
Airway Epithelial Cell Junctions as Targets for Pathogens and Antimicrobial Therapy

Abstract
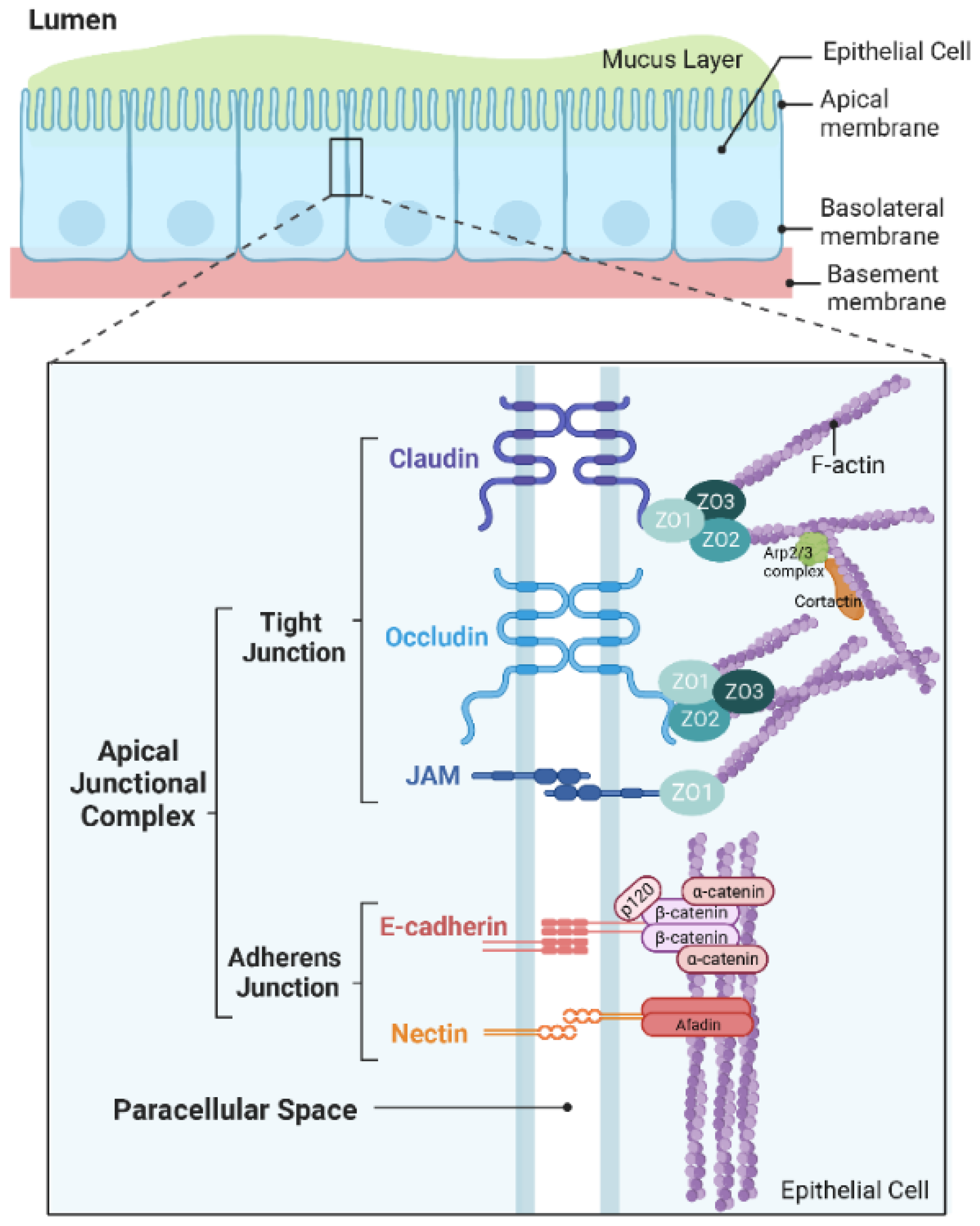
:1. Introduction
2. Bacteria
2.1. Staphylococcus aureus
2.2. Streptococcus pneumonia
2.3. Pseudomonas aeruginosa
2.4. Burkholderia
2.5. Haemophilus Influenzae
3. Viruses
3.1. Respiratory Syncytial Virus
3.2. Human Rhinovirus
3.3. Influenza Viruses
3.4. Human Parainfluenza Virus
3.5. Coronaviruses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 16HBE | 16HBE14o- human bronchial epithelial |
| ADAM10 | metalloproteinase domain-containing protein 10 |
| AECs | airway epithelial cells |
| ALI | air–liquid interface |
| AJ | adherens junction |
| AJC | apical junctional complex |
| BSA | bovine serum albumin |
| cAMP | cyclic adenosine monophosphate |
| CAR | coxsackie and adenovirus receptor |
| CDC | Centers for Disease Control and Prevention |
| CF | cystic fibrosis |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| COPD | chronic obstructive pulmonary disease |
| COVID-19 | Coronavirus disease 2019 |
| FAK | focal adhesion kinase |
| FITC | fluorescein isothiocyanate |
| HBECs | human bronchial epithelial cells |
| HNECs | human nasal epithelial cells |
| HPIV | human parainfluenza virus |
| HRV | human rhinovirus |
| IAV | influenza A virus |
| ICU | intensive care unit |
| IFN | interferon |
| JAM | junctional adhesion molecule |
| MAPK | mitogen-activated protein kinase |
| MDCK | Madin–Darby canine kidney |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MRSA | methicillin-resistant Staphylococcus aureus |
| NHBE | normal human bronchial epithelial |
| NOX | NADPH oxidase |
| PALS1 | Proteins Associated with Lin Seven 1 |
| PI3K | phosphoinositide 3-kinase |
| PKD | protein kinase D |
| PTK | Protein–tyrosine kinase |
| ROS | reactive oxygen species |
| RSV | respiratory syncytial virus |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| Src | steroid receptor coactivator |
| TEER | trans-epithelial electrical resistance |
| TLR | Toll-like receptor |
| TJ | tight junction |
| WHO | World Health Organization |
| ZO | zonula occludens |
References
- Linfield, D.T.; Raduka, A.; Aghapour, M.; Rezaee, F. Airway tight junctions as targets of viral infections. Tissue Barriers 2021, 9, 1883965. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; Georas, S.N. Breaking barriers. New insights into airway epithelial barrier function in health and disease. Am. J. Respir. Cell Mol. Biol. 2014, 50, 857–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.D.; Wypych, T.P. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 2021, 14, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.A.; Nelson, W.J.; Chavez, N. Cell-Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb. Perspect. Biol. 2018, 10, a029181. [Google Scholar] [CrossRef] [Green Version]
- Rusu, A.D.; Georgiou, M. The multifarious regulation of the apical junctional complex. Open Biol. 2020, 10, 190278. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Fanning, A.S.; Bridges, A.; Anderson, J.M. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 2009, 20, 3930–3940. [Google Scholar] [CrossRef]
- Fanning, A.S.; Van Itallie, C.M.; Anderson, J.M. Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol. Biol. Cell 2012, 23, 577–590. [Google Scholar] [CrossRef]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The Tight Junction Protein ZO-1 Establishes a Link between the Transmembrane Protein Occludin and the Actin Cytoskeleton*. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [Green Version]
- Wittchen, E.S.; Haskins, J.; Stevenson, B.R. Protein Interactions at the Tight Junction: Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 AND ZO-3*. J. Biol. Chem. 1999, 274, 35179–35185. [Google Scholar] [CrossRef] [Green Version]
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef]
- Smallcombe, C.C.; Linfield, D.T.; Harford, T.J.; Bokun, V.; Ivanov, A.I.; Piedimonte, G.; Rezaee, F. Disruption of the airway epithelial barrier in a murine model of respiratory syncytial virus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L358–L368. [Google Scholar] [CrossRef]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Pharo, E.A.; Williams, S.M.; Boyd, V.; Sundaramoorthy, V.; Durr, P.A.; Baker, M.L. Host–Pathogen Responses to Pandemic Influenza H1N1pdm09 in a Human Respiratory Airway Model. Viruses 2020, 12, 679. [Google Scholar] [CrossRef]
- Rezaee, F.; DeSando, S.A.; Ivanov, A.I.; Chapman, T.J.; Knowlden, S.A.; Beck, L.A.; Georas, S.N. Sustained Protein Kinase D Activation Mediates Respiratory Syncytial Virus-Induced Airway Barrier Disruption. J. Virol. 2013, 87, 11088–11095. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-C.; Shi, Y.-F.; Yang, J.-J.; Hsiao, Y.-C.; Tzang, B.-S.; Hsu, T.-C. Effects of Human Parvovirus B19 and Bocavirus VP1 Unique Region on Tight Junction of Human Airway Epithelial A549 Cells. PLoS ONE 2014, 9, e107970. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Häcker, H.; Chi, L.; Tuomanen, E.; Redecke, V. Type I Interferon Protects against Pneumococcal Invasive Disease by Inhibiting Bacterial Transmigration across the Lung. PLoS Pathog. 2013, 9, e1003727. [Google Scholar] [CrossRef]
