
Torsemide Pharmacometrics in Healthy Adult Populations Including CYP2C9 Genetic Polymorphisms and Various Patient Groups through Physiologically Based Pharmacokinetic-Pharmacodynamic Modeling

Abstract
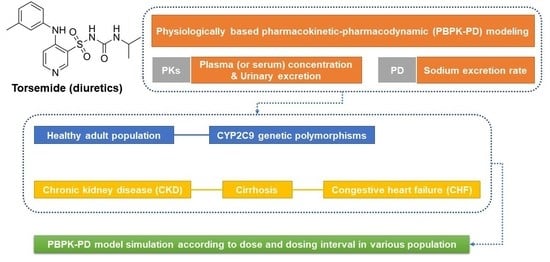
:
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Workflow of Torsemide PBPK-PD Modeling
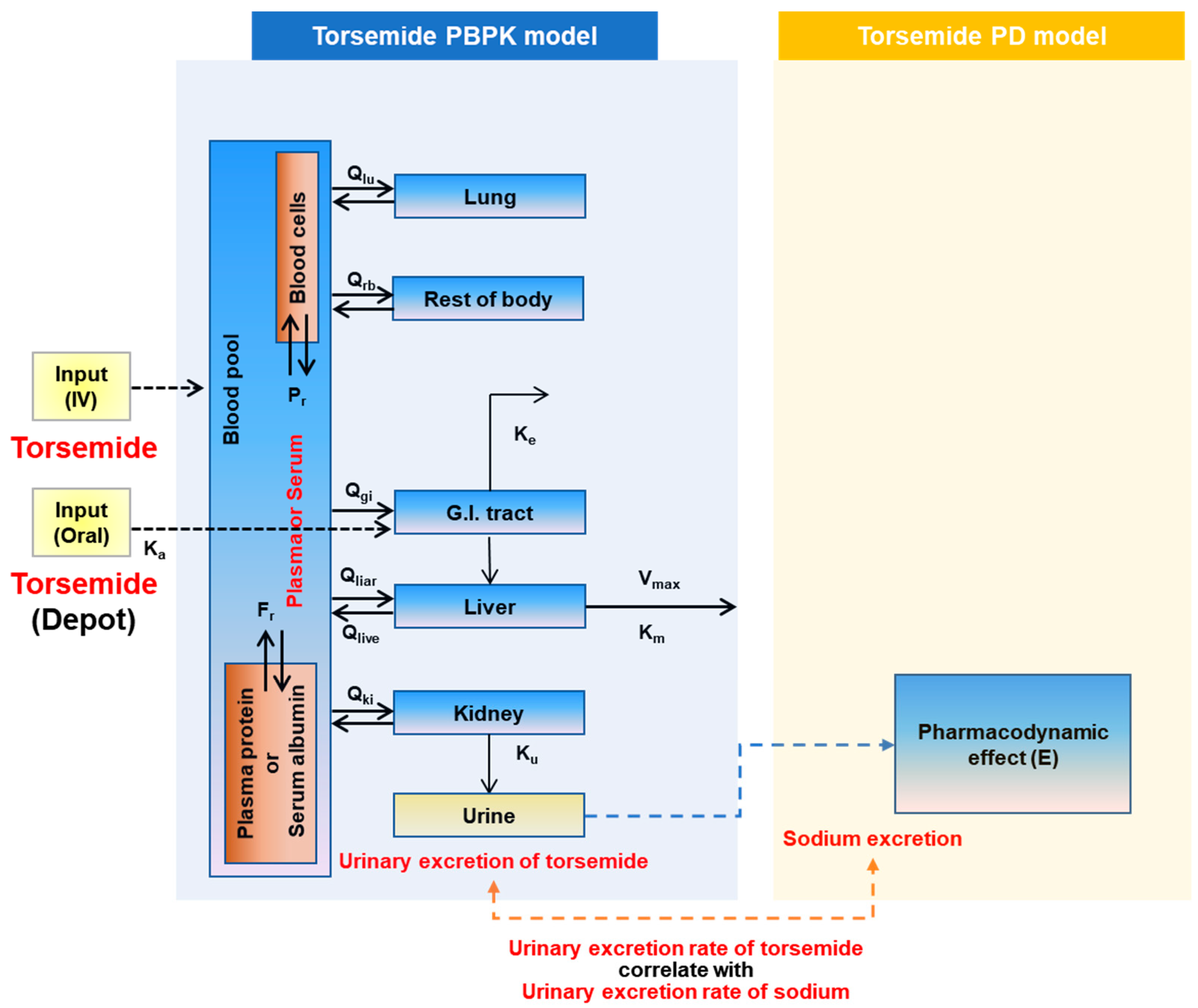
2.3. PBPK-PD Model Construction
2.4. PBPK-PD Model Parameterization
