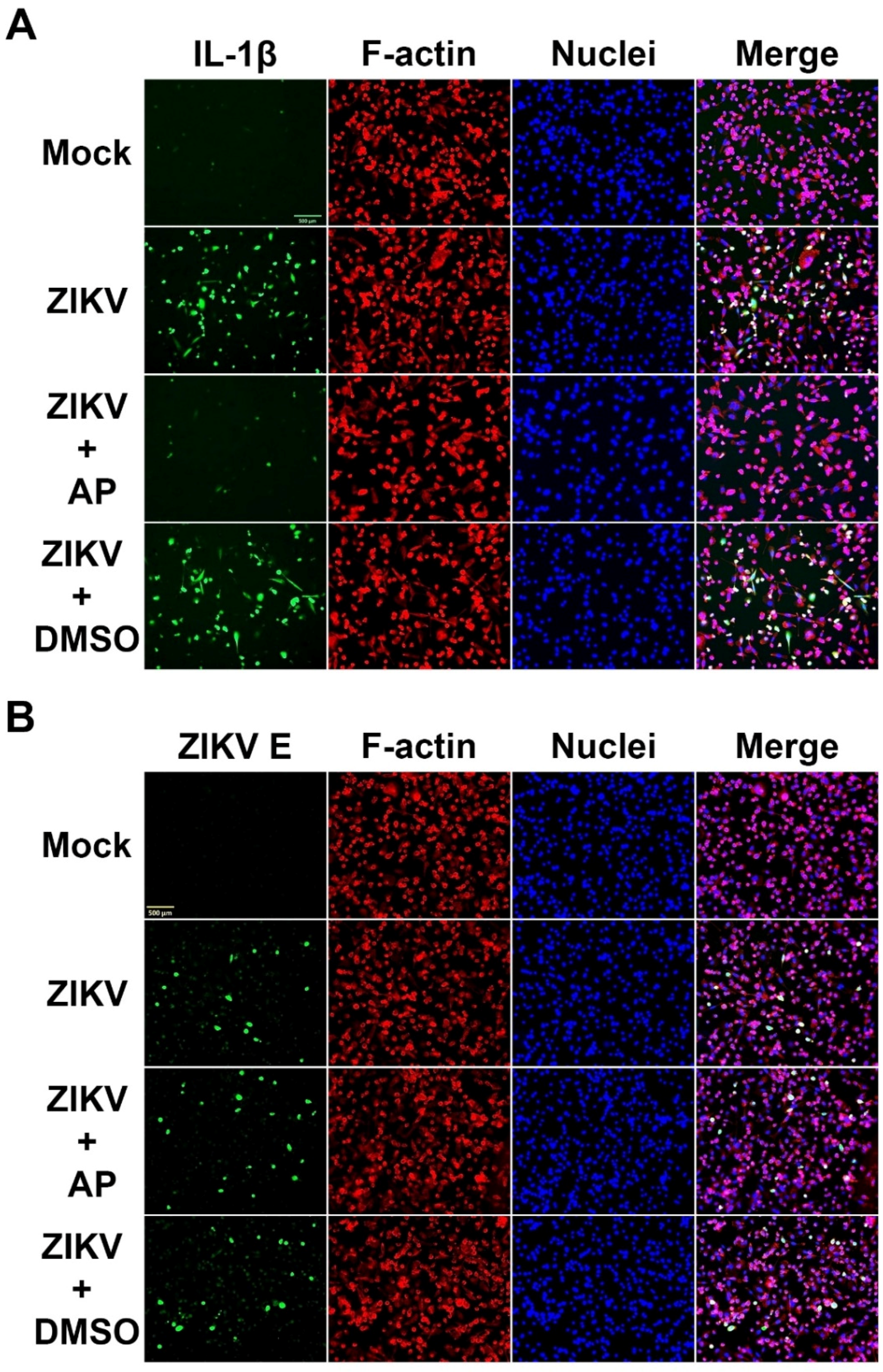
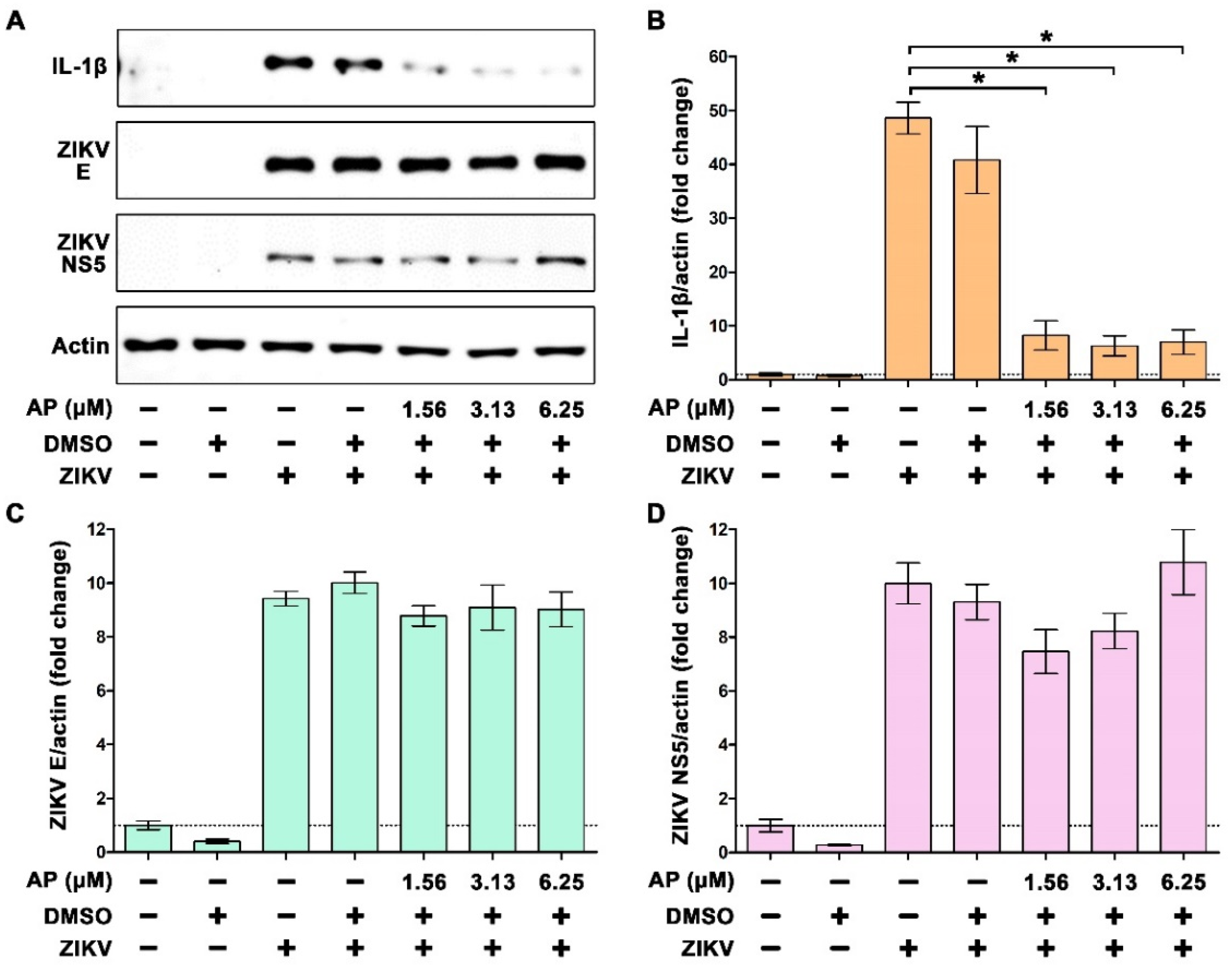
3.2. AP Reduces ZIKV-Induced Secretion of Interleukin 1β (IL-1β) from THP-1 Macrophages
3.3. AP Reduces ZIKV-Induced Production of IL-1β in THP-1 Macrophages
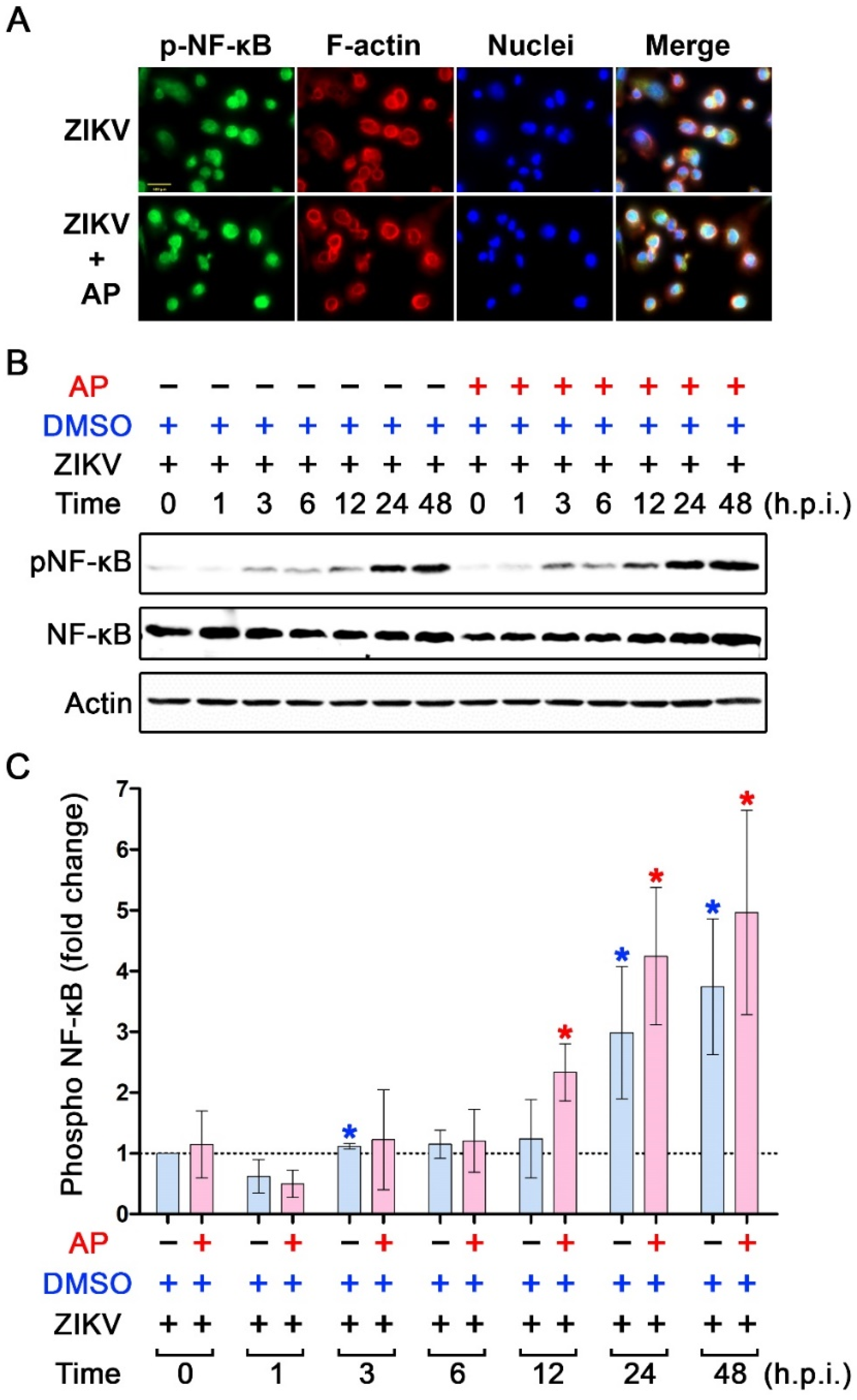
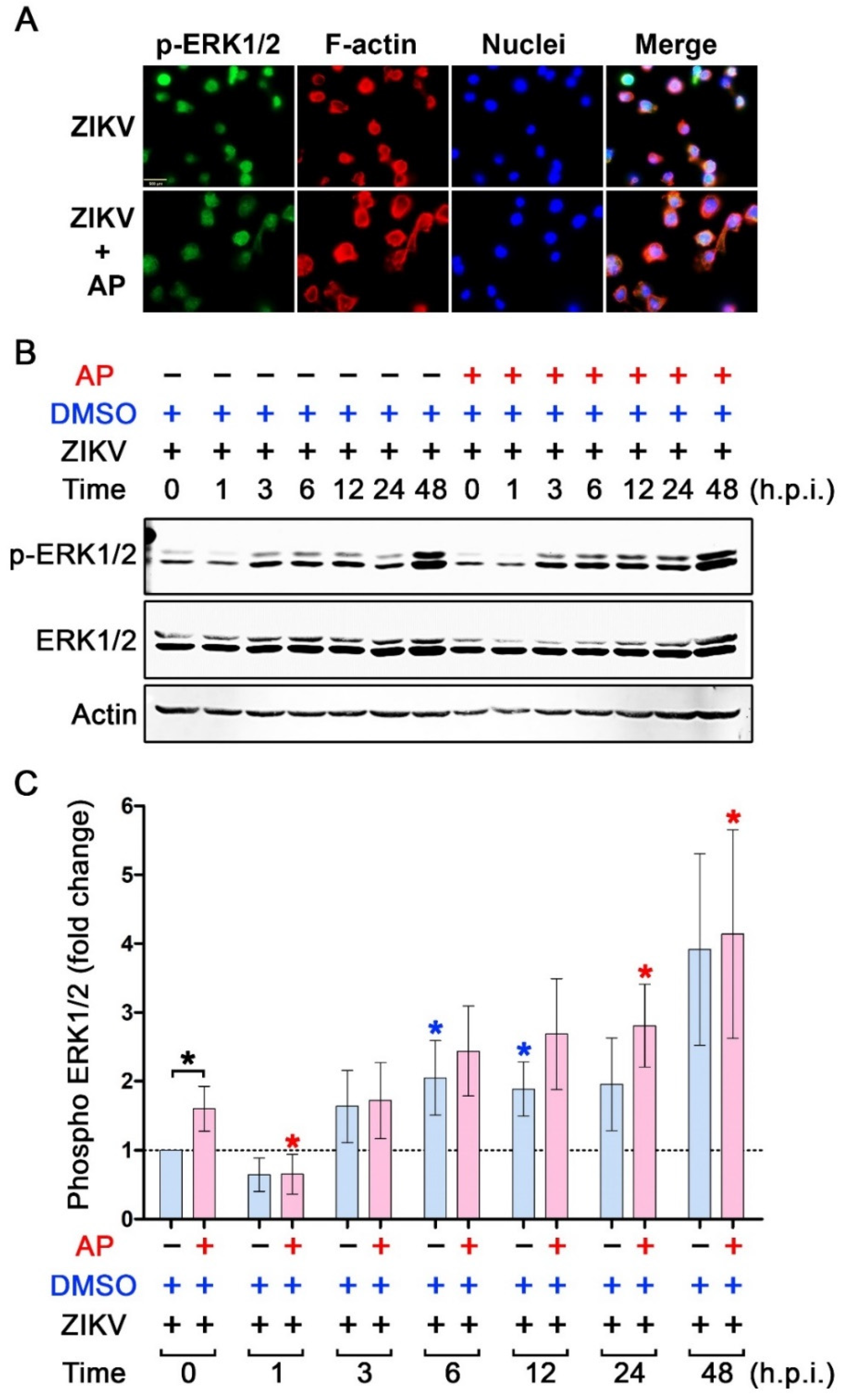
3.4. Inhibitory Effects of AP on ZIKV-Induced IL-1β Is Not Regulated through the Inhibition of NF-κB Signaling
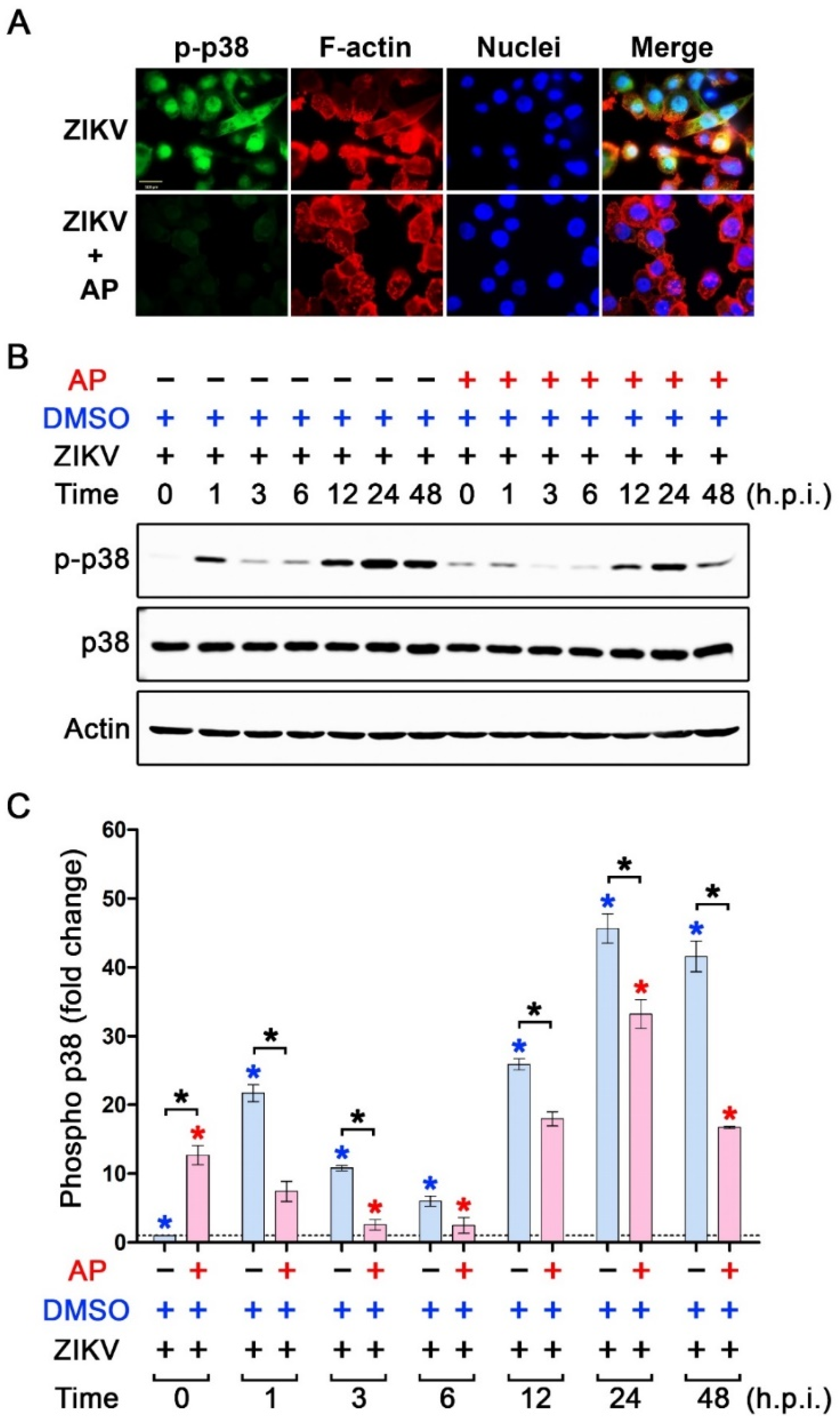
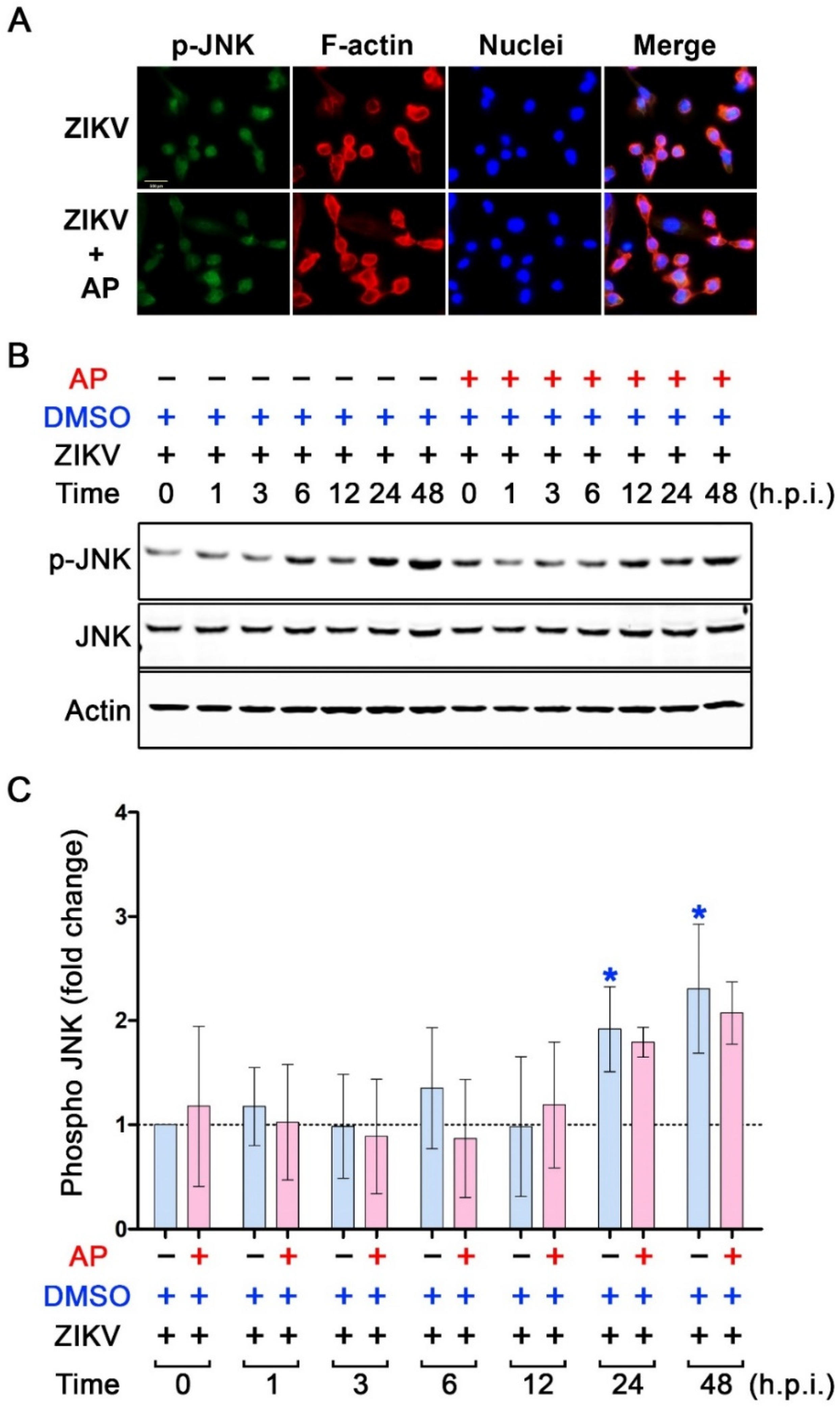
3.5. AP Selectively Suppresses ZIKV-Induced Activation of p38 MAPK
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes, E.B. Zika virus outside Africa. Emerg. Infect. Dis. 2009, 15, 1347–1350. [Google Scholar] [CrossRef] [PubMed]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martinez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N. Zika Virus Infection as a Cause of Congenital Brain Abnormalities and Guillain-Barre Syndrome: Systematic Review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetro, J.A. Zika and microcephaly: Causation, correlation, or coincidence? Microbes Infect. 2016, 18, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Leis, A.A.; Stokic, D.S. Zika Virus and Guillain-Barre Syndrome: Is There Sufficient Evidence for Causality? Front. Neurol. 2016, 7, 170. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.; Ali, S.; Ahamad, S.; Malik, M.Z.; Ishrat, R. From ZikV genome to vaccine: In silico approach for the epitope-based peptide vaccine against Zika virus envelope glycoprotein. Immunology 2016, 149, 386–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Costa, H.; Gouilly, J.; Mansuy, J.M.; Chen, Q.; Levy, C.; Cartron, G.; Veas, F.