
Potent Platinum(IV) Prodrugs That Incorporate a Biotin Moiety to Selectively Target Cancer Cells
 , , ,
, , , 
Abstract
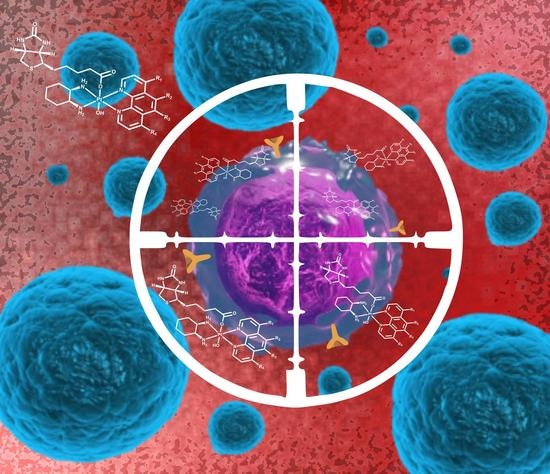
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of [Pt(PL)(SSDACH)]Cl2 (PHENSS(II), 56MESS(II), 47OMESS(II), and 5MESS(II)) and [Pt(PL)(SSDACH)(OH)2](NO3)2 (PHENSS(IV), 56MESS(IV), 47OMESS(IV), and 5MESS(IV)) (Where PL = Phen, 56Me2Phen, 47O2Me2Phen or 5MePhen)
2.3. Synthesis of Biotin-NHS Ester
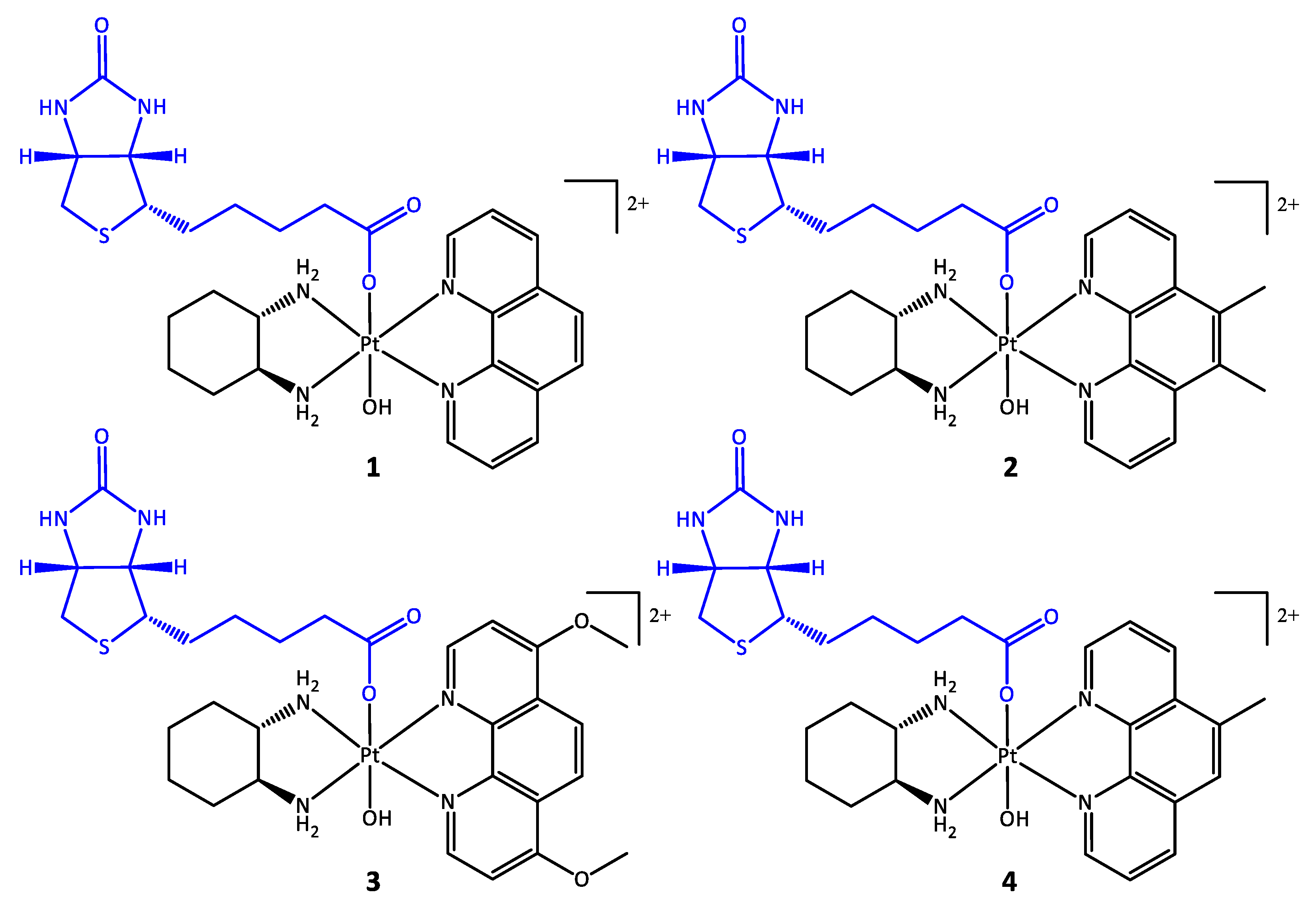
2.4. Synthesis of [Pt(PL)(SSDACH)(Biotin)(OH)](NO3)2 (PL = Phen (1), 56Me2Phen (2), 47O2Me2Phen (3), or 5MePhen (4))
2.4.1. [Pt(Phen)(SSDACH)(Biotin)(OH)](NO3)2 (1)
2.4.2. [Pt(56Me2Phen)(SSDACH)(Biotin)(OH)](NO3)2 (2)
2.4.3. [Pt(47O2Me2Phen)(SSDACH)(Biotin)(OH)](NO3)2 (3)
2.4.4. [Pt(5MePhen)(SSDACH)(Biotin)(OH)](NO3)2 (4)
2.5. NMR Spectroscopy
2.6. Flash Chromatography
2.7. Ultraviolet (UV) Absorption Spectroscopy
2.8. Circular Dichroism (CD) Spectroscopy
2.9. Mass Spectrometry (MS)
2.10. High Performance Liquid Chromatography (HPLC) and Lipophilicity Measurements
2.11. In Vitro Cytotoxicity Assay
2.12. Cellular Accumulation
2.13. Platinum Analysis
3. Results and Discussion
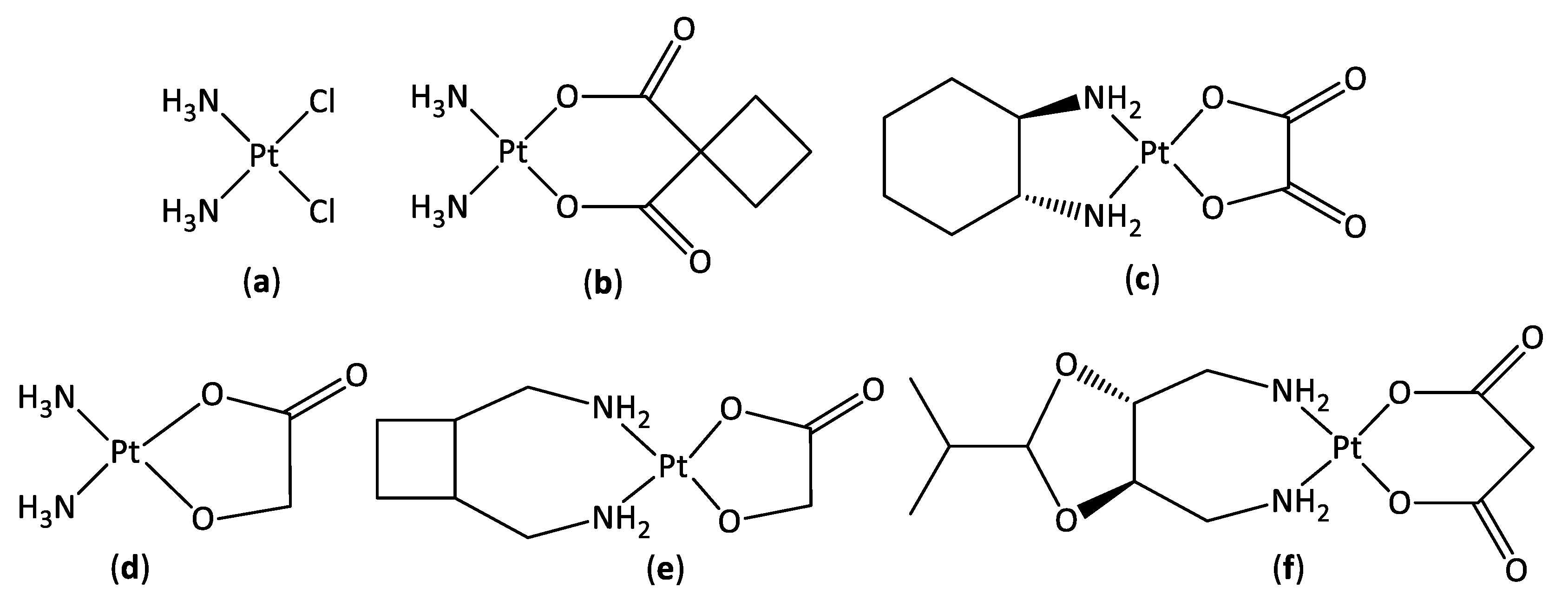
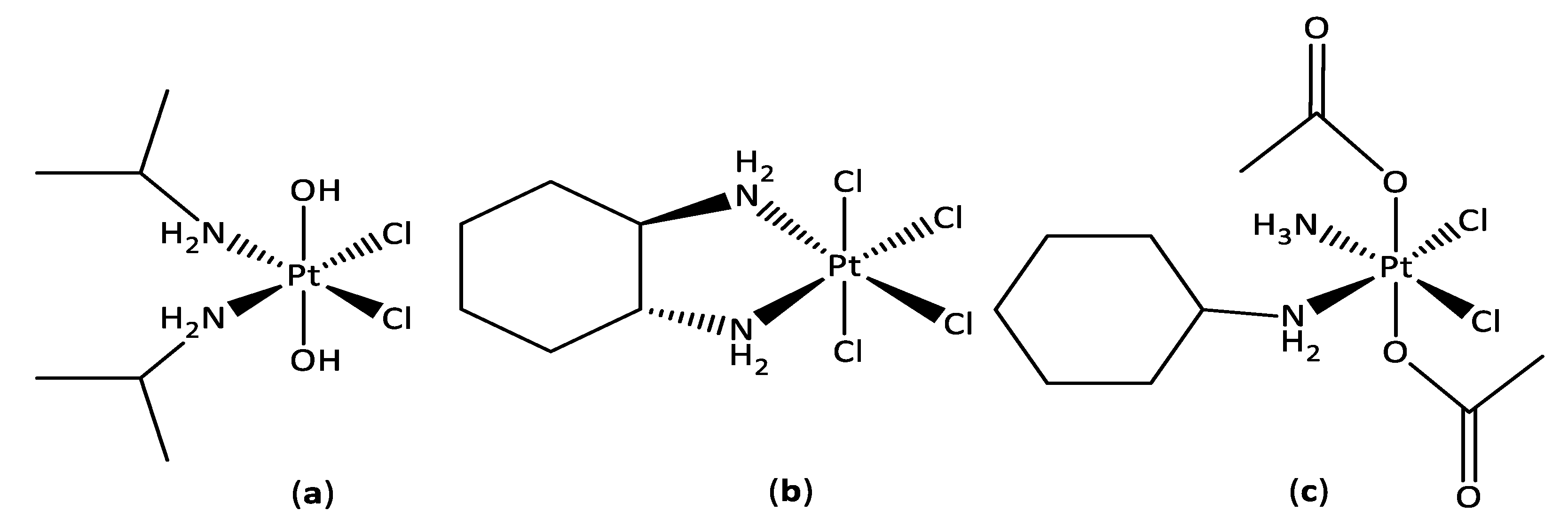
3.1. Synthesis and Characterisation
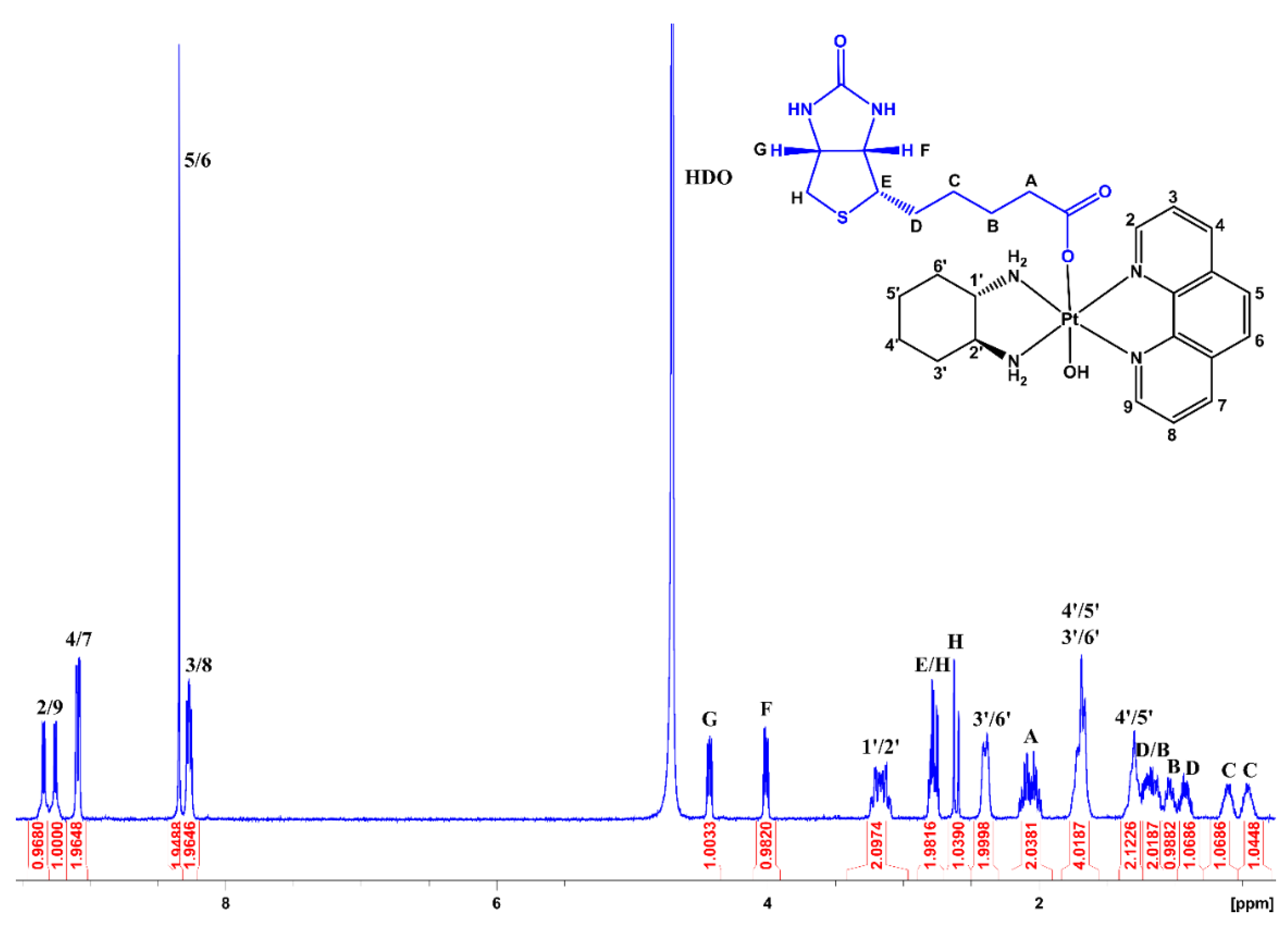
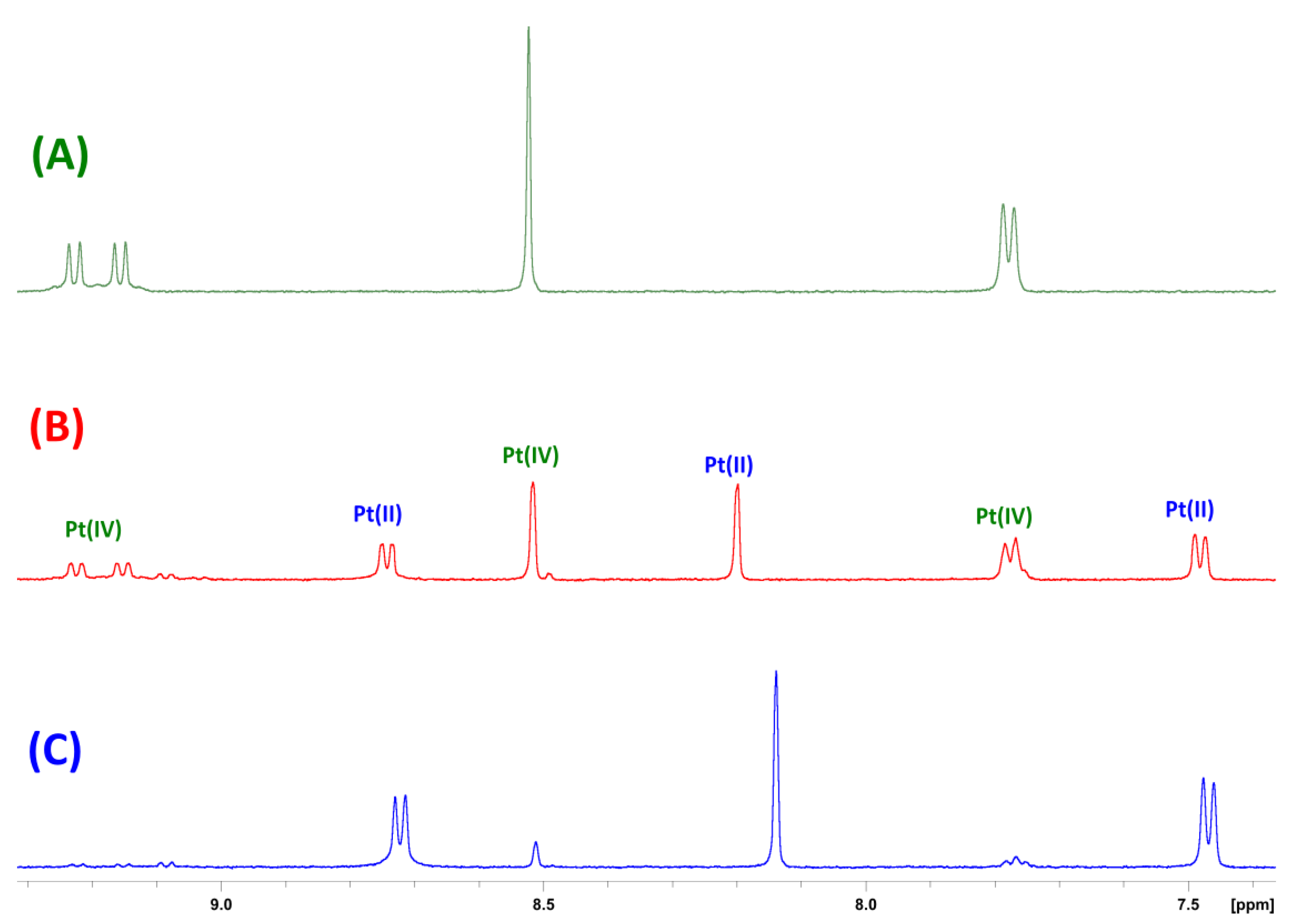
3.2. NMR Spectroscopy
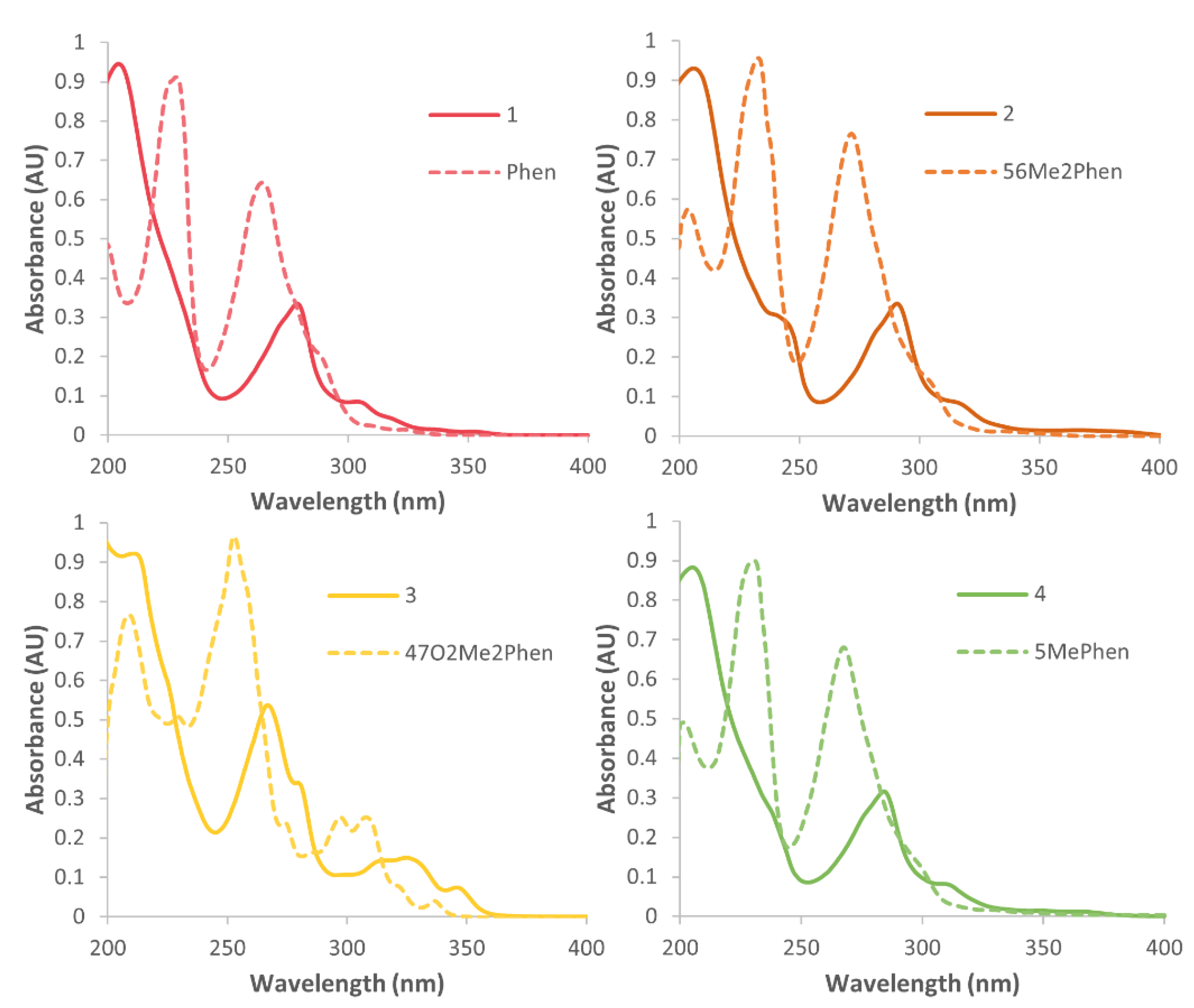
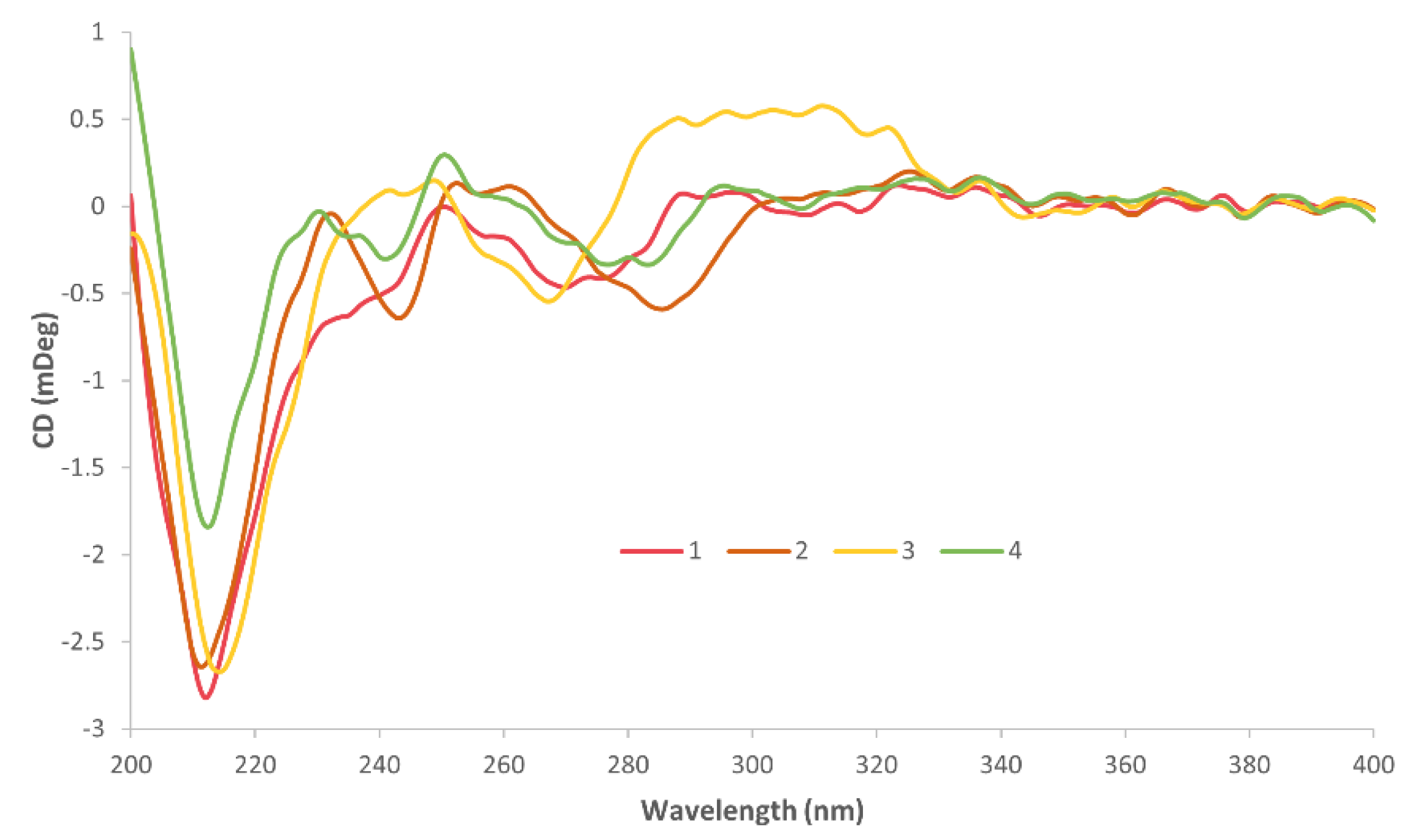
3.3. Electronic Spectra
3.4. Reduction Studies via 1H NMR
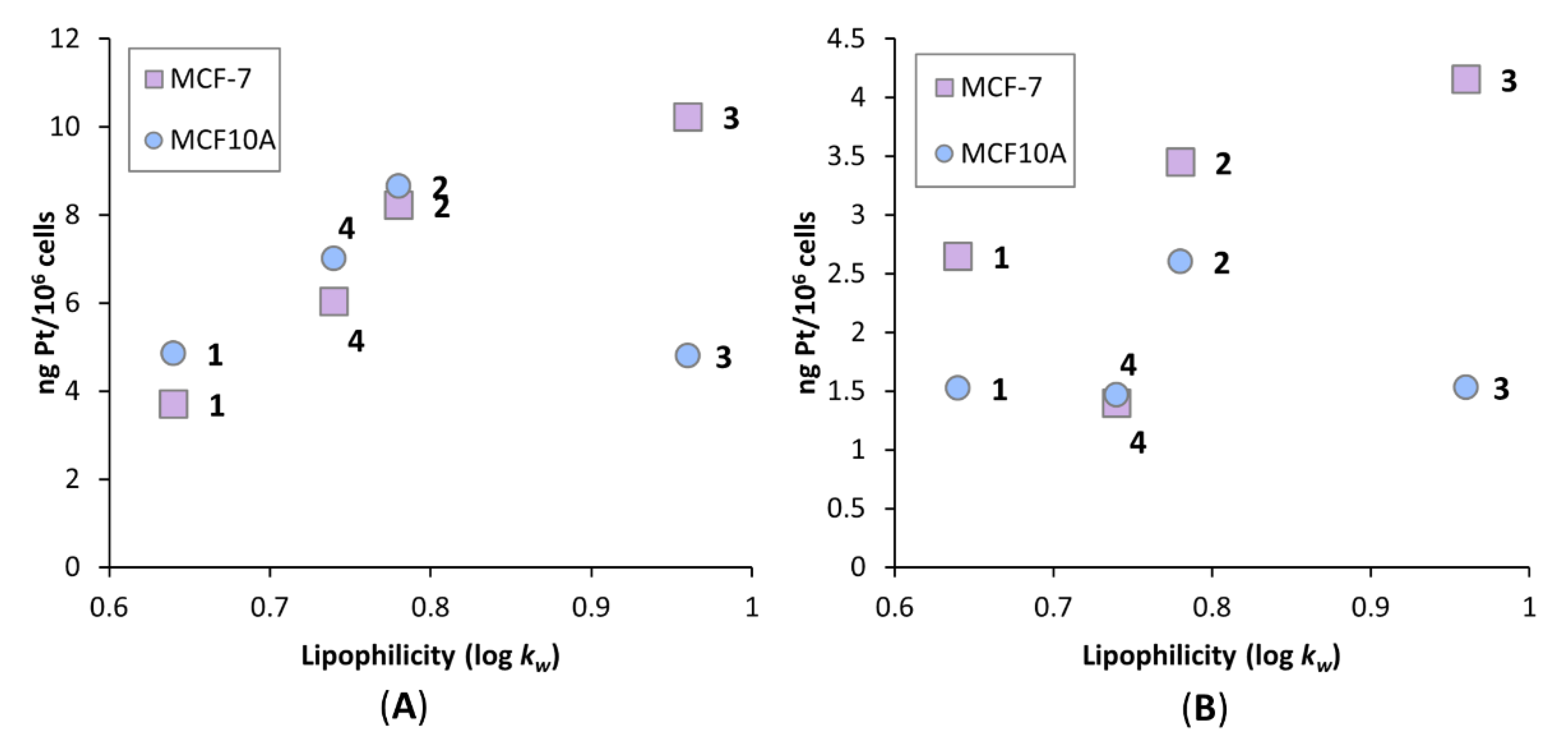