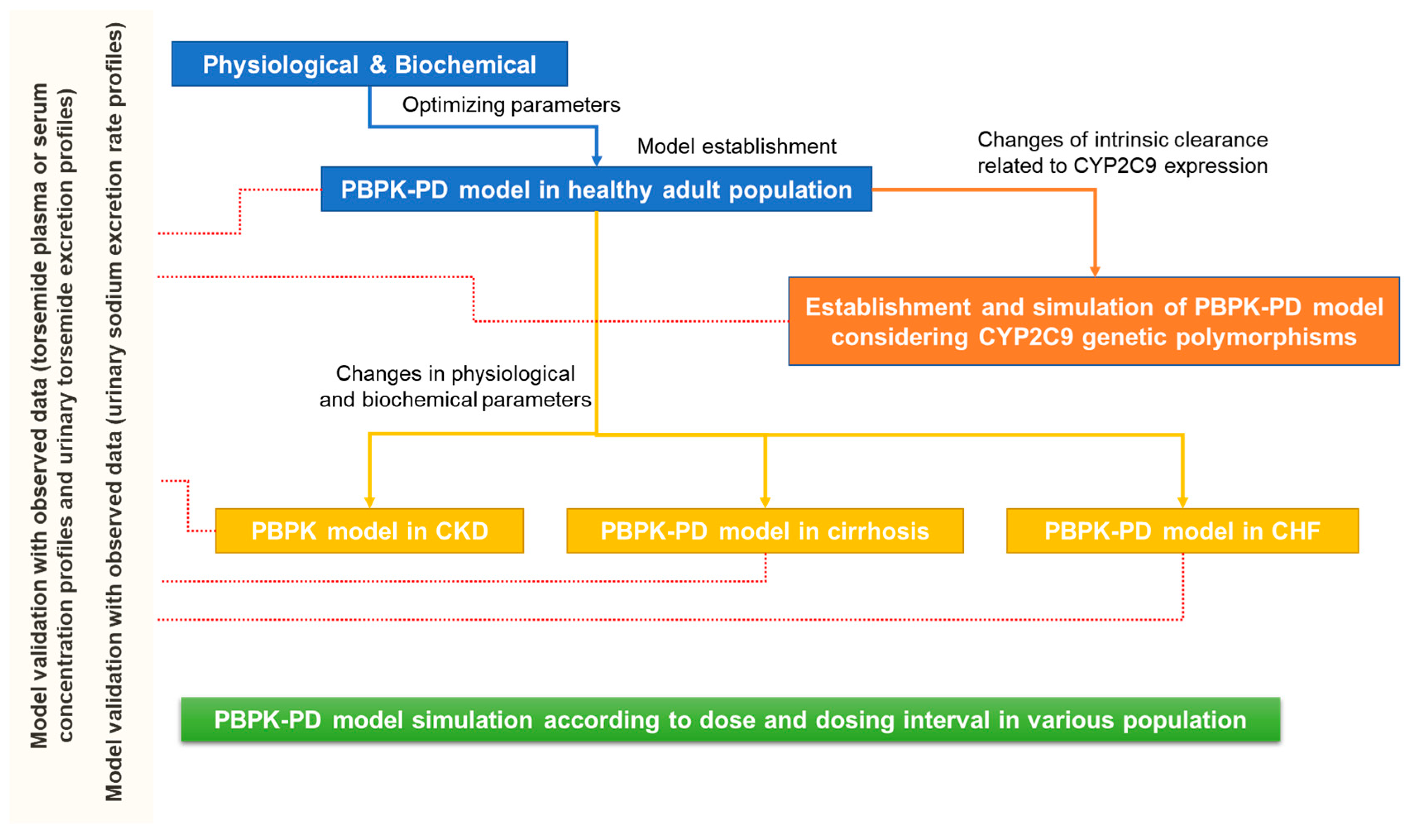
2.5. PBPK-PD Model Validation
2.6. PBPK-PD Model Application
2.6.1. PK and PD Simulations of Torsemide According to Different CYP2C9 Phenotypes
2.6.2. Prediction of Torsemide PKs and PDs in Cirrhosis Patients
2.6.3. Prediction of Torsemide PKs in CKD Patients
2.6.4. Prediction of Torsemide PKs and PDs in CHF Patients
3. Results and Discussion
3.1. Establishment of the Torsemide PD Model