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N. ZIKA virus reveals broad tissue and cell tropism during the first trimester of pregnancy. Sci. Rep. 2016, 6, 35296. [Google Scholar] [CrossRef] [Green Version]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Agrelli, A.; de Moura, R.R.; Crovella, S.; Brandao, L.A.C. ZIKA virus entry mechanisms in human cells. Infect. Genet. Evol. 2019, 69, 22–29. [Google Scholar] [CrossRef]
- Alcendor, D.J. Zika Virus Infection of the Human Glomerular Cells: Implications for Viral Reservoirs and Renal Pathogenesis. J. Infect. Dis. 2017, 216, 162–171. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.F.; Chen, J.; Zhou, X.; Dong, Z.; Chen, T.; Yang, Y.; Zou, P.; Jiang, B.; Hu, Y.; et al. Zika virus infects renal proximal tubular epithelial cells with prolonged persistency and cytopathic effects. Emerg. Microbes Infect. 2017, 6, e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaiboullina, S.F.; Uppal, T.; Sarkar, R.; Gorzalski, A.; St Jeor, S.; Verma, S.C. ZIKV infection regulates inflammasomes pathway for replication in monocytes. Sci. Rep. 2017, 7, 16050. [Google Scholar] [CrossRef] [Green Version]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappe, D.; Perez-Giron, J.V.; Zammarchi, L.; Rissland, J.; Ferreira, D.F.; Jaenisch, T.; Gomez-Medina, S.; Gunther, S.; Bartoloni, A.; Munoz-Fontela, C.; et al. Cytokine kinetics of Zika virus-infected patients from acute to reconvalescent phase. Med. Microbiol. Immunol. 