- Martens, K.; Seys, S.F.; Alpizar, Y.A.; Schrijvers, R.; Bullens, D.M.A.; Breynaert, C.; Lebeer, S.; Steelant, B. Staphylococcus aureus enterotoxin B disrupts nasal epithelial barrier integrity. Clin. Exp. Allergy 2021, 51, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, A.P.; Orihuela, C.J.; Bogaert, D. Bacterial-Host Interactions: Physiology and Pathophysiology of Respiratory Infection. Physiol. Rev. 2018, 98, 781–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisele, N.A.; Anderson, D.M. Host Defense and the Airway Epithelium: Frontline Responses That Protect against Bacterial Invasion and Pneumonia. J. Pathog. 2011, 2011, 249802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maginnis, M.S. Virus-Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef] [PubMed]
- Torres-Flores, J.M.; Arias, C.F. Tight Junctions Go Viral! Viruses 2015, 7, 5145–5154. [Google Scholar] [CrossRef]
- Malik, Z.; Roscioli, E.; Murphy, J.; Ou, J.; Bassiouni, A.; Wormald, P.-J.; Vreugde, S. Staphylococcus aureus impairs the airway epithelial barrier in vitro. Int. Forum Allergy Rhinol. 2015, 5, 551–556. [Google Scholar] [CrossRef]
- Murphy, J.; Ramezanpour, M.; Stach, N.; Dubin, G.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Staphylococcus aureus V8 protease disrupts the integrity of the airway epithelial barrier and impairs IL-6 production in vitro. Laryngoscope 2018, 128, E8–E15. [Google Scholar] [CrossRef]
- Kalsi, K.K.; Garnett, J.P.; Patkee, W.; Weekes, A.; Dockrell, M.E.; Baker, E.H.; Baines, D.L. Metformin attenuates the effect of Staphylococcus aureus on airway tight junctions by increasing PKCζ-mediated phosphorylation of occludin. J. Cell. Mol. Med. 2019, 23, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Wardenburg, J.B. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [Green Version]
- Wilke, G.A.; Wardenburg, J.B. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef]
- Ziesemer, S.; Eiffler, I.; Schönberg, A.; Müller, C.; Hochgräfe, F.; Beule, A.G.; Hildebrandt, J.P. Staphylococcus aureus α-Toxin Induces Actin Filament Remodeling in Human Airway Epithelial Model Cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 482–491. [Google Scholar] [CrossRef]
- Clarke, T.B.; Francella, N.; Huegel, A.; Weiser, J.N. Invasive Bacterial Pathogens Exploit TLR-Mediated Downregulation of Tight Junction Components to Facilitate Translocation across the Epithelium. Cell Host Microbe 2011, 9, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Rayner, C.F.; Jackson, A.D.; Rutman, A.; Dewar, A.; Mitchell, T.J.; Andrew, P.W.; Cole, P.J.; Wilson, R. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect. Immun. 1995, 63, 442–447. [Google Scholar] [CrossRef] [Green Version]
- Mo, D.; Xu, S.; Rosa, J.P.; Hasan, S.; Adams, W. Dynamic Python-Based Method Provides Quantitative Analysis of Intercellular Junction Organization During S. pneumoniae Infection of the Respiratory Epithelium. Front. Cell. Infect. Microbiol. 2022, 12, 865528. [Google Scholar] [CrossRef]
- Peter, A.; Fatykhova, D.; Kershaw, O.; Gruber, A.D.; Rueckert, J.; Neudecker, J.; Toennies, M.; Bauer, T.T.; Schneider, P.; Schimek, M.; et al. Localization and pneumococcal alteration of junction proteins in the human alveolar-capillary compartment. Histochem. Cell Biol. 2017, 147, 707–719. [Google Scholar] [CrossRef]
- Attali, C.; Durmort, C.; Vernet, T.; Di Guilmi, A.M. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect. Immun. 2008, 76, 5350–5356. [Google Scholar] [CrossRef] [Green Version]
- Patkee, W.R.; Carr, G.; Baker, E.H.; Baines, D.L.; Garnett, J.P. Metformin prevents the effects of Pseudomonas aeruginosa on airway epithelial tight junctions and restricts hyperglycaemia-induced bacterial growth. J. Cell. Mol. Med. 2016, 20, 758–764. [Google Scholar] [CrossRef] [Green Version]
- Rejman, J.; Di Gioia, S.; Bragonzi, A.; Conese, M. Pseudomonas aeruginosa infection destroys the barrier function of lung epithelium and enhances polyplex-mediated transfection. Hum. Gene Ther. 2007, 18, 642–652. [Google Scholar] [CrossRef]
- Nomura, K.; Obata, K.; Keira, T.; Miyata, R.; Hirakawa, S.; Takano, K.-i.; Kohno, T.; Sawada, N.; Himi, T.; Kojima, T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir. Res. 2014, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.A.; Thomas, L.K.; Azghani, A.O. Inhibition of protein kinase C attenuates Pseudomonas aeruginosa elastase-induced epithelial barrier disruption. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1263–1271. [Google Scholar] [CrossRef]
- Zulianello, L.; Canard, C.; Kohler, T.; Caille, D.; Lacroix, J.S.; Meda, P. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, G.; Parker, D.; Magargee, M.; Prince, A.S. The Type III Toxins of Pseudomonas aeruginosa Disrupt Epithelial Barrier Function. J. Bacteriol. 2008, 190, 2814–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Sajjan, U.S.; Krasan, G.P.; LiPuma, J.J. Disruption of tight junctions during traversal of the respiratory epithelium by Burkholderia cenocepacia. Infect. Immun. 2005, 73, 7107–7112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjan, U.; Corey, M.; Humar, A.; Tullis, E.; Cutz, E.; Ackerley, C.; Forstner, J. Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J. Med. Microbiol. 2001, 50, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Schwab, U.; Leigh, M.; Ribeiro, C.; Yankaskas, J.; Burns, K.; Gilligan, P.; Sokol, P.; Boucher, R. Patterns of epithelial cell invasion by different species of the Burkholderia cepacia complex in well-differentiated human airway epithelia. Infect. Immun. 2002, 70, 4547–4555. [Google Scholar] [CrossRef] [Green Version]