3.2. PBPK-PD Modeling for Healthy Adult Populations
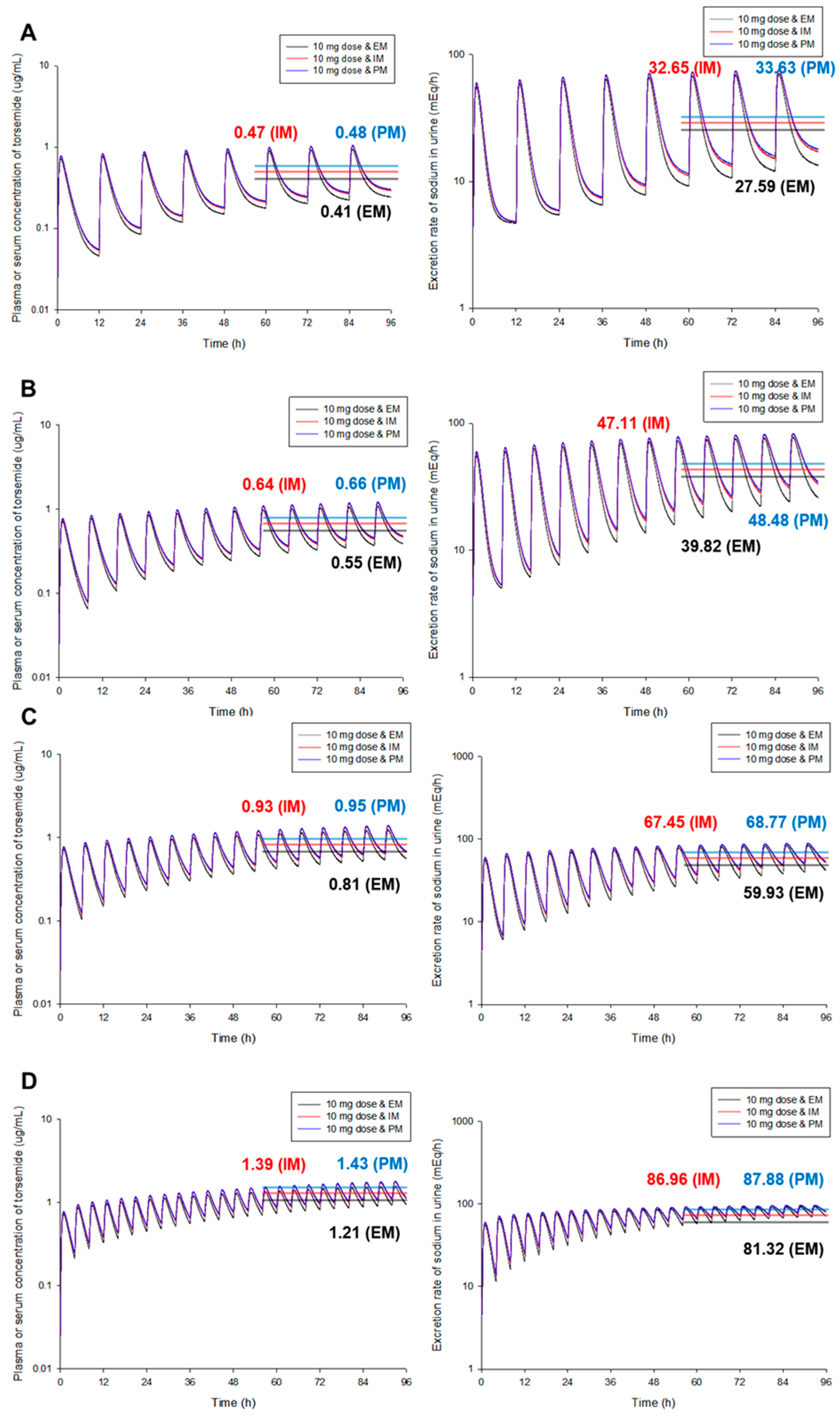
3.3. PBPK-PD Model Simulation According to Different CYP2C9 Phenotypes
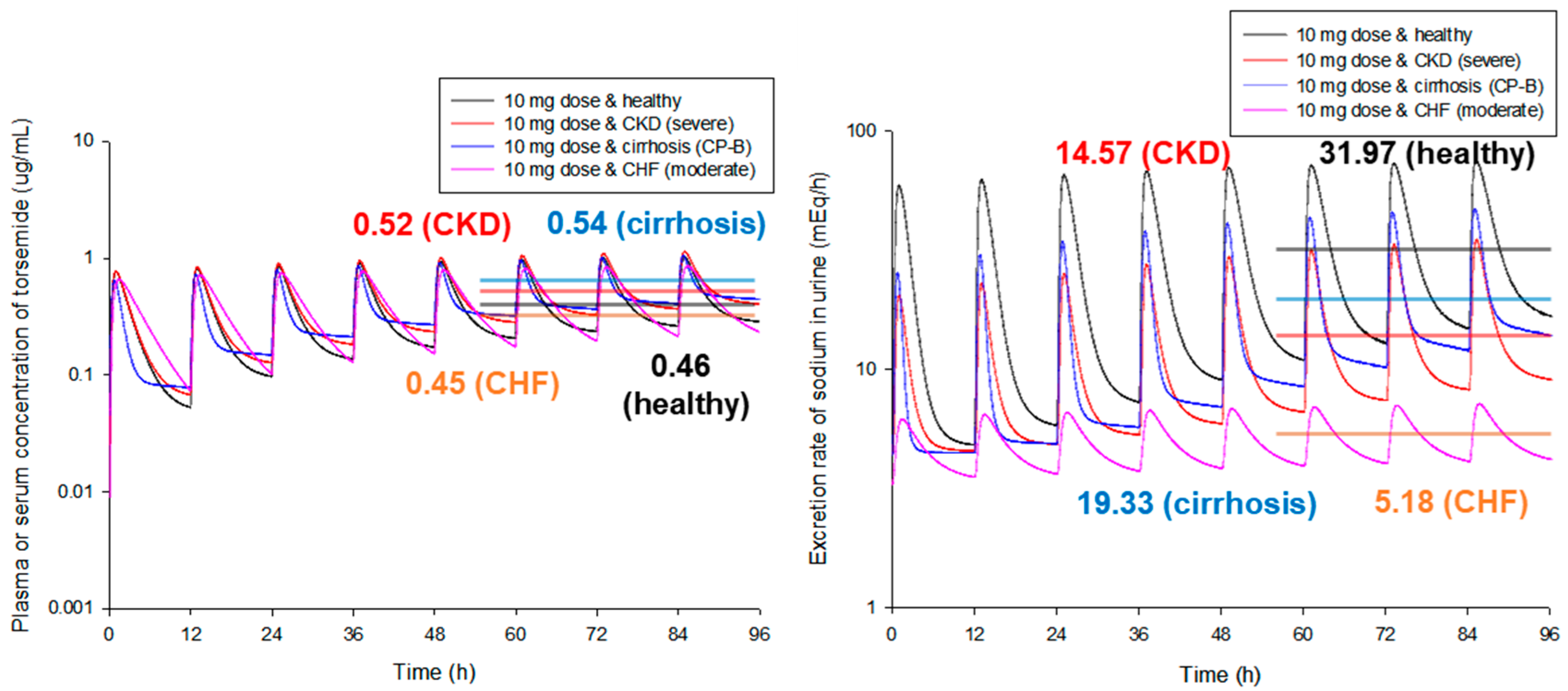
3.4. Simulation of the PBPK-PD Model for Cirrhosis Patient Groups
3.5. Simulation of the PBPK Model for CKD Patient Groups
3.6. Simulation of the PBPK-PD Model for CHF Patient Groups
3.7. Torsemide PK-PD Relationship
3.8. Model Validation