2016, 205, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoro, T.; Dandekar, A.; Jennelle, L.T.; Bajaj, R.; Lipkowitz, G.; Angelucci, A.R.; Bessong, P.O.; Hahn, Y.S. IL-1beta/TNF-alpha/IL-6 inflammatory cytokines promote STAT1-dependent induction of CH25H in Zika virus-infected human macrophages. J. Biol. Chem. 2019, 294, 14591–14602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.L.; Kim, M.O.; Morgello, S.; Lee, S.C. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J. Neuroimmunol. 2001, 115, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mishra, M.K.; Ghosh, J.; Basu, A. Japanese Encephalitis Virus infection induces IL-18 and IL-1beta in microglia and astrocytes: Correlation with in vitro cytokine responsiveness of glial cells and subsequent neuronal death. J. Neuroimmunol. 2008, 195, 60–72. [Google Scholar] [CrossRef]
- Guo, S.; Yang, C.; Diao, B.; Huang, X.; Jin, M.; Chen, L.; Yan, W.; Ning, Q.; Zheng, L.; Wu, Y.; et al. The NLRP3 Inflammasome and IL-1beta Accelerate Immunologically Mediated Pathology in Experimental Viral Fulminant Hepatitis. PLoS Pathog. 2015, 11, e1005155. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, G.; De, W.; Luo, Z.; Pan, P.; Tian, M.; Wang, Y.; Xiao, F.; Li, A.; Wu, K.; et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat. Commun. 2018, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Chen, J.; Zhu, X.; An, S.; Dong, X.; Yu, J.; Zhang, S.; Wu, Y.; Li, G.; Zhang, Y.; et al. NLRP3 Inflammasome Activation Mediates Zika Virus-Associated Inflammation. J. Infect. Dis. 2018, 217, 1942–1951. [Google Scholar] [CrossRef]
- Liu, T.; Tang, L.; Tang, H.; Pu, J.; Gong, S.; Fang, D.; Zhang, H.; Li, Y.P.; Zhu, X.; Wang, W.; et al. Zika Virus Infection Induces Acute Kidney Injury Through Activating NLRP3 Inflammasome Via Suppressing Bcl-2. Front. Immunol. 