- Duff, C.; Murphy, P.G.; Callaghan, M.; McClean, S. Differences in invasion and translocation of Burkholderia cepacia complex species in polarised lung epithelial cells in vitro. Microb. Pathog. 2006, 41, 183–192. [Google Scholar] [CrossRef]
- Blume, C.; David, J.; Bell, R.E.; Laver, J.R.; Read, R.C.; Clark, G.C.; Davies, D.E.; Swindle, E.J. Modulation of Human Airway Barrier Functions during Burkholderia thailandensis and Francisella tularensis Infection Running Title: Airway Barrier Functions during Bacterial Infections. Pathogens 2016, 5, 53. [Google Scholar] [CrossRef]
- Bevivino, A.; Pirone, L.; Pilkington, R.; Cifani, N.; Dalmastri, C.; Callaghan, M.; Ascenzioni, F.; McClean, S. Interaction of environmental Burkholderia cenocepacia strains with cystic fibrosis and non-cystic fibrosis bronchial epithelial cells in vitro. Microbiology 2012, 158, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.S.; Silva, I.N.; Fernandes, F.; Pilkington, R.; Callaghan, M.; McClean, S.; Moreira, L.M. The tyrosine kinase BceF and the phosphotyrosine phosphatase BceD of Burkholderia contaminans are required for efficient invasion and epithelial disruption of a cystic fibrosis lung epithelial cell line. Infect. Immun. 2015, 83, 812–821. [Google Scholar] [CrossRef] [Green Version]
- Kaufhold, I.; Osbahr, S.; Shima, K.; Marwitz, S.; Rohmann, K.; Dromann, D.; Goldmann, T.; Dalhoff, K.; Rupp, J. Nontypeable Haemophilus influenzae (NTHi) directly interfere with the regulation of E-cadherin in lung epithelial cells. Microbes Infect. 2017, 19, 560–566. [Google Scholar] [CrossRef]
- Rezaee, F.; Harford, T.J.; Linfield, D.T.; Altawallbeh, G.; Midura, R.J.; Ivanov, A.I.; Piedimonte, G. cAMP-dependent activation of protein kinase A attenuates respiratory syncytial virus-induced human airway epithelial barrier disruption. PLoS ONE 2017, 12, e0181876. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; McCann, K.L.; Imani, F. MAPK and heat shock protein 27 activation are associated with respiratory syncytial virus induction of human bronchial epithelial monolayer disruption. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L436–L445. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Raduka, A.; Rezaee, F. Respiratory syncytial virus disrupts the airway epithelial barrier by decreasing cortactin and destabilizing F-actin. J. Cell Sci. 2022, 135, jcs259871. [Google Scholar] [CrossRef]
- Kast, J.I.; McFarlane, A.J.; Głobińska, A.; Sokolowska, M.; Wawrzyniak, P.; Sanak, M.; Schwarze, J.; Akdis, C.A.; Wanke, K. Respiratory syncytial virus infection influences tight junction integrity. Clin. Exp. Immunol. 2017, 190, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Masaki, T.; Kojima, T.; Okabayashi, T.; Ogasawara, N.; Ohkuni, T.; Obata, K.; Takasawa, A.; Murata, M.; Tanaka, S.; Hirakawa, S.; et al. A nuclear factor-κB signaling pathway via protein kinase C δ regulates replication of respiratory syncytial virus in polarized normal human nasal epithelial cells. Mol. Biol. Cell 2011, 22, 2144–2156. [Google Scholar] [CrossRef]
- Kilani, M.M.; Mohammed, K.A.; Nasreen, N.; Hardwick, J.A.; Kaplan, M.H.; Tepper, R.S.; Antony, V.B. Respiratory syncytial virus causes increased bronchial epithelial permeability. Chest 2004, 126, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Faris, A.N.; Ganesan, S.; Chattoraj, A.; Chattoraj, S.S.; Comstock, A.T.; Unger, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus Delays Cell Repolarization in a Model of Injured/Regenerating Human Airway Epithelium. Am. J. Respir. Cell Mol. Biol. 2016, 55, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Waltl, E.E.; Selb, R.; Eckl-Dorna, J.; Mueller, C.A.; Cabauatan, C.R.; Eiwegger, T.; Resch-Marat, Y.; Niespodziana, K.; Vrtala, S.; Valenta, R.; et al. Betamethasone prevents human rhinovirus- and cigarette smoke- induced loss of respiratory epithelial barrier function. Sci. Rep. 2018, 8, 9688. [Google Scholar] [CrossRef] [Green Version]
- Michi, A.N.; Yipp, B.G.; Dufour, A.; Lopes, F.; Proud, D. PGC-1α mediates a metabolic host defense response in human airway epithelium during rhinovirus infections. Nat. Commun. 2021, 12, 3669. [Google Scholar] [CrossRef]
- Looi, K.; Troy, N.M.; Garratt, L.W.; Iosifidis, T.; Bosco, A.; Buckley, A.G.; Ling, K.-M.; Martinovich, K.M.; Kicic-Starcevich, E.; Shaw, N.C.; et al. Effect of human rhinovirus infection on airway epithelium tight junction protein disassembly and transepithelial permeability. Exp. Lung Res. 2016, 42, 380–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, N.K.; Jang, Y.J. Rhinovirus infection-induced alteration of tight junction and adherens junction components in human nasal epithelial cells. Laryngoscope 2010, 120, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Jung, J.H.; Kang, I.G.; Choi, Y.S.; Kim, S.T. ROS Is Involved in Disruption of Tight Junctions of Human Nasal Epithelial Cells Induced by HRV16. Laryngoscope 2018, 128, E393–E401. [Google Scholar] [CrossRef] [PubMed]