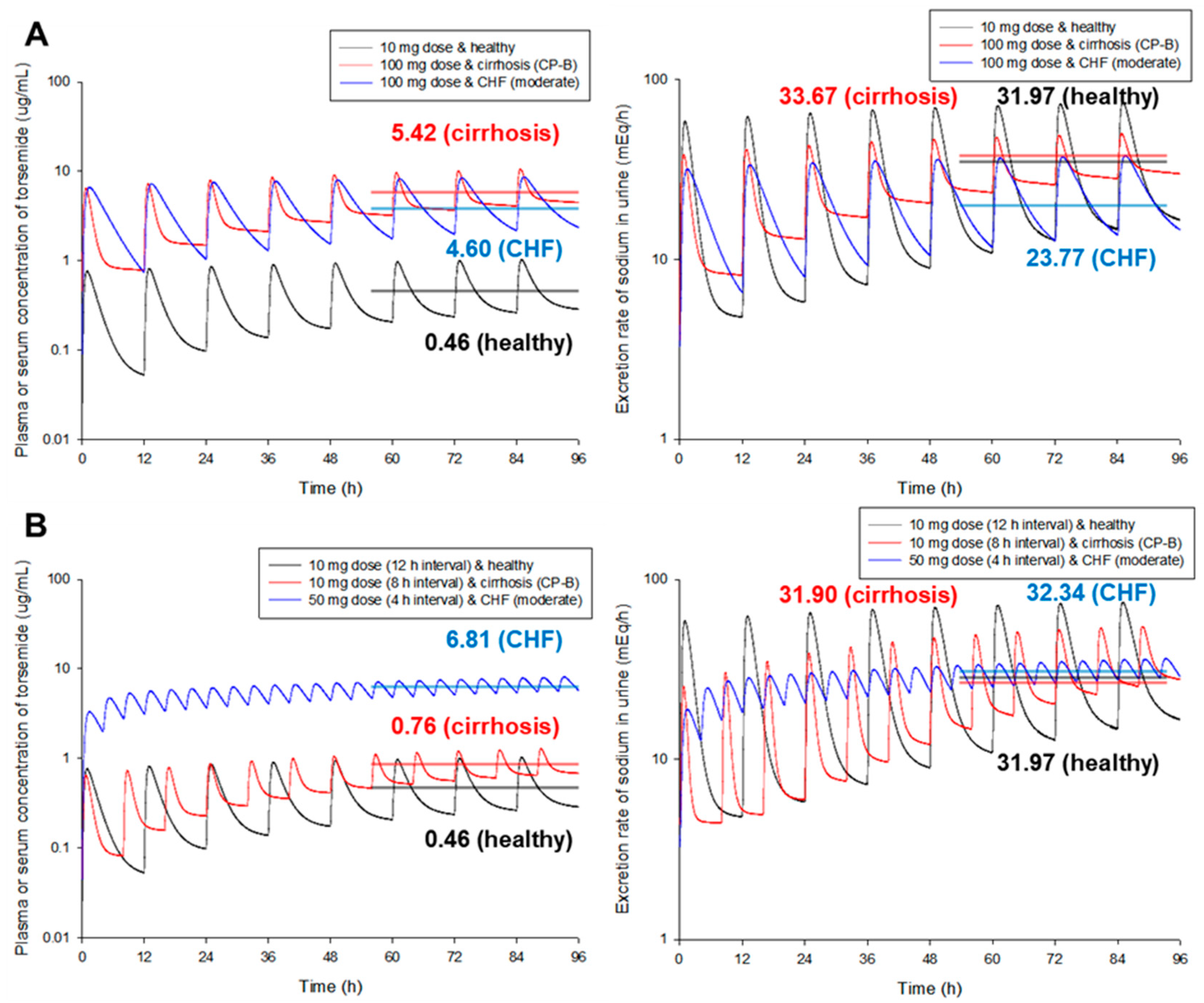
3.9. Model Simulation by Scenario for Various Groups
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
- Blood pool compartment:
- For intravenous injection:
- For the oral compartment:
- GI-tract compartment (oral dosing):
- Liver compartment:
- Kidney compartment:
- Urinary excretion compartment:
- Lung compartment:
- Rest of body compartment: including adipose, heart, spleen, muscle, etc.