3.5. Lipophilicity Studies
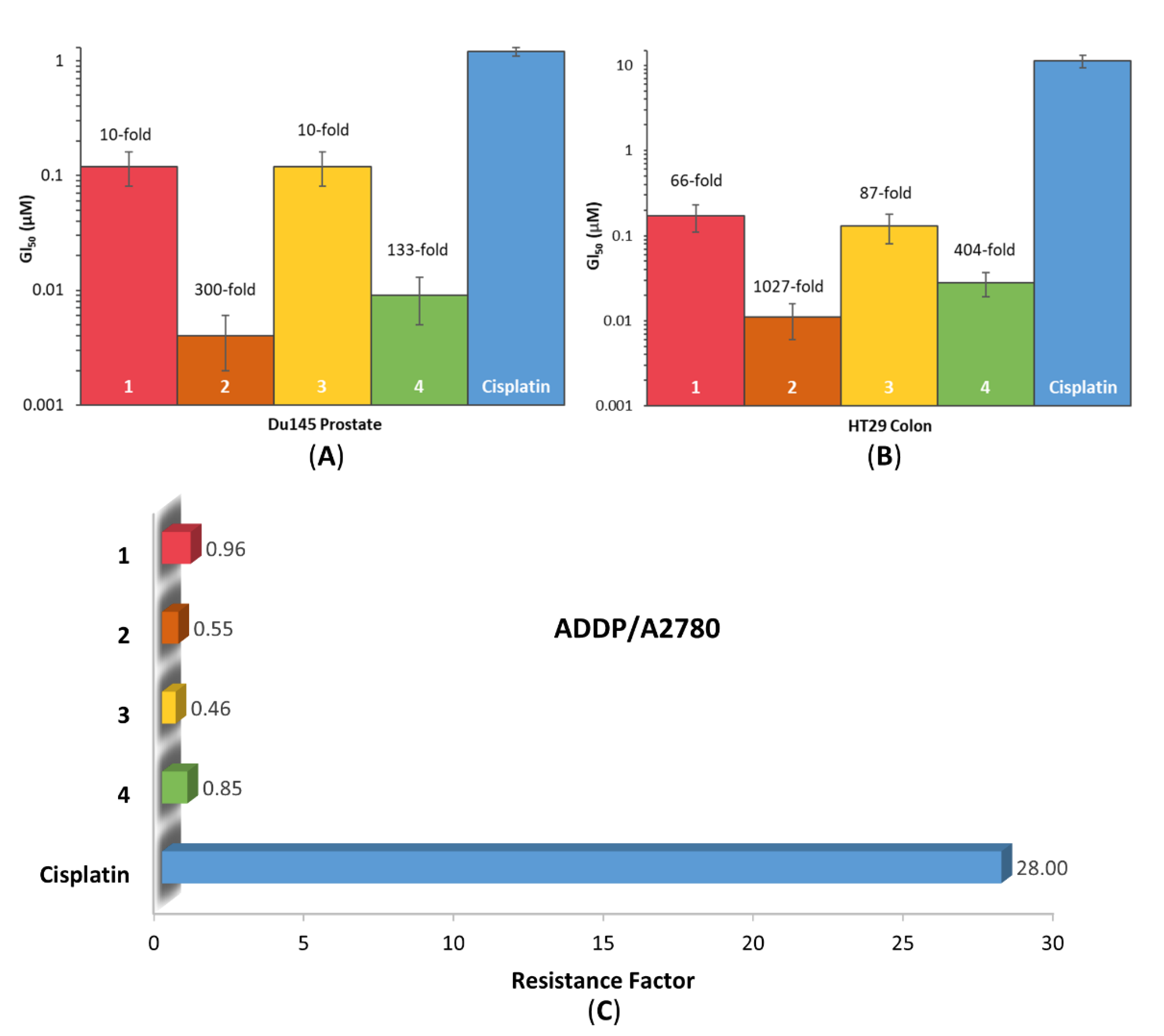
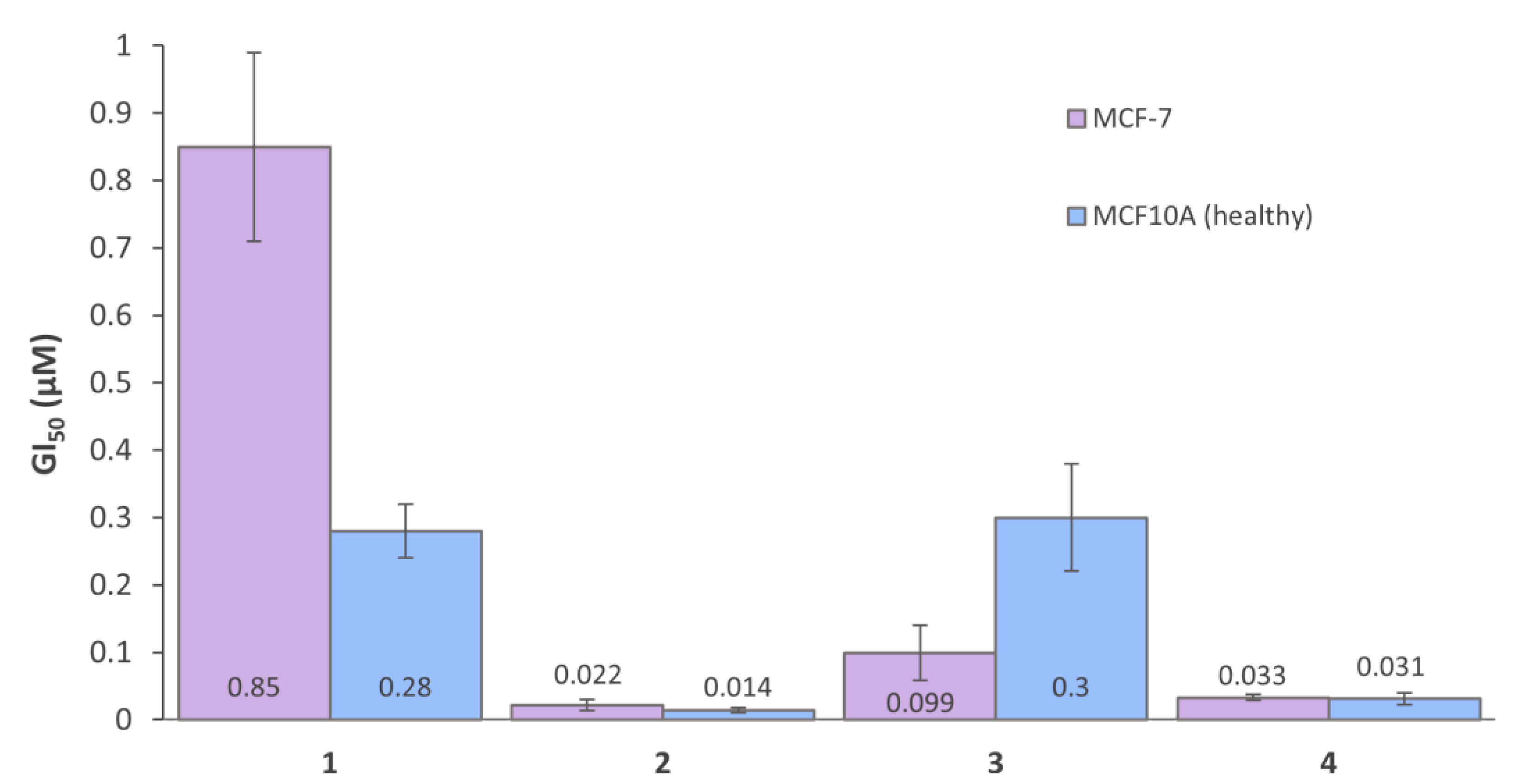
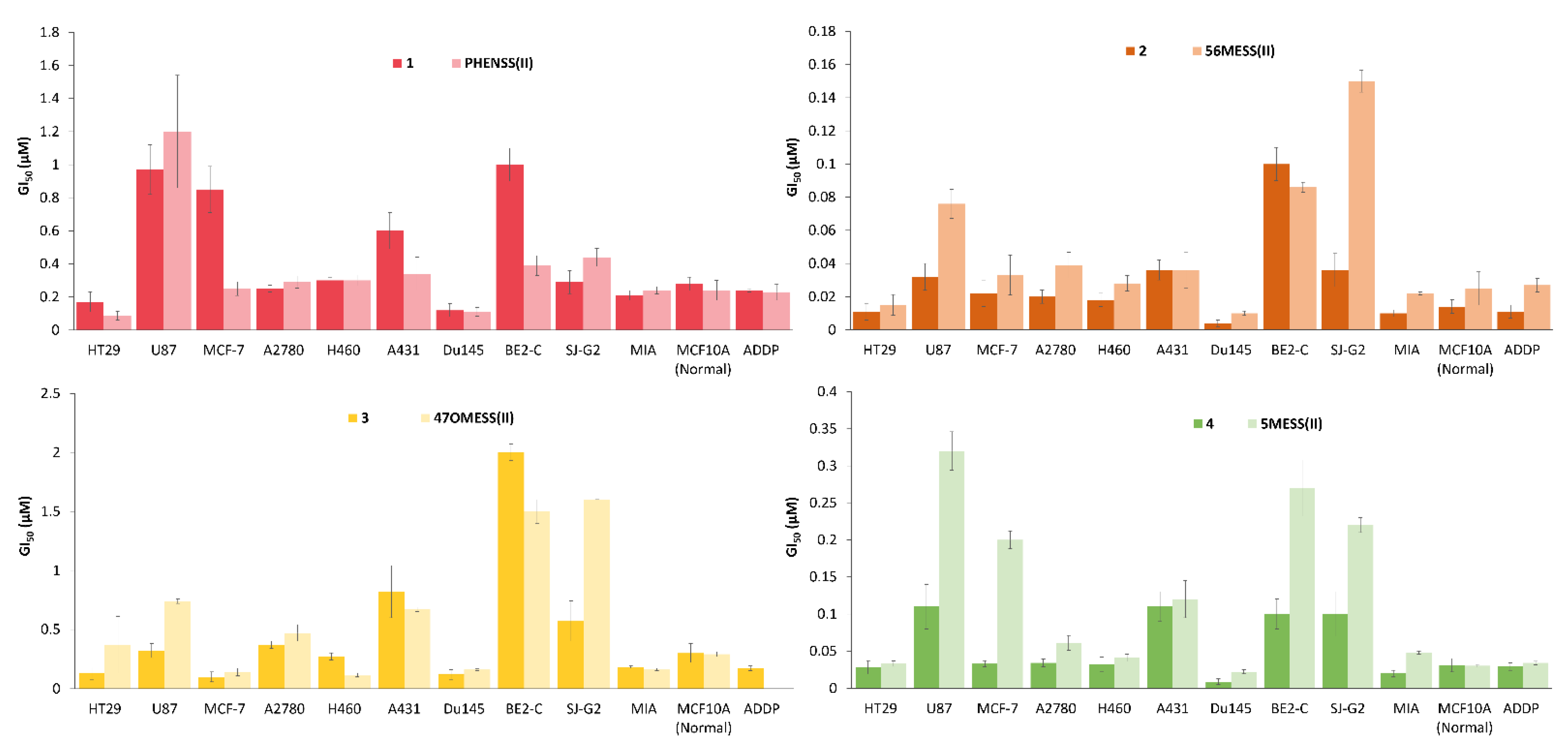
3.6. In Vitro Cytotoxicity
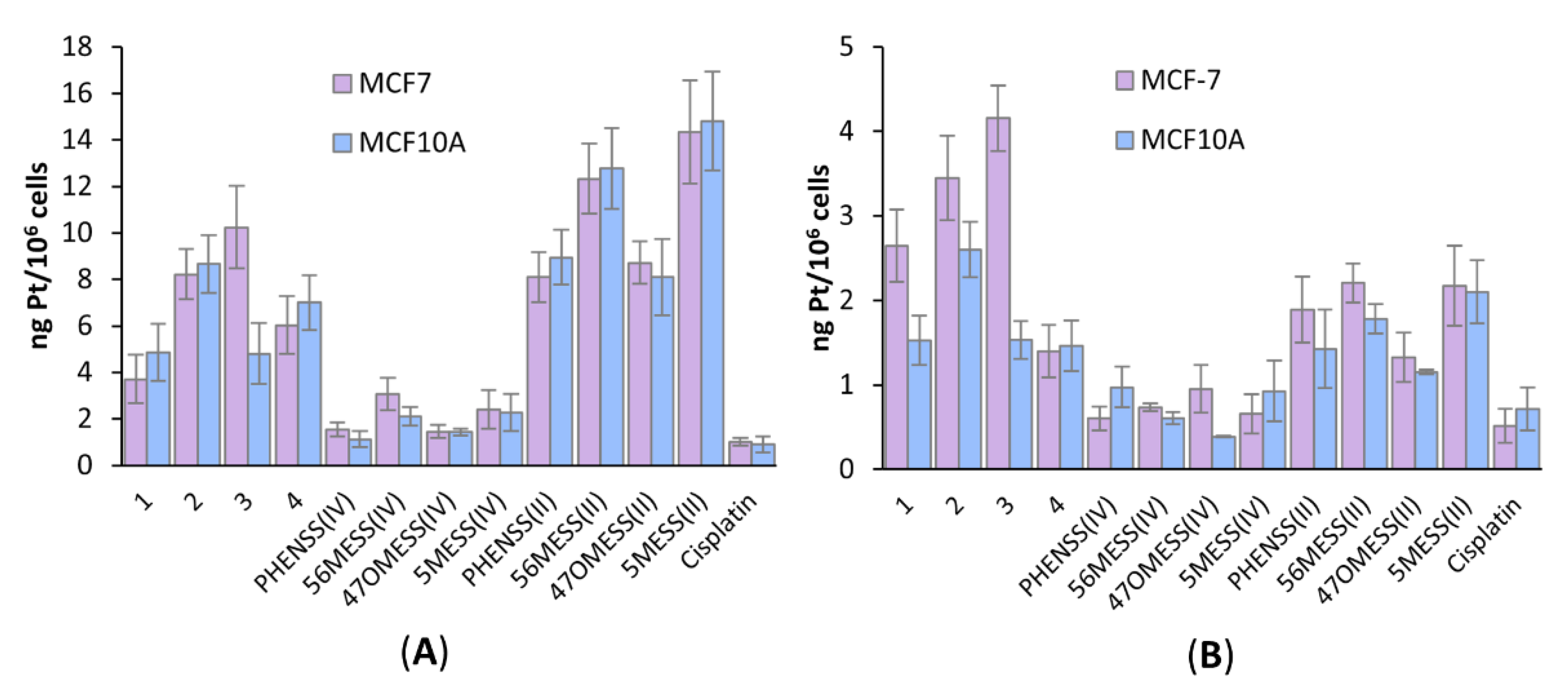
3.7. Platinum Uptake Studies
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Van Camp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, recognition, and processing of cisplatin− DNA adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Deo, K.M.; Ang, D.L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.; Bjelosevic, A.; Pages, B.; Gordon, C. Platinum coordination compounds with potent anticancer activity. Coord. Chem. Rev. 2018, 375, 148–163. [Google Scholar] [CrossRef]
- Khoury, A.; Deo, K.M.; Aldrich-Wright, J.R. Recent advances in platinum-based chemotherapeutics that exhibit inhibitory and targeted mechanisms of action. J. Inorg. Biochem. 2020, 207, 111070. [Google Scholar] [CrossRef]
- Ivanova, S. Comparative assessment of clinical trials, indications, pharmacokinetic parameters and side effects of approved platinum drugs. Pharmacia 2022, 69, 1–7. [Google Scholar] [CrossRef]
- Boulikas, T.; Pantos, A.; Bellis, E.; Christofis, P. Designing platinum compounds in cancer: Structures and mechanisms. Cancer Ther. 2007, 5, 537–583. [Google Scholar]
- Wang, X.; Guo, Z. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheate, N.J.; Taleb, R.I.; Krause-Heuer, A.M.; Cook, R.L.; Wang, S.; Higgins, V.J.; Aldrich-Wright, J.R. Novel platinum(II)-based anticancer complexes and molecular hosts as their drug delivery vehicles. Dalton Trans. 