2019, 10, 1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Blocking IL-1 in systemic inflammation. J. Exp. Med. 2005, 201, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Vermillion, M.S.; Jia, B.; Xie, H.; Xie, L.; McLane, M.W.; Sheffield, J.S.; Pekosz, A.; Brown, A.; Klein, S.L.; et al. IL-1 receptor antagonist therapy mitigates placental dysfunction and perinatal injury following Zika virus infection. JCI Insight 2019, 4, e122678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Tong, Y.; Luan, F.; Zhu, W.; Zhan, C.; Qin, T.; An, W.; Zeng, N. Alpinetin: A Review of Its Pharmacology and Pharmacokinetics. Front. Pharm. 2022, 13, 814370. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Chen, N.; Chi, G.; Yuan, X.; Guan, S.; Li, H.; Zhong, W.; Guo, W.; Soromou, L.W.; Gao, R.; et al. Traditional medicine alpinetin inhibits the inflammatory response in Raw 264.7 cells and mouse models. Int. Immunopharmacol. 2012, 12, 241–248. [Google Scholar] [CrossRef]
- He, X.; Wei, Z.; Wang, J.; Kou, J.; Liu, W.; Fu, Y.; Yang, Z. Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci. Rep. 2016, 6, 28370. [Google Scholar] [CrossRef]
- Hu, K.; Yang, Y.; Tu, Q.; Luo, Y.; Ma, R. Alpinetin inhibits LPS-induced inflammatory mediator response by activating PPAR-gamma in THP-1-derived macrophages. Eur. J. Pharm. 2013, 721, 96–102. [Google Scholar] [CrossRef]
- Tan, Y.; Zheng, C. Effects of Alpinetin on Intestinal Barrier Function, Inflammation and Oxidative Stress in Dextran Sulfate Sodium-Induced Ulcerative Colitis Mice. Am. J. Med. Sci. 2018, 355, 377–386. [Google Scholar] [CrossRef]