References
- Knauf, H.; Mutschler, E. Clinical pharmacokinetics and pharmacodynamics of torasemide. Clin. Pharmacokinet. 1998, 34, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Z.I.; Raušl, D.; Radošević, S.; Filić, D.; Danilovski, A.; Dumić, M.; Knežević, Z. Classification of torasemide based on the Biopharmaceutics Classification System and evaluation of the FDA biowaiver provision for generic products of Class I drugs. J. Pharm. Pharmacol. 2006, 58, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Wittner, M.; Di Stefano, A.; Wangemann, P.; Greger, R. How do loop diuretics act? Drugs 1991, 41, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Rees, D.; Valente, L.; Veronese, M.E.; Birkett, D.J. Human hepatic cytochrome P450 2C9 catalyzes the rate-limiting pathway of torsemide metabolism. J. Pharmacol. Exp. Ther. 1995, 272, 1076–1081. [Google Scholar] [PubMed]
- Vormfelde, S.V.; Engelhardt, S.; Zirk, A.; Meineke, I.; Tuchen, F.; Kirchheiner, J.; Brockmöller, J. CYP2C9 polymorphisms and the interindividual variability in pharmacokinetics and pharmacodynamics of the loop diuretic drug torsemide. Clin. Pharmacol. Ther. 2004, 76, 557–566. [Google Scholar] [CrossRef]
- Miners, J.O.; Coulter, S.; Birkett, D.J.; Goldstein, J.A. Torsemide metabolism by CYP2C9 variants and other human CYP2C subfamily enzymes. Pharmacogenet. Genomics 2000, 10, 267–270. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jang, J.-H.; Cho, H.-Y.; Lee, Y.-B. Population pharmacokinetic (Pop-PK) analysis of torsemide in healthy Korean males considering CYP2C9 and OATP1B1 genetic polymorphisms. Pharmaceutics 2022, 14, 771. [Google Scholar] [CrossRef]
- Lee, D.Y.; Kim, J.Y.; Kim, Y.C.; Kwon, J.W.; Kim, W.B.; Lee, M.G. Dose-independent pharmacokinetics of torasemide after intravenous and oral administration to rats. Biopharm. Drug Dispos. 2005, 26, 173–182. [Google Scholar] [CrossRef]
- Kramer, W. Pharmacokinetics and pharmacodynamics of torasemide in congestive heart failure. Cardiology 1994, 84, 108–114. [Google Scholar] [CrossRef]
- Schwartz, S.; Brater, D.C.; Pound, D.; Green, P.K.; Kramer, W.G.; Rudy, D. Bioavailability, pharmacokinetics, and pharmacodynamics of torsemide in patients with cirrhosis. Clin. Pharmacol. Ther. 1993, 54, 90–97. [Google Scholar] [CrossRef]
- Vargo, D.L.; Kramer, W.G.; Black, P.K.; Smith, W.B.; Serpas, T.; Brater, D.C. Bioavailability, pharmacokinetics, and pharmacodynamics of torsemide and furosemide in patients with congestive heart failure. Clin. Pharmacol. Ther. 1995, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Bleske, B.E.; Welage, L.S.; Kramer, W.G.; Nicklas, J.M. Pharmacokinetics of torsemide in patients with decompensated and compensated congestive heart failure. J. Clin. Pharmacol. 1998, 38, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Vargo, D.; Kramer, W.G.; Black, P.K.; Smith, W.B.; Serpas, T.; Brater, D.C. The pharmacodynamics of torsemide in patients with congestive heart failure. Clin. Pharmacol. Ther. 1994, 56, 48–54. [Google Scholar] [PubMed]
- Rudy, D.W.; Gehr, T.W.; Matzke, G.R.; Kramer, W.G.; Sica, D.A.; Brater, D.C. The pharmacodynamics of intravenous and oral torsemide in patients with chronic renal insufficiency. Clin. Pharmacol. Ther. 1994, 56, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Hariman, R.J.; Bremner, S.; Louie, E.K.; Rogers, W.J.; Kostis, J.B.; Nocero, M.A.; Jones, J.P. Dose-response study of intravenous torsemide in congestive heart failure. Am. Heart J. 1994, 128, 352–357. [Google Scholar] [CrossRef]
- Barr, W.; Smith, H.; Karnes, H.; Sica, D.; Vetticaden, S.; Purich, E.; Prasad, V.; Schary, W.; Kramer, W.; Linberg, S. Comparison of bioavailability, pharmacokinetics and pharmacodynamics of torasemide in young and elderly healthy volunteers. Prog. Pharmacol. Clin. Pharmacol. 1990, 8, 15–28. [Google Scholar]
- Kang, H.-A.; Yoon, H.; Lee, Y.-B. Bioequivalence of Torad tablet 5 mg to Torem tablet 5 mg (torasemide 5 mg). J. Pharm. Investig. 2013, 43, 153–159. [Google Scholar] [CrossRef]
- Barbanoj, M.; Ballester, M.; Antonijoan, R.; Gich, I.; Pelagio, P.; Gropper, S.; Santos, B.; Guglietta, A. Comparison of repeated-dose pharmacokinetics of prolonged-release and immediate-release torasemide formulations in healthy young volunteers. Fundam. Clin. Pharmacol. 2009, 23, 115–125. [Google Scholar] [CrossRef]