- Looi, K.; Buckley, A.G.; Rigby, P.J.; Garratt, L.W.; Iosifidis, T.; Zosky, G.R.; Larcombe, A.N.; Lannigan, F.J.; Ling, K.M.; Martinovich, K.M.; et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin. Exp. Allergy 2018, 48, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Wronski, S.; Beinke, S.; Obernolte, H.; Belyaev, N.N.; Saunders, K.A.; Lennon, M.G.; Schaudien, D.; Braubach, P.; Jonigk, D.; Warnecke, G.; et al. Rhinovirus-induced Human Lung Tissue Responses Mimic Chronic Obstructive Pulmonary Disease and Asthma Gene Signatures. Am. J. Respir. Cell Mol. Biol. 2021, 65, 544–554. [Google Scholar] [CrossRef]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-Induced Barrier Dysfunction in Polarized Airway Epithelial Cells Is Mediated by NADPH Oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [Green Version]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-Like Receptor X-1 Is Required for Rhinovirus-Induced Barrier Dysfunction in Airway Epithelial Cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.A.; Jung, J.H.; Choi, Y.S.; Kim, S.T. Ginsenoside Re Protects Rhinovirus-Induced Disruption of Tight Junction Through Inhibition Of ROS-Mediated Phosphatases Inactivation In Human Nasal Epithelial Cells. J. Allergy Clin. Immunol. 2018, 141, AB175. [Google Scholar] [CrossRef]
- Kim, K.A.; Jung, J.H.; Choi, Y.S.; Kim, S.T. Wogonin inhibits tight junction disruption via suppression of inflammatory response and phosphorylation of AKT/NF-κB and ERK1/2 in rhinovirus-infected human nasal epithelial cells. Inflamm. Res. 2022, 71, 357–368. [Google Scholar] [CrossRef]
- Travanty, E.; Zhou, B.; Zhang, H.; Di, Y.P.; Alcorn, J.F.; Wentworth, D.E.; Mason, R.; Wang, J. Differential Susceptibilities of Human Lung Primary Cells to H1N1 Influenza Viruses. J. Virol. 2015, 89, 11935–11944. [Google Scholar] [CrossRef]
- Ruan, T.; Sun, J.; Liu, W.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. H1N1 Influenza Virus Cross-Activates Gli1 to Disrupt the Intercellular Junctions of Alveolar Epithelial Cells. Cell Rep. 2020, 31, 107801. [Google Scholar] [CrossRef]
- Ruan, T.; Sun, Y.; Zhang, J.; Sun, J.; Liu, W.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. H5N1 infection impairs the alveolar epithelial barrier through intercellular junction proteins via Itch-mediated proteasomal degradation. Commun. Biol. 2022, 5, 186. [Google Scholar] [CrossRef]
- Zhang, L.; Collins, P.L.; Lamb, R.A.; Pickles, R.J. Comparison of differing cytopathic effects in human airway epithelium of parainfluenza virus 5 (W3A), parainfluenza virus type 3, and respiratory syncytial virus. Virology 2011, 421, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Yumine, N.; Matsumoto, Y.; Ohta, K.; Fukasawa, M.; Nishio, M. Claudin-1 inhibits human parainfluenza virus type 2 dissemination. Virology 2019, 531, 93–99. [Google Scholar] [CrossRef]
- Teoh, K.-T.; Siu, Y.-L.; Chan, W.-L.; Schlüter, M.A.; Liu, C.-J.; Peiris, J.S.M.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS Coronavirus E Protein Interacts with PALS1 and Alters Tight Junction Formation and Epithelial Morphogenesis. Mol. Biol. Cell 2010, 21, 3838–3852. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020, 11, 3910. [Google Scholar] [CrossRef]
- Robinot, R.; Hubert, M.; de Melo, G.D.; Lazarini, F.; Bruel, T.; Smith, N.; Levallois, S.; Larrous, F.; Fernandes, J.; Gellenoncourt, S.; et al. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat. Commun. 2021, 12, 4354. [Google Scholar] [CrossRef]
- Deinhardt-Emmer, S.; Böttcher, S.; Häring, C.; Giebeler, L.; Henke, A.; Zell, R.; Jungwirth, J.; Jordan, P.M.; Werz, O.; Hornung, F.; et al. SARS-CoV-2 Causes Severe Epithelial Inflammation and Barrier Dysfunction. J. Virol. 2021, 95, e00110–e00121. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef]
- Gnanamani, A.; Hariharan, P.; Paul-Satyaseela, M. Staphylococcus aureus: Overview of Bacteriology, Clinical Diseases, Epidemiology, Antibiotic Resistance and Therapeutic Approach. In Frontiers in Staphylococcus aureus; Enany, S., Alexander, L., Eds.; IntechOpen: London, UK, 2017. [Google Scholar]
- Chmielowiec-Korzeniowska, A.; Tymczyna, L.; Wlazło, Ł.; Nowakowicz-Dębek, B.; Trawińska, B. Staphylococcus aureus carriage state in healthy adult population and phenotypic and genotypic properties of isolated strains. Adv. Dermatol. Allergol. 2020, 37, 184–189. [Google Scholar] [CrossRef]
- Sakr, A.; Brégeon, F.; Mège, J.-L.; Rolain, J.-M.; Blin, O. Staphylococcus aureus Nasal Colonization: An Update on Mechanisms, Epidemiology, Risk Factors, and Subsequent Infections. Front. Microbiol. 2018, 9, 2419. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, H.F.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Laux, C.; Peschel, A.; Krismer, B. Staphylococcus aureus Colonization of the Human Nose and Interaction with Other Microbiome Members. Microbiol. Spectr. 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- David, M.Z.; Daum, R.S. Treatment of Staphylococcus aureus Infections. In Staphylococcus aureus: Microbiology, Pathology, Immunology, Therapy and Prophylaxis; Bagnoli, F., Rappuoli, R., Grandi, G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 325–383. [Google Scholar]
- Guo, Y.; Song, G.; Sun, M.; Wang, J.; Wang, Y. Prevalence and Therapies of Antibiotic-Resistance in Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2020, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- CDC. 2019 Antibiotic Resistance Threats Report. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html#pse (accessed on 3 November 2022).