- Jitsatja, A.; Ramphan, S.; Promma, P.; Kuadkitkan, A.; Wikan, N.; Uiprasertkul, M.; Phatihattakorn, C.; Smith, D.R. Comparative analysis of a Thai congenital-Zika-syndrome-associated virus with a Thai Zika-fever-associated virus. Arch. Virol. 2020, 165, 1791–1801. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with image. Biophotonics Int. 2004, 11, 36–41. [Google Scholar]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome--case report, French Polynesia, December 2013. Eurosurveill 2014, 19, 20720. [Google Scholar] [CrossRef]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.K.; Wollebo, H.S.; David Beckham, J.; Tyler, K.L.; Khalili, K. Zika virus: An emergent neuropathological agent. Ann. Neurol. 2016, 80, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Paula Freitas, B.; de Oliveira Dias, J.R.; Prazeres, J.; Sacramento, G.A.; Ko, A.I.; Maia, M.; Belfort, R., Jr. Ocular Findings in Infants With Microcephaly Associated With Presumed Zika Virus Congenital Infection in Salvador, Brazil. JAMA Ophthalmol. 2016, 134, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, C.V.; Maia, M.; Travassos, S.B.; Martins, T.T.; Patriota, F.; Nunes, M.E.; Agra, C.; Torres, V.L.; van der Linden, V.; Ramos, R.C.; et al. Risk Factors Associated With the Ophthalmoscopic Findings Identified in Infants With Presumed Zika Virus Congenital Infection. JAMA Ophthalmol. 2016, 134, 912–918. [Google Scholar] [CrossRef]
- Olmo, I.G.; Carvalho, T.G.; Costa, V.V.; Alves-Silva, J.; Ferrari, C.Z.; Izidoro-Toledo, T.C.; da Silva, J.F.; Teixeira, A.L.; Souza, D.G.; Marques, J.T.; et al. Zika Virus Promotes Neuronal Cell Death in a Non-Cell Autonomous Manner by Triggering the Release of Neurotoxic Factors. Front. Immunol. 2017, 8, 1016. [Google Scholar] [CrossRef] [Green Version]