- Shah, S.; Pitt, B.; Brater, D.C.; Feig, P.U.; Shen, W.; Khwaja, F.S.; Wilcox, C.S. Sodium and fluid excretion with torsemide in healthy subjects is limited by the short duration of diuretic action. J. Am. Heart Assoc. 2017, 6, e006135. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-Y.; Kang, H.-A.; Park, C.-H.; Kim, S.-M.; Kim, D.-H.; Park, S.; Kim, K.-R.; Hur, H.; Lee, Y.-B. Bioequivalence of Boryung torsemide tablet to Torem tablet (torasemide 10 mg) by high performance liquid chromatography/UV detector. J. Pharm. Investig. 2005, 35, 323–328. [Google Scholar]
- Spahn, H.; Knauf, H.; Mutschler, E. Pharmacokinetics of torasemide and its metabolites in healthy controls and in chronic renal failure. Eur. J. Clin. Pharmacol. 1990, 39, 345–348. [Google Scholar] [CrossRef]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied concepts in PBPK modeling: How to build a PBPK/PD model. Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef]
- Jameson, J.L.; Longo, D.L. Precision medicine—Personalized, problematic, and promising. Obstet. Gynecol. Surv. 2015, 70, 612–614. [Google Scholar] [CrossRef]
- Gehr, T.W.; Rudy, D.W.; Matzke, G.R.; Kramer, W.G.; Sica, D.A.; Brater, D.C. The pharmacokinetics of intravenous and oral torsemide in patients with chronic renal insufficiency. Clin. Pharmacol. Ther. 1994, 56, 31–38. [Google Scholar] [CrossRef]
- Ballester, M.R.; Roig, E.; Gich, I.; Puntes, M.; Delgadillo, J.; Santos, B.; Antonijoan, R.M. Randomized, open-label, blinded-endpoint, crossover, single-dose study to compare the pharmacodynamics of torasemide-PR 10 mg, torasemide-IR 10 mg, and furosemide-IR 40 mg, in patients with chronic heart failure. Drug Des. Dev. Ther. 2015, 9, 4291–4302. [Google Scholar] [CrossRef] [Green Version]
- Kramer, W.G.; Smith, W.B.; Ferguson, J.; Serpas, T.; Grant III, A.G.; Black, P.K.; Brater, D.C. Pharmacodynamics of torsemide administered as an intravenous injection and as a continuous infusion to patients with congestive heart failure. J. Clin. Pharmacol. 1996, 36, 265–270. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Igari, Y.; Sugiyama, Y.; Sawada, Y.; Iga, T.; Hanano, M. Prediction of diazepam disposition in the rat and man by a physiologically based pharmacokinetic model. J. Pharmacokinet. Biopharm. 1983, 11, 577–593. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jang, J.-H.; Cho, H.-Y.; Lee, Y.-B. Risk assessment for humans using physiologically based pharmacokinetic model of diethyl phthalate and its major metabolite, monoethyl phthalate. Arch. Toxicol. 2020, 94, 2377–2400. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jang, J.-H.; Cho, H.-Y.; Lee, Y.-B. Human risk assessment of di-isobutyl phthalate through the application of a developed physiologically based pharmacokinetic model of di-isobutyl phthalate and its major metabolite mono-isobutyl phthalate. Arch. Toxicol. 2021, 95, 2385–2402. [Google Scholar] [CrossRef]
- Thiel, C.; Schneckener, S.; Krauss, M.; Ghallab, A.; Hofmann, U.; Kanacher, T.; Zellmer, S.; Gebhardt, R.; Hengstler, J.G.; Kuepfer, L. A systematic evaluation of the use of physiologically based pharmacokinetic modeling for cross-species extrapolation. J. Pharm. Sci. 2015, 104, 191–206. [Google Scholar] [CrossRef]
- Khurshid, S.; Păsăreanu, C.S.; Visser, W. Generalized symbolic execution for model checking and testing. In Proceedings of the 9th International Conference on Tools and Algorithms for the Construction and Analysis of Systems, Warsaw, Poland, 7–11 April 2003; pp. 553–568. [Google Scholar]
- Kirchheiner, J.; Tsahuridu, M.; Jabrane, W.; Roots, I.; Brockmöller, J. The CYP2C9 polymorphism: From enzyme kinetics to clinical dose recommendations. Pers. Med. 2004, 1, 63–84. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Kang, P.; Cho, C.K.; Jung, E.H.; Park, H.-J.; Lee, Y.J.; Bae, J.W.; Jang, C.-G.; Lee, S.-Y. Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch. Pharm. Res. 2021, 44, 713–724. [Google Scholar] [CrossRef]
- Cho, C.K.; Park, H.-J.; Kang, P.; Moon, S.; Lee, Y.J.; Bae, J.W.; Jang, C.-G.; Lee, S.-Y. Physiologically based pharmacokinetic (PBPK) modeling of meloxicam in different CYP2C9 genotypes. Arch. Pharm. Res. 2021, 44, 1076–1090. [Google Scholar] [CrossRef]
- Elbekai, R.H.; Korashy, H.M.; El-Kadi, A.O. The effect of liver cirrhosis on the regulation and expression of drug metabolizing enzymes. Curr. Drug Metab. 2004, 5, 157–167. [Google Scholar] [CrossRef]
- Chen, Y.; Ke, M.; Xu, J.; Lin, C. Simulation of the pharmacokinetics of oseltamivir and its active metabolite in normal populations and patients with hepatic cirrhosis using physiologically based pharmacokinetic modeling. AAPS PharmSciTech 2020, 21, 98. [Google Scholar] [CrossRef]