2007, 43, 5055–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodie, C.R.; Collins, J.G.; Aldrich-Wright, J.R. DNA binding and biological activity of some platinum(II) intercalating compounds containing methyl-substituted 1, 10-phenanthrolines. Dalton Trans. 2004, 8, 1145–1152. [Google Scholar] [CrossRef]
- Kostrhunova, H.; Zajac, J.; Novohradsky, V.; Kasparkova, J.; Malina, J.; Aldrich-Wright, J.R.; Petruzzella, E.; Sirota, R.; Gibson, D.; Brabec, V. A subset of new platinum antitumor agents kills cells by a multimodal mechanism of action also involving changes in the organization of the microtubule cytoskeleton. J. Med. Chem. 2019, 62, 5176–5190. [Google Scholar] [CrossRef]
- Moretto, J.; Chauffert, B.; Ghiringhelli, F.; Aldrich-Wright, J.R.; Bouyer, F. Discrepancy between in vitro and in vivo antitumor effect of a new platinum(II) metallointercalator. Investig. New Drugs 2011, 29, 1164–1176. [Google Scholar] [CrossRef]
- Macias, F.J.; Deo, K.M.; Wormell, P.; Clegg, J.K.; Zhang, Y.; Li, F.; Zheng, G.; Sakoff, J.; Gilbert, J.; Aldrich-Wright, J.R. Synthesis and analysis of the structure, diffusion and cytotoxicity of heterocyclic platinum(IV) complexes. Chem. A Eur. J. 2015, 21, 16990–17001. [Google Scholar] [CrossRef]
- Harper, B.W.; Petruzzella, E.; Sirota, R.; Faccioli, F.F.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. Synthesis, characterization and in vitro and in vivo anticancer activity of Pt(IV) derivatives of [Pt (1 S, 2 S-DACH)(5, 6-dimethyl-1, 10-phenanthroline)]. Dalton Trans. 2017, 46, 7005–7019. [Google Scholar] [CrossRef]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007, 50, 3403–3411. [Google Scholar] [CrossRef]