- Lum, F.M.; Low, D.K.; Fan, Y.; Tan, J.J.; Lee, B.; Chan, J.K.; Renia, L.; Ginhoux, F.; Ng, L.F. Zika Virus Infects Human Fetal Brain Microglia and Induces Inflammation. Clin. Infect. Dis. 2017, 64, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, P.M.; Caracciolo, I.; Crovella, S.; D’Agaro, P. Zika virus induces inflammasome activation in the glial cell line U87-MG. Biochem. Biophys. Res. Commun. 2017, 492, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Manangeeswaran, M.; Ireland, D.D.; Verthelyi, D. Zika (PRVABC59) Infection Is Associated with T cell Infiltration and Neurodegeneration in CNS of Immunocompetent Neonatal C57Bl/6 Mice. PLoS Pathog. 2016, 12, e1006004. [Google Scholar] [CrossRef]
- Shao, Q.; Herrlinger, S.; Yang, S.L.; Lai, F.; Moore, J.M.; Brindley, M.A.; Chen, J.F. Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 2016, 143, 4127–4136. [Google Scholar] [CrossRef] [Green Version]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Current status of therapeutic and vaccine approaches against Zika virus. Eur. J. Intern. Med. 2017, 44, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Basile, K.; Kok, J.; Dwyer, D.E. Zika virus: What, where from and where to? Pathology 2017, 49, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Attaway, D.F.; Waters, N.M.; Geraghty, E.M.; Jacobsen, K.H. Zika virus: Endemic and epidemic ranges of Aedes mosquito transmission. J. Infect. Public Health 2017, 10, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumtaz, N.; van Kampen, J.J.; Reusken, C.B.; Boucher, C.A.; Koopmans, M.P. Zika Virus: Where Is the Treatment? Curr. Treat. Options Infect. Dis. 2016, 8, 