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef]
- Garnett, J.P.; Baker, E.H.; Naik, S.; Lindsay, J.A.; Knight, G.M.; Gill, S.; Tregoning, J.S.; Baines, D.L. Metformin reduces airway glucose permeability and hyperglycaemia-induced Staphylococcus aureus load independently of effects on blood glucose. Thorax 2013, 68, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Popov, L.M.; Marceau, C.D.; Starkl, P.M.; Lumb, J.H.; Shah, J.; Guerrera, D.; Cooper, R.L.; Merakou, C.; Bouley, D.M.; Meng, W.; et al. The adherens junctions control susceptibility to Staphylococcus aureus α-toxin. Proc. Natl. Acad. Sci. USA 2015, 112, 14337–14342. [Google Scholar] [CrossRef]
- Shah, J.; Rouaud, F.; Guerrera, D.; Vasileva, E.; Popov, L.M.; Kelley, W.L.; Rubinstein, E.; Carette, J.E.; Amieva, M.R.; Citi, S. A Dock-and-Lock Mechanism Clusters ADAM10 at Cell-Cell Junctions to Promote α-Toxin Cytotoxicity. Cell Rep. 2018, 25, 2132–2147.e7. [Google Scholar] [CrossRef] [Green Version]
- Vuori, K.; Hirai, H.; Aizawa, S.; Ruoslahti, E. Introduction of p130cas signaling complex formation upon integrin-mediated cell adhesion: A role for Src family kinases. Mol. Cell. Biol. 1996, 16, 2606–2613. [Google Scholar] [CrossRef] [Green Version]
- Nojima, Y.; Morino, N.; Mimura, T.; Hamasaki, K.; Furuya, H.; Sakai, R.; Sato, T.; Tachibana, K.; Morimoto, C.; Yazaki, Y.; et al. Integrin-mediated cell adhesion promotes tyrosine phosphorylation of p130Cas, a Src homology 3-containing molecule having multiple Src homology 2-binding motifs. J. Biol. Chem. 1995, 270, 15398–15402. [Google Scholar] [CrossRef] [Green Version]
- Carlier, M.F.; Ressad, F.; Pantaloni, D. Control of actin dynamics in cell motility. Role of ADF/cofilin. J. Biol. Chem. 1999, 274, 33827–33830. [Google Scholar] [CrossRef] [Green Version]
- Agerer, F.; Michel, A.; Ohlsen, K.; Hauck, C.R. Integrin-mediated Invasion of Staphylococcus aureus into Human Cells Requires Src Family Protein-tyrosine Kinases. J. Biol. Chem. 2003, 278, 42524–42531. [Google Scholar] [CrossRef] [Green Version]
- Weiser, J.N.; Ferreira, D.M.; Paton, J.C. Streptococcus pneumoniae: Transmission, colonization and invasion. Nat. Rev. Microbiol. 2018, 16, 355–367. [Google Scholar] [CrossRef]
- Henriques-Normark, B.; Tuomanen, E.I. The pneumococcus: Epidemiology, microbiology, and pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a010215. [Google Scholar] [CrossRef]
- Subramanian, K.; Henriques-Normark, B.; Normark, S. Emerging concepts in the pathogenesis of the Streptococcus pneumoniae: From nasopharyngeal colonizer to intracellular pathogen. Cell. Microbiol. 2019, 21, e13077. [Google Scholar] [CrossRef] [Green Version]
- Brooks, L.R.K.; Mias, G.I. Streptococcus pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front. Immunol. 2018, 9, 1366. [Google Scholar] [CrossRef] [Green Version]
- Evans, W.; Hansman, D. Tetracycline-Resistant Pneumococcus. Lancet 1963, 281, 451. [Google Scholar] [CrossRef]
- Andrejko, K.; Ratnasiri, B.; Hausdorff, W.P.; Laxminarayan, R.; Lewnard, J.A. Antimicrobial resistance in paediatric Streptococcus pneumoniae isolates amid global implementation of pneumococcal conjugate vaccines: A systematic review and meta-regression analysis. Lancet Microbe 2021, 2, e450–e460. [Google Scholar] [CrossRef] [PubMed]
- Cherazard, R.; Epstein, M.; Doan, T.-L.; Salim, T.; Bharti, S.; Smith, M.A. Antimicrobial Resistant Streptococcus pneumoniae: Prevalence, Mechanisms, and Clinical Implications. Am. J. Ther. 2017, 24, e361–e369. [Google Scholar] [CrossRef]
- Sader, H.S.; Mendes, R.E.; Le, J.; Denys, G.; Flamm, R.K.; Jones, R.N. Antimicrobial Susceptibility of Streptococcus pneumoniae from North America, Europe, Latin America, and the Asia-Pacific Region: Results From 20 Years of the SENTRY Antimicrobial Surveillance Program (1997–2016). Open Forum Infect. Dis. 2019, 6, S14–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klugman, K.P.; Rodgers, G.L. Time for a third-generation pneumococcal conjugate vaccine. Lancet Infect. Dis. 2021, 21, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Dhoubhadel, B.G.; Ishifuji, T.; Yasunami, M.; Yaegashi, M.; Asoh, N.; Ishida, M.; Hamaguchi, S.; Aoshima, M.; Ariyoshi, K.; et al. Serotype-specific effectiveness of 23-valent pneumococcal polysaccharide vaccine against pneumococcal pneumonia in adults aged 65 years or older: A multicentre, prospective, test-negative design study. Lancet Infect. Dis. 2017, 17, 313–321. [Google Scholar] [CrossRef]
- Ganaie, F.; Saad, J.S.; McGee, L.; van Tonder, A.J.; Bentley, S.D.; Lo, S.W.; Gladstone, R.A.; Turner, P.; Keenan, J.D.; Breiman, R.F.; et al. A New Pneumococcal Capsule Type, 10D, is the 100th Serotype and Has a Large cps Fragment from an Oral Streptococcus. mBio 2020, 11, 32430472. [Google Scholar] [CrossRef]
- Feikin, D.R.; Kagucia, E.W.; Loo, J.D.; Link-Gelles, R.; Puhan, M.A.; Cherian, T.; Levine, O.S.; Whitney, C.G.; O’Brien, K.L.; Moore, M.R. Serotype-specific changes in invasive pneumococcal disease after pneumococcal conjugate vaccine introduction: A pooled analysis of multiple surveillance sites. PLoS Med. 2013, 10, e1001517. [Google Scholar] [CrossRef] [Green Version]
- Ladhani, S.N.; Collins, S.; Djennad, A.; Sheppard, C.L.; Borrow, R.; Fry, N.K.; Andrews, N.J.; Miller, E.; Ramsay, M.E. Rapid increase in non-vaccine serotypes causing invasive pneumococcal disease in England and Wales, 2000–2017: A prospective national observational cohort study. Lancet Infect. Dis. 2018, 18, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Galanis, I.; Lindstrand, A.; Darenberg, J.; Browall, S.; Nannapaneni, P.; Sjöström, K.; Morfeldt, E.; Naucler, P.; Blennow, M.; Örtqvist, Å.; et al. Effects of PCV7 and PCV13 on invasive pneumococcal disease and carriage in Stockholm, Sweden. Eur. Respir. J. 2016, 47, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- LeMessurier, K.S.; Tiwary, M.; Morin, N.P.; Samarasinghe, A.E. Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol. 2020, 11, 3. [Google Scholar] [CrossRef]
- Fliegauf, M.; Sonnen, A.F.P.; Kremer, B.; Henneke, P. Mucociliary Clearance Defects in a Murine In Vitro Model of Pneumococcal Airway Infection. PLoS ONE 2013, 8, e59925. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, D.; Kollef, M. The Epidemiology and Pathogenesis and Treatment of Pseudomonas aeruginosa Infections: An Update. Drugs 2021, 81, 2117–2131. [Google Scholar] [CrossRef]
- Morin, C.D.; Deziel, E.; Gauthier, J.; Levesque, R.C.; Lau, G.W. An Organ System-Based Synopsis of Pseudomonas aeruginosa Virulence. Virulence 2021, 12, 1469–1507. [Google Scholar] [CrossRef]