- Hall, M.D.; Hambley, T.W. Platinum(IV) antitumour compounds: Their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Gibson, D. Platinum(IV) anticancer prodrugs–hypotheses and facts. Dalton Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Zhang, J.Z.; Aitken, J.B.; Hambley, T.W. Influence of equatorial and axial carboxylato ligands on the kinetic inertness of platinum(IV) complexes in the presence of ascorbate and cysteine and within DLD-1 cancer cells. J. Med. Chem. 2013, 56, 8757–8764. [Google Scholar] [CrossRef]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Wright, J.R.A. Synthesis, characterisation and influence of lipophilicity on cellular accumulation and cytotoxicity of unconventional platinum (IV) prodrugs as potent anticancer agents. Dalton Trans. 2019, 48, 17228–17240. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, T.C.; Wilson, J.J.; Lippard, S.J. Monofunctional and higher-valent platinum anticancer agents. Inorg. Chem. 2013, 52, 12234–12249. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.T.; Er, H.M.; Hambley, T.W. The influence of the axial ligands of a series of platinum(IV) anti-cancer complexes on their reduction to platinum(II) and reaction with DNA. Aust. J. Chem. 1995, 48, 793–806. [Google Scholar] [CrossRef]
- Weaver, E.L.; Bose, R.N. Platinum(II) catalysis and radical intervention in reductions of platinum(IV) antitumor drugs by ascorbic acid. J. Inorg. Biochem. 2003, 95, 231–239. [Google Scholar] [CrossRef]