- Rasool, M.F.; Khalil, F.; Läer, S. Optimizing the clinical use of carvedilol in liver cirrhosis using a physiologically based pharmacokinetic modeling approach. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 383–396. [Google Scholar] [CrossRef]
- Kalam, M.N.; Rasool, M.F.; Alqahtani, F.; Imran, I.; Rehman, A.U.; Ahmed, N. Development and evaluation of a physiologically based pharmacokinetic drug-disease model of propranolol for suggesting model informed dosing in liver cirrhosis patients. Drug Des. Devel. Ther. 2021, 15, 1195–1211. [Google Scholar] [CrossRef]
- Johnson, T.N.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.T.; Rostami-Hodjegan, A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 2010, 49, 189–206. [Google Scholar] [CrossRef]
- Barry, M.; Keeling, P.W.; Weir, D.; Feely, J. Severity of cirrhosis and the relationship of α1-acid glycoprotein concentration to plasma protein binding of lidocaine. Clin. Pharmacol. Ther. 1990, 47, 366–370. [Google Scholar] [CrossRef]
- Albers, I.; Hartmann, H.; Bircher, J.; Creutzfeldt, W. Superiority of the Child-Pugh classification to quantitative liver function tests for assessing prognosis of liver cirrhosis. Scand. J. Gastroenterol. 1989, 24, 269–276. [Google Scholar] [CrossRef]
- Li, G.-F.; Yu, G.; Li, Y.; Zheng, Y.; Zheng, Q.-S.; Derendorf, H. Quantitative estimation of plasma free drug fraction in patients with varying degrees of hepatic impairment: A methodological evaluation. J. Pharm. Sci. 2018, 107, 1948–1956. [Google Scholar] [CrossRef]
- Suchy-Dicey, A.M.; Laha, T.; Hoofnagle, A.; Newitt, R.; Sirich, T.L.; Meyer, T.W.; Thummel, K.E.; Yanez, N.D.; Himmelfarb, J.; Weiss, N.S. Tubular secretion in CKD. J. Am. Soc. Nephrol. 2016, 27, 2148–2155. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.L.; Zhao, P.; Zhang, L.; Ho, Y.F.; Varma, M.V.; Neuhoff, S.; Nolin, T.D.; Galetin, A.; Huang, S.M. Use of physiologically based pharmacokinetic modeling to evaluate the effect of chronic kidney disease on the disposition of hepatic CYP 2C8 and OATP 1B drug substrates. Clin. Pharmacol. Ther. 2019, 105, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, C.H.; Hsu, V.; Zhao, P.; Zhang, L.; Giacomini, K.; Huang, S.M. PBPK modeling of the effect of reduced kidney function on the pharmacokinetics of drugs excreted renally by organic anion transporters. Clin. Pharmacol. Ther. 2018, 103, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-F.; Wang, K.; Chen, R.; Zhao, H.-R.; Yang, J.; Zheng, Q.-S. Simulation of the pharmacokinetics of bisoprolol in healthy adults and patients with impaired renal function using whole-body physiologically based pharmacokinetic modeling. Acta Pharmacol. Sin. 2012, 33, 1359–1371. [Google Scholar] [CrossRef] [Green Version]
- Decreased, G. Definition and classification of CKD. Kidney Int. 2013, 3, 19–62. [Google Scholar]
- Samsky, M.D.; Patel, C.B.; DeWald, T.A.; Smith, A.D.; Felker, G.M.; Rogers, J.G.; Hernandez, A.F. Cardiohepatic interactions in heart failure: An overview and clinical implications. J. Am. Coll. Cardiol. 2013, 61, 2397–2405. [Google Scholar] [CrossRef] [Green Version]
- Rasool, M.F.; Khalil, F.; Läer, S. A physiologically based pharmacokinetic drug-disease model to predict carvedilol exposure in adult and Paediatric heart failure patients by incorporating pathophysiological changes in hepatic and renal blood flows. Clin. Pharmacokinet. 2015, 54, 943–962. [Google Scholar] [CrossRef] [Green Version]
- Rasool, M.F.; Khalil, F.; Läer, S. Predicting stereoselective disposition of carvedilol in adult and pediatric chronic heart failure patients by incorporating pathophysiological changes in organ blood flows–A physiologically based pharmacokinetic approach. Drug Metab. Dispos. 2016, 44, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Rostagno, C.; Galanti, G.; Comeglio, M.; Boddi, V.; Olivo, G.; Serneri, G.G.N. Comparison of different methods of functional evaluation in patients with chronic heart failure. Eur. J. Heart Fail. 2000, 2, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepner, G.W.; Vesell, E.S.; Tantum, K.R. Reduced drug elimination in congestive heart failure: Studies using aminopyrine as a model drug. Am. J. Med. 1978, 65, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-llama, P.; Turner, R.; Dibona, G.; Knepper, M.A. Renal expression of aquaporins in liver cirrhosis induced by chronic common bile duct ligation in rats. J. Am. Soc. Nephrol. 1999, 10, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.S.; Brater, D.C. Loop diuretics: From the Na-K-2Cl transporter to clinical use. Am. J. Physiol. Renal Physiol. 2003, 284, F11–F21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, M.; Matsuoka, H.; Miyazaki, H.; Ueda, S.; Okuda, S.; Imaizumi, T. Increased endogenous nitric oxide synthase inhibitor in patients with congestive heart failure. Life Sci. 1998, 62, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Rodighiero, V. Effects of liver disease on pharmacokinetics. Clin. Pharmacokinet. 1999, 37, 399–431. [Google Scholar] [CrossRef] [PubMed]