- Norbury, W.; Herndon, D.N.; Tanksley, J.; Jeschke, M.G.; Finnerty, C.C. Infection in Burns. Surg. Infect. (Larchmt) 2016, 17, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Philips, B.J.; Meguer, J.X.; Redman, J.; Baker, E.H. Factors determining the appearance of glucose in upper and lower respiratory tract secretions. Intensive Care Med. 2003, 29, 2204–2210. [Google Scholar] [CrossRef]
- Baker, E.H.; Janaway, C.H.; Philips, B.J.; Brennan, A.L.; Baines, D.L.; Wood, D.M.; Jones, P.W. Hyperglycaemia is associated with poor outcomes in patients admitted to hospital with acute exacerbations of chronic obstructive pulmonary disease. Thorax 2006, 61, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Garnett, J.P.; Gray, M.A.; Tarran, R.; Brodlie, M.; Ward, C.; Baker, E.H.; Baines, D.L. Elevated paracellular glucose flux across cystic fibrosis airway epithelial monolayers is an important factor for Pseudomonas aeruginosa growth. PLoS ONE 2013, 8, e76283. [Google Scholar] [CrossRef] [Green Version]
- Horna, G.; Ruiz, J. Type 3 secretion system of Pseudomonas aeruginosa. Microbiol. Res. 2021, 246, 126719. [Google Scholar] [CrossRef]
- Morrow, K.A.; Frank, D.W.; Balczon, R.; Stevens, T. The Pseudomonas aeruginosa Exoenzyme Y: A Promiscuous Nucleotidyl Cyclase Edema Factor and Virulence Determinant. Handb. Exp. Pharmacol. 2017, 238, 67–85. [Google Scholar] [CrossRef]
- Mancl, J.M.; Suarez, C.; Liang, W.G.; Kovar, D.R.; Tang, W.J. Pseudomonas aeruginosa exoenzyme Y directly bundles actin filaments. J. Biol. Chem. 2020, 295, 3506–3517. [Google Scholar] [CrossRef]
- Wagener, B.M.; Anjum, N.; Christiaans, S.C.; Banks, M.E.; Parker, J.C.; Threet, A.T.; Walker, R.R.; Isbell, K.D.; Moser, S.A.; Stevens, T.; et al. Exoenzyme Y Contributes to End-Organ Dysfunction Caused by Pseudomonas aeruginosa Pneumonia in Critically Ill Patients: An Exploratory Study. Toxins 2020, 12, 369. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.; Goldufsky, J.; Shafikhani, S.H. Pseudomonas aeruginosa ExoT Induces Atypical Anoikis Apoptosis in Target Host Cells by Transforming Crk Adaptor Protein into a Cytotoxin. PLoS Pathog. 2015, 11, e1004934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, S.J.; Goldufsky, J.W.; Bello, D.; Masood, S.; Shafikhani, S.H. Pseudomonas aeruginosa ExoT Induces Mitochondrial Apoptosis in Target Host Cells in a Manner That Depends on Its GTPase-activating Protein (GAP) Domain Activity. J. Biol. Chem. 2015, 290, 29063–29073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, I.; Wooten, R.M. Interactions Between Pathogenic Burkholderia and the Complement System: A Review of Potential Immune Evasion Mechanisms. Front. Cell. Infect. Microbiol. 2021, 11, 701362. [Google Scholar] [CrossRef] [PubMed]
- Coenye, T.; Vandamme, P.; Govan, J.R.; LiPuma, J.J. Taxonomy and identification of the Burkholderia cepacia complex. J. Clin. Microbiol. 2001, 39, 3427–3436. [Google Scholar] [CrossRef] [Green Version]
- Huse, H.K.; Lee, M.J.; Wootton, M.; Sharp, S.E.; Traczewski, M.; LiPuma, J.J.; Jorth, P. Evaluation of Antimicrobial Susceptibility Testing Methods for Burkholderia cenocepacia and Burkholderia multivorans Isolates from Cystic Fibrosis Patients. J. Clin. Microbiol. 2021, 59, e0144721. [Google Scholar] [CrossRef]
- Bressler, A.M.; Kaye, K.S.; LiPuma, J.J.; Alexander, B.D.; Moore, C.M.; Reller, L.B.; Woods, C.W. Risk factors for Burkholderia cepacia complex bacteremia among intensive care unit patients without cystic fibrosis: A case-control study. Infect. Control Hosp. Epidemiol. 2007, 28, 951–958. [Google Scholar] [CrossRef]
- Rhodes, K.A.; Schweizer, H.P. Antibiotic resistance in Burkholderia species. Drug Resist. Updates 2016, 28, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Aziz, A.; Sarovich, D.S.; Nosworthy, E.; Beissbarth, J.; Chang, A.B.; Smith-Vaughan, H.; Price, E.P.; Harris, T.M. Molecular Signatures of Non-typeable Haemophilus influenzae Lung Adaptation in Pediatric Chronic Lung Disease. Front. Microbiol. 2019, 10, 1622. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.C.; Jalalvand, F.; Thegerstrom, J.; Riesbeck, K. The Interplay Between Immune Response and Bacterial Infection in COPD: Focus Upon Non-typeable Haemophilus influenzae. Front. Immunol. 2018, 9, 2530. [Google Scholar] [CrossRef]
- Kadry, N.A.; Porsch, E.A.; Shen, H.; St Geme, J.W., 3rd. Immunization with HMW1 and HMW2 adhesins protects against colonization by heterologous strains of nontypeable Haemophilus influenzae. Proc. Natl. Acad. Sci. USA 2021, 118, e2019923118. [Google Scholar] [CrossRef]
- Ahearn, C.P.; Gallo, M.C.; Murphy, T.F. Insights on persistent airway infection by non-typeable Haemophilus influenzae in chronic obstructive pulmonary disease. Pathog. Dis. 2017, 75, ftx042. [Google Scholar] [CrossRef] [Green Version]
- Sajjan, U.S.; Jia, Y.; Newcomb, D.C.; Bentley, J.K.; Lukacs, N.W.; LiPuma, J.J.; Hershenson, M.B. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 2006, 20, 2121–2123. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Jono, H.; Han, J.; Lim, D.J.; Li, J.D. Synergistic activation of NF-kappaB by nontypeable Haemophilus influenzae and tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 2004, 101, 3563–3568. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Schlingmann, B.; Stecenko, A.A.; Guidot, D.M.; Koval, M. NF-kappaB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers 2015, 3, e982424. [Google Scholar] [CrossRef]
- Rokkam, D.; Lafemina, M.J.; Lee, J.W.; Matthay, M.A.; Frank, J.A. Claudin-4 levels are associated with intact alveolar fluid clearance in human lungs. Am. J. Pathol. 2011, 179, 1081–1087. [Google Scholar] [CrossRef]
- Wang, F.; Daugherty, B.; Keise, L.L.; Wei, Z.; Foley, J.P.; Savani, R.C.; Koval, M. Heterogeneity of claudin expression by alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2003, 29, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Koval, M. Claudin heterogeneity and control of lung tight junctions. Annu. Rev. Physiol. 2013, 75, 551–567. [Google Scholar] [CrossRef]
- LaFemina, M.J.; Sutherland, K.M.; Bentley, T.; Gonzales, L.W.; Allen, L.; Chapin, C.J.; Rokkam, D.; Sweerus, K.A.; Dobbs, L.G.; Ballard, P.L.; et al. Claudin-18 deficiency results in alveolar barrier dysfunction and impaired alveologenesis in mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Milara, J.; Peiro, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef]