- Choi, S.; Filotto, C.; Bisanzo, M.; Delaney, S.; Lagasee, D.; Whitworth, J.L.; Jusko, A.; Li, C.; Wood, N.A.; Willingham, J.; et al. Reduction and anticancer activity of platinum(IV) complexes. Inorg. Chem. 1998, 37, 2500–2504. [Google Scholar] [CrossRef]
- Czapla-Masztafiak, J.; Kubas, A.; Kayser, Y.; Fernandes, D.L.A.; Kwiatek, W.M.; Lipiec, E.; Deacon, G.B.; Al-Jorani, K.; Wood, B.R.; Szlachetko, J.; et al. Mechanism of hydrolysis of a platinum(IV) complex discovered by atomic telemetry. J. Inorg. Biochem. 2018, 187, 56–61. [Google Scholar] [CrossRef]
- Galanski, M.; Jakupec, M.A.; Keppler, B.K. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Roh, J.K.; Wolpert-DeFilippes, M.K.; Goldin, A.; Venditti, J.M.; Woolley, P.V. Therapeutic and pharmacological studies of tetrachloro (d, l-trans) 1, 2-diaminocyclohexane platinum (IV)(tetraplatin), a new platinum analogue. Cancer Res. 1988, 48, 1745–1752. [Google Scholar]
- Chaney, S.G.; Wyrick, S.; Till, G.K. In vitro biotransformations of tetrachloro (d, l-trans)-1, 2-diaminocyclohexaneplatinum (IV)(tetraplatin) in rat plasma. Cancer Res. 1990, 50, 4539–4545. [Google Scholar]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, C.N.; Petrylak, D.P.; Sartor, O.; Witjes, J.A.; Demkow, T.; Ferrero, J.-M.; Eymard, J.-C.; Falcon, S.; Calabrò, F.; James, N. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: The SPARC trial. J. Clin. Oncol. 