- Gerner, B.; Scherf-Clavel, O. Physiologically based pharmacokinetic modelling of cabozantinib to simulate enterohepatic recirculation, drug-drug interaction with rifampin and liver impairment. Pharmaceutics 2021, 13, 778. [Google Scholar] [CrossRef]
- Hammarlund-Udenaes, M.; Benet, L.Z. Furosemide pharmacokinetics and pharmacodynamics in health and disease—An update. J. Pharmacokinet. Biopharm. 1989, 17, 1–46. [Google Scholar] [CrossRef]
- Jusko, W.J.; Ko, H.C. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin. Pharmacol. Ther. 1994, 56, 406–419. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Parameters | Value | Unit | Source |
|---|---|---|---|---|
| Maximum metabolic rate constant for EM | Vmax, EM | 2.216 | μg/mL/h | Estimated * |
| Michaelis-Menten constant for EM | Km, EM | 1.951 | μg/mL | Estimated * |
| Maximum metabolic rate constant for IM | Vmax, IM | 0.886 | μg/mL/h | Estimated * |
| Michaelis-Menten constant for IM | Km, IM | 5.073 | μg/mL | Estimated * |
| Maximum metabolic rate constant for PM | Vmax, PM | 0.222 | μg/mL/h | Estimated * |
| Michaelis-Menten constant for PM | Km, PM | 8.242 | μg/mL | Estimated * |
| Parameters | Control | Child-Pugh (CP) Class | ||
|---|---|---|---|---|
| A | B | C | ||
| Liver volume fraction * | 1 | 0.81 | 0.65 | 0.53 |
| Cardiac output fraction * | 1 | 1.16 | 1.32 | 1.41 |
| Kidney blood flow rate fraction * | 1 | 0.70 | 0.58 | 0.55 |
| Portal blood flow rate fraction * | 1 | 0.91 | 0.63 | 0.55 |
| Hepatic arterial blood flow rate fraction * | 1 | 1.41 | 1.62 | 1.91 |
| Villous blood flow rate fraction * | 1 | 1.29 | 1.52 | 1.99 |
| Hematocrit (%) ** | 43 | 38.46 | 34.51 | 33.51 |
| Albumin & α1-acid glycoprotein (g/L) ** | 50.34 | 46.33 | 38.18 | 29.69 |
| CYP2C9 (pmol/mg) * | 73 | 50.4 | 38.0 | 24.1 |
| Parameters | Control | CKD Class | ||
|---|---|---|---|---|
| Mild | Moderate | Severe | ||
| Kidney volume fraction * | 1 | 0.99 | 0.93 | 0.87 |
| Kidney blood flow rate fraction * | 1 | 0.99 | 0.86 | 0.72 |
| Liver intrinsic clearance fraction * | 1 | 1.00 | 0.69 | 0.52 |
| Renal elimination rate fraction * | 1 | 0.99 | 0.68 | 0.37 |
| Parameters | Control | CHF Class | ||
|---|---|---|---|---|
| Mild | Moderate | Severe | ||
| Cardiac output fraction * | 1 | 0.69 | 0.50 | 0.42 |
| Kidney blood flow rate fraction * | 1 | 0.78 | 0.55 | 0.63 |
| Portal blood flow rate fraction * | 1 | 0.76 | 0.54 | 0.46 |
| Hepatic arterial blood flow rate fraction * | 1 | 0.76 | 0.54 | 0.46 |
| Other tissues blood flow rate fraction * | 1 | 0.57 | 0.44 | 0.28 |
| Liver intrinsic clearance fraction * | 1 | 0.76 | 0.54 | 0.46 |
| Baseline Sigmoid Emax Model | Populations | ||
|---|---|---|---|
| Healthy * | Cirrhosis ** | CHF *** | |
| Equation | |||
| Parameters | |||
| E0 (mEq/h) | 4.39 | 4.33 | 3.25 |
| Emax (mEq/h) | 105.1 | 65.24 | 83.45 |
| EC50 (μg/h) | 345.5 | 388.8 | 3985 |
| P | −2.13 | −2.82 | −1.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.-H.; Jang, J.-H.; Lee, Y.-B. Torsemide Pharmacometrics in Healthy Adult Populations Including CYP2C9 Genetic Polymorphisms and Various Patient Groups through Physiologically Based Pharmacokinetic-Pharmacodynamic Modeling. Pharmaceutics 2022, 14, 2720. https://doi.org/10.3390/pharmaceutics14122720
Jeong S-H, Jang J-H, Lee Y-B. Torsemide Pharmacometrics in Healthy Adult Populations Including CYP2C9 Genetic Polymorphisms and Various Patient Groups through Physiologically Based Pharmacokinetic-Pharmacodynamic Modeling. Pharmaceutics. 2022; 14(12):2720. https://doi.org/10.3390/pharmaceutics14122720
Chicago/Turabian StyleJeong, Seung-Hyun, Ji-Hun Jang, and Yong-Bok Lee. 2022. "Torsemide Pharmacometrics in Healthy Adult Populations Including CYP2C9 Genetic Polymorphisms and Various Patient Groups through Physiologically Based Pharmacokinetic-Pharmacodynamic Modeling" Pharmaceutics 14, no. 12: 2720. https://doi.org/10.3390/pharmaceutics14122720