- Glockner, M.; Marwitz, S.; Rohmann, K.; Watz, H.; Nitschkowski, D.; Rupp, J.; Dalhoff, K.; Goldmann, T.; Dromann, D. Haemophilus influenzae causes cellular trans-differentiation in human bronchial epithelia. Innate Immun. 2021, 27, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Schleimer, R.P.; Berdnikovs, S. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J. Allergy Clin. Immunol. 2017, 139, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, P.; Surette, M.G.; Virchow, J.C. Neutrophilic asthma: Misconception or misnomer? Lancet Respir. Med. 2021, 9, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Jabeen, M.; Bharj, G.; Hinks, T.S.C. Non-typeable Haemophilus influenzae airways infection: The next treatable trait in asthma? Eur. Respir. Rev. 2022, 31, 220008. [Google Scholar] [CrossRef]
- Gibson, P.G.; Yang, I.A.; Upham, J.W.; Reynolds, P.N.; Hodge, S.; James, A.L.; Jenkins, C.; Peters, M.J.; Marks, G.B.; Baraket, M.; et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Brusselle, G.G.; Vanderstichele, C.; Jordens, P.; Deman, R.; Slabbynck, H.; Ringoet, V.; Verleden, G.; Demedts, I.K.; Verhamme, K.; Delporte, A.; et al. Azithromycin for prevention of exacerbations in severe asthma (AZISAST): A multicentre randomised double-blind placebo-controlled trial. Thorax 2013, 68, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Serisier, D.J. Risks of population antimicrobial resistance associated with chronic macrolide use for inflammatory airway diseases. Lancet Respir. Med. 2013, 1, 262–274. [Google Scholar] [CrossRef]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV Virus Taxonomy Profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef]
- Grayson, S.A.; Griffiths, P.S.; Perez, M.K.; Piedimonte, G. Detection of airborne respiratory syncytial virus in a pediatric acute care clinic. Pediatr. Pulmonol. 2017, 52, 684–688. [Google Scholar] [CrossRef]
- Hall, C.B. Respiratory syncytial virus: Its transmission in the hospital environment. Yale J. Biol. Med. 1982, 55, 219–223. [Google Scholar]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, Detection, and New Options for Prevention and Treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [Green Version]
- Rezaee, F.; Linfield, D.T.; Harford, T.J.; Piedimonte, G. Ongoing developments in RSV prophylaxis: A clinician’s analysis. Curr. Opin. Virol. 2017, 24, 70–78. [Google Scholar] [CrossRef]
- Li, Y.; Johnson, E.K.; Shi, T.; Campbell, H.; Chaves, S.S.; Commaille-Chapus, C.; Dighero, I.; James, S.L.; Mahé, C.; Ooi, Y.; et al. National burden estimates of hospitalisations for acute lower respiratory infections due to respiratory syncytial virus in young children in 2019 among 58 countries: A modelling study. Lancet Respir. Med. 2021, 9, 175–185. [Google Scholar] [CrossRef]
- Srikantiah, P.; Vora, P.; Klugman, K.P. Assessing the Full Burden of Respiratory Syncytial Virus in Young Infants in Low- and Middle-Income Countries: The Importance of Community Mortality Studies. Clin. Infect. Dis. 2021, 73, S177–S179. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis. Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef]
- Carbonell-Estrany, X.; Pérez-Yarza, E.G.; García, L.S.; Guzmán Cabañas, J.M.; Bòria, E.V.; Atienza, B.B.; Group, I.S. Long-Term Burden and Respiratory Effects of Respiratory Syncytial Virus Hospitalization in Preterm Infants—The SPRING Study. PLoS ONE 2015, 10, e0125422. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Hartert, T.V. Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Rev. Anti Infec. Ther. 2011, 9, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Sigurs, N.; Aljassim, F.; Kjellman, B.; Robinson, P.D.; Sigurbergsson, F.; Bjarnason, R.; Gustafsson, P.M. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 2010, 65, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Jartti, T.; Gern, J.E. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 2017, 140, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.A.; Al Thani, A.A.; Al Ansari, K.; Yassine, H.M. Human respiratory syncytial virus: Pathogenesis, immune responses, and current vaccine approaches. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Belderbos, M.E.; Houben, M.L.; Wilbrink, B.; Lentjes, E.; Bloemen, E.M.; Kimpen, J.L.; Rovers, M.; Bont, L. Cord blood vitamin D deficiency is associated with respiratory syncytial virus bronchiolitis. Pediatrics 2011, 127, e1513–e1520. [Google Scholar] [CrossRef] [PubMed]
- Smallcombe, C.C.; Harford, T.J.; Linfield, D.T.; Lechuga, S.; Bokun, V.; Piedimonte, G.; Rezaee, F. Titanium dioxide nanoparticles exaggerate respiratory syncytial virus-induced airway epithelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L481–L496. [Google Scholar] [CrossRef]
- Nicolai, A.; Frassanito, A.; Nenna, R.; Cangiano, G.; Petrarca, L.; Papoff, P.; Pierangeli, A.; Scagnolari, C.; Moretti, C.; Midulla, F. Risk Factors for Virus-induced Acute Respiratory Tract Infections in Children Younger Than 3 Years and Recurrent Wheezing at 36 Months Follow-Up After Discharge. Pediatr. Infect. Dis. J. 2017, 36, 179–183. [Google Scholar] [CrossRef]
- Karr, C.J.; Rudra, C.B.; Miller, K.A.; Gould, T.R.; Larson, T.; Sathyanarayana, S.; Koenig, J.Q. Infant exposure to fine particulate matter and traffic and risk of hospitalization for RSV bronchiolitis in a region with lower ambient air pollution. Environ. Res. 2009, 109, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.P.; Bacharier, L.B.; Bonfiglio, J.; Schechtman, K.B.; Strunk, R.; Storch, G.; Castro, M. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics 2005, 115, e7–e14. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, S.; Yoshida, H.; Akashi, T.; Komemoto, K.; Ueda, T.; Ikarashi, Y.; Miyauchi, A.; Konno, K.; Yamanaka, S.; Hirose, A.; et al. Titanium dioxide nanoparticles exacerbate pneumonia in respiratory syncytial virus (RSV)-infected mice. Environ. Toxicol. Pharmacol. 2015, 39, 879–886. [Google Scholar] [CrossRef]
- Xing, Y.; Proesmans, M. New therapies for acute RSV infections: Where are we? Eur. J. Pediatr. 2019, 178, 131–138. [Google Scholar] [CrossRef]
- Mammas, I.N.; Drysdale, S.B.; Rath, B.; Theodoridou, M.; Papaioannou, G.; Papatheodoropoulou, A.; Koutsounaki, E.; Koutsaftiki, C.; Kozanidou, E.; Achtsidis, V.; et al. Update on current views and advances on RSV infection. Int. J. Mol. Med. 2020, 46, 509–520. [Google Scholar] [CrossRef]
- Behzadi, M.A.; Leyva-Grado, V.H. Overview of Current Therapeutics and Novel Candidates Against Influenza, Respiratory Syncytial Virus, and Middle East Respiratory Syndrome Coronavirus Infections. Front. Microbiol. 2019, 10, 1327. [Google Scholar] [CrossRef]