2009, 27, 5431–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theiner, S.; Varbanov, H.P.; Galanski, M.; Egger, A.E.; Berger, W.; Heffeter, P.; Keppler, B.K. Comparative in vitro and in vivo pharmacological investigation of platinum(IV) complexes as novel anticancer drug candidates for oral application. JBIC J. Biol. Inorg. Chem. 2015, 20, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef]
- Saha, S.; Majumdar, R.; Hussain, A.; Dighe, R.R.; Chakravarty, A.R. Biotin-conjugated tumour-targeting photocytotoxic iron(III) complexes. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2013, 371, 20120190. [Google Scholar] [CrossRef]
- Russell-Jones, G.; McTavish, K.; McEwan, J.; Rice, J.; Nowotnik, D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J. Inorg. Biochem. 2004, 98, 1625–1633. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S.S.; Ojima, I. Mechanism-based tumor-targeting drug delivery system. Validation of efficient vitamin receptor-mediated endocytosis and drug release. Bioconjugate Chem. 2010, 21, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Tripodo, G.; Mandracchia, D.; Collina, S.; Rui, M.; Rossi, D. New perspectives in cancer therapy: The biotin-antitumor molecule conjugates. Med. Chem. 2014, 8, 1–4. [Google Scholar]
- Muhammad, N.; Sadia, N.; Zhu, C.; Luo, C.; Guo, Z.; Wang, X. Biotin-tagged platinum(IV) complexes as targeted cytostatic agents against breast cancer cells. Chem. Commun. 2017, 53, 9971–9974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, K.; Shettar, A.; Kondaiah, P.; Chakravarty, A.R. Biotinylated platinum(II) ferrocenylterpyridine complexes for targeted photoinduced cytotoxicity. Inorg. Chem. 2016, 55, 5612–5622. [Google Scholar] [CrossRef]