208–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, S.; Tremblay, L.; Lepage, M.; Sebire, G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 2010, 184, 3997–4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, K.; Al Shammary, M.; McLane, M.; Johnston, M.V.; Elovitz, M.A.; Burd, I. IL-1 receptor blockade prevents fetal cortical brain injury but not preterm birth in a mouse model of inflammation-induced preterm birth and perinatal brain injury. Am. J. Reprod. Immunol. 2014, 71, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, J.M.; Lei, J.; Burd, I. Interleukin-1 receptor blockade in perinatal brain injury. Front. Pediatr. 2014, 2, 108. [Google Scholar] [CrossRef]
- Martinon, F. Mechanisms of uric acid crystal-mediated autoinflammation. Immunol. Rev. 2010, 233, 218–232. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lee, H.; Hwangbo, H.; Kim, S.Y.; Ji, S.Y.; Kim, M.Y.; Park, S.K.; Park, S.H.; Kim, M.Y.; Kim, G.Y.; et al. Particulate matter 2.5 promotes inflammation and cellular dysfunction via reactive oxygen species/p38 MAPK pathway in primary rat corneal epithelial cells. Cutan. Ocul. Toxicol. 2022, 41, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Luo, H.; Liu, H.; Ha, Y.; Mays, E.R.; Lawrence, R.E.; Winkelmann, E.; Barrett, A.D.; Smith, S.B.; Wang, M.; et al. p38MAPK plays a critical role in induction of a pro-inflammatory phenotype of retinal Muller cells following Zika virus infection. Antivir. Res. 2017, 145, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yip, A.; Seah, P.G.; Blasco, F.; Shi, P.Y.; Herve, M. Modulation of inflammation and pathology during dengue virus infection by p38 MAPK inhibitor SB203580. Antivir. Res. 2014, 110, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Sreekanth, G.P.; Chuncharunee, A.; Sirimontaporn, A.; Panaampon, J.; Noisakran, S.; Yenchitsomanus, P.T.; Limjindaporn, T. SB203580 Modulates p38 MAPK Signaling and Dengue Virus-Induced Liver Injury by Reducing MAPKAPK2, HSP27, and ATF2 Phosphorylation. PLoS ONE 2016, 11, e0149486. [Google Scholar] [CrossRef]
- Yu, J.W.; Farias, A.; Hwang, I.; Fernandes-Alnemri, T.; Alnemri, E.S. Ribotoxic stress through p38 mitogen-activated protein kinase activates in vitro the human pyrin inflammasome. J. Biol. Chem. 2013, 288, 11378–11383. [Google Scholar] [CrossRef]
- Baldassare, J.J.; Bi, Y.; Bellone, C.J. The role of p38 mitogen-activated protein kinase in IL-1 beta transcription. J. Immunol. 1999, 162, 5367–5373. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wikan, N.; Potikanond, S.; Hankittichai, P.; Thaklaewphan, P.; Monkaew, S.; Smith, D.R.; Nimlamool, W. Alpinetin Suppresses Zika Virus-Induced Interleukin-1β Production and Secretion in Human Macrophages. Pharmaceutics 2022, 14, 2800. https://doi.org/10.3390/pharmaceutics14122800
Wikan N, Potikanond S, Hankittichai P, Thaklaewphan P, Monkaew S, Smith DR, Nimlamool W. Alpinetin Suppresses Zika Virus-Induced Interleukin-1β Production and Secretion in Human Macrophages. Pharmaceutics. 2022; 14(12):2800. https://doi.org/10.3390/pharmaceutics14122800
Chicago/Turabian StyleWikan, Nitwara, Saranyapin Potikanond, Phateep Hankittichai, Phatarawat Thaklaewphan, Sathit Monkaew, Duncan R. Smith, and Wutigri Nimlamool. 2022. "Alpinetin Suppresses Zika Virus-Induced Interleukin-1β Production and Secretion in Human Macrophages" Pharmaceutics 14, no. 12: 2800. https://doi.org/10.3390/pharmaceutics14122800