- Zhao, J.; Hua, W.; Xu, G.; Gou, S. Biotinylated platinum(IV) complexes designed to target cancer cells. J. Inorg. Biochem. 2017, 176, 175–180. [Google Scholar] [CrossRef]
- Ren, W.X.; Han, J.; Uhm, S.; Jang, Y.J.; Kang, C.; Kim, J.-H.; Kim, J.S. Recent development of biotin conjugation in biological imaging, sensing, and target delivery. Chem. Commun. 2015, 51, 10403–10418. [Google Scholar] [CrossRef] [PubMed]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Wright, J.R.A. Synthesis, characterisation and potent cytotoxicity of unconventional platinum (IV) complexes with modified lipophilicity. Dalton Trans. 2019, 48, 17217–17227. [Google Scholar] [CrossRef] [PubMed]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. Med. Chem. Commun. 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Scott, K.F.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Cyclooxygenase-inhibiting platinum(IV) prodrugs with potent anticancer activity. Pharmaceutics 2022, 14, 787. [Google Scholar] [CrossRef] [PubMed]
- Khoury, A.; Elias, E.; Mehanna, S.; Shebaby, W.; Deo, K.M.; Mansour, N.; Khalil, C.; Sayyed, K.; Sakoff, J.A.; Gilbert, J.; et al. Novel Platinum(II) and Platinum(IV) Antitumor Agents that Exhibit Potent Cytotoxicity and Selectivity. J. Med. Chem. 2022. [Google Scholar] [CrossRef]
- Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Potent Chlorambucil-Platinum (IV) Prodrugs. Int. J. Mol. Sci. 2022, 23, 10471. [Google Scholar] [CrossRef]
- Garbutcheon-Singh, K.B.; Leverett, P.; Myers, S.; Aldrich-Wright, J.R. Cytotoxic platinum(II) intercalators that incorporate 1 R, 2 R-diaminocyclopentane. Dalton Trans. 2013, 42, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Aldrich-Wright, J.R. Platinum intercalators of DNA as anticancer agents. Eur. J. Inorg. Chem. 2017, 2017, 1613–1624. [Google Scholar]
- Oldfield, S.P.; Hall, M.D.; Platts, J.A. Calculation of lipophilicity of a large, diverse dataset of anticancer platinum complexes and the relation to cellular uptake. J. Med. Chem. 2007, 50, 5227–5237. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | PL | Average GI50 (µM) | |
|---|---|---|---|
| 1 | 0.64 | Phen | 0.455 |
| 4 | 0.74 | 5MePhen | 0.055 |
| 2 | 0.78 | 56Me2Phen | 0.027 |
| 3 | 0.96 | 47O2Me2Phen | 0.459 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoury, A.; Sakoff, J.A.; Gilbert, J.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Potent Platinum(IV) Prodrugs That Incorporate a Biotin Moiety to Selectively Target Cancer Cells. Pharmaceutics 2022, 14, 2780. https://doi.org/10.3390/pharmaceutics14122780
Khoury A, Sakoff JA, Gilbert J, Karan S, Gordon CP, Aldrich-Wright JR. Potent Platinum(IV) Prodrugs That Incorporate a Biotin Moiety to Selectively Target Cancer Cells. Pharmaceutics. 2022; 14(12):2780. https://doi.org/10.3390/pharmaceutics14122780
Chicago/Turabian StyleKhoury, Aleen, Jennette A. Sakoff, Jayne Gilbert, Shawan Karan, Christopher P. Gordon, and Janice R. Aldrich-Wright. 2022. "Potent Platinum(IV) Prodrugs That Incorporate a Biotin Moiety to Selectively Target Cancer Cells" Pharmaceutics 14, no. 12: 2780. https://doi.org/10.3390/pharmaceutics14122780