- Krilov, L.R. Safety issues related to the administration of ribavirin. Pediatr. Infect. Dis. J. 2002, 21, 479–481. [Google Scholar] [CrossRef]
- Nicholson, E.G.; Munoz, F.M. A Review of Therapeutics in Clinical Development for Respiratory Syncytial Virus and Influenza in Children. Clin. Ther. 2018, 40, 1268–1281. [Google Scholar] [CrossRef] [Green Version]
- Frogel, M.; Nerwen, C.; Cohen, A.; VanVeldhuisen, P.; Harrington, M.; Boron, M. Prevention of hospitalization due to respiratory syncytial virus: Results from the Palivizumab Outcomes Registry. J. Perinatol. 2008, 28, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Shahabi, A.; Peneva, D.; Incerti, D.; McLaurin, K.; Stevens, W. Assessing Variation in the Cost of Palivizumab for Respiratory Syncytial Virus Prevention in Preterm Infants. PharmacoEcon.-Open 2018, 2, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; Abu Raya, B.; Baraldi, E.; Flanagan, K.; Martinon Torres, F.; Tsolia, M.; Zielen, S. RSV Prevention in All Infants: Which Is the Most Preferable Strategy? Front. Immunol. 2022, 13, 880368. [Google Scholar] [CrossRef]
- Zhang, L.; Peeples, M.E.; Boucher, R.C.; Collins, P.L.; Pickles, R.J. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 2002, 76, 5654–5666. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Gonzales, R.A.; Olson, S.J.; Wright, P.F.; Graham, B.S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2007, 20, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Spacova, I.; De Boeck, I.; Bron, P.A.; Delputte, P.; Lebeer, S. Topical Microbial Therapeutics against Respiratory Viral Infections. Trends Mol. Med. 2021, 27, 538–553. [Google Scholar] [CrossRef]
- Bruewer, M.; Utech, M.; Ivanov, A.I.; Hopkins, A.M.; Parkos, C.A.; Nusrat, A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005, 19, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Saatian, B.; Rezaee, F.; Desando, S.; Emo, J.; Chapman, T.; Knowlden, S.; Georas, S.N. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 2013, 1, e24333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utech, M.; Ivanov, A.I.; Samarin, S.N.; Bruewer, M.; Turner, J.R.; Mrsny, R.J.; Parkos, C.A.; Nusrat, A. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: Myosin II-dependent vacuolarization of the apical plasma membrane. Mol. Biol. Cell 2005, 16, 5040–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Rozengurt, E. Protein Kinase D Signaling: Multiple Biological Functions in Health and Disease. Physiology 2011, 26, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Connelly, J.; Chao, Y.; Wang, Q.J. Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases. Biomolecules 2021, 11, 483. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Barisic, S.; Hausser, A. Multi-level control of actin dynamics by protein kinase D. Cell. Signal. 2013, 25, 1739–1747. [Google Scholar] [CrossRef]
- Eiseler, T.; Hausser, A.; De Kimpe, L.; Van Lint, J.; Pfizenmaier, K. Protein Kinase D Controls Actin Polymerization and Cell Motility through Phosphorylation of Cortactin. J. Biol. Chem. 2010, 285, 18672–18683. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell Functions of a Multifaceted Actin-Binding Protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef]
- Cosen-Binker, L.I.; Kapus, A. Cortactin: The Gray Eminence of the Cytoskeleton. Physiology 2006, 21, 352–361. [Google Scholar] [CrossRef]
- Weed, S.A.; Parsons, J.T. Cortactin: Coupling membrane dynamics to cortical actin assembly. Oncogene 2001, 20, 6418–6434. [Google Scholar] [CrossRef]
- Bougnères, L.; Girardin, S.p.E.; Weed, S.A.; Karginov, A.V.; Olivo-Marin, J.-C.; Parsons, J.T.; Sansonetti, P.J.; Van Nhieu, G.T. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J. Cell Biol. 2004, 166, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, Y.; González, L.; Noguera, L.; González, P.A.; Riedel, C.A.; Bertrand, P.; Bueno, S.M. Cytokines in the Respiratory Airway as Biomarkers of Severity and Prognosis for Respiratory Syncytial Virus Infection: An Update. Front. Immunol. 2019, 10, 1154. [Google Scholar] [CrossRef]
- McNamara, P.S.; Ritson, P.; Selby, A.; Hart, C.A.; Smyth, R.L. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 2003, 88, 922–926. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Herbert, J.A.; Smith, C.M.; Smyth, R.L. An in vitro transepithelial migration assay to evaluate the role of neutrophils in Respiratory Syncytial Virus (RSV) induced epithelial damage. Sci. Rep. 2018, 8, 6777. [Google Scholar] [CrossRef] [Green Version]
- Price, W.H. The Isolation of a New Virus Associated with Respiratory Clinical Disease in Humans. Proc. Natl. Acad. Sci. USA 1956, 42, 892–896. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.P.; Papadopoulos, N.G.; Skevaki, C. Respiratory Viral Pathogens. In Encyclopedia of Respiratory Medicine, 2nd ed.; Janes, S.M., Ed.; Academic Press: Oxford, UK, 2022; pp. 129–137. [Google Scholar]
- Stobart, C.C.; Nosek, J.M.; Moore, M.L. Rhinovirus Biology, Antigenic Diversity, and Advancements in the Design of a Human Rhinovirus Vaccine. Front. Microbiol. 2017, 8, 2412. [Google Scholar] [CrossRef]
- Kutter, J.S.; Spronken, M.I.; Fraaij, P.L.; Fouchier, R.A.M.; Herfst, S. Transmission routes of respiratory viruses among humans. Curr. Opin. Virol. 2018, 28, 142–151. [Google Scholar] [CrossRef]
- Monto, A.S. The seasonality of rhinovirus infections and its implications for clinical recognition. Clin. Ther. 2002, 24, 1987–1997. [Google Scholar] [CrossRef]
- Morikawa, S.; Kohdera, U.; Hosaka, T.; Ishii, K.; Akagawa, S.; Hiroi, S.; Kase, T. Seasonal variations of respiratory viruses and etiology of human rhinovirus infection in children. J. Clin. Virol. 2015, 73, 14–19. [Google Scholar] [CrossRef]
- Moriyama, M.; Hugentobler, W.J.; Iwasaki, A. Seasonality of Respiratory Viral Infections. Annu. Rev. Virol. 2020, 7, 83–101. [Google Scholar] [CrossRef]
- Kennedy, J.L.; Turner, R.B.; Braciale, T.; Heymann, P.W.; Borish, L. Pathogenesis of rhinovirus infection. Curr. Opin. Virol. 2012, 2, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Bizot, E.; Bousquet, A.; Charpié, M.; Coquelin, F.; Lefevre, S.; Le Lorier, J.; Patin, M.; Sée, P.; Sarfati, E.; Walle, S.; et al. Rhinovirus: A Narrative Review on Its Genetic Characteristics, Pediatric Clinical Presentations, and Pathogenesis. Front. Pediatr. 2021, 9, 643219. [Google Scholar] [CrossRef]
- Jain, S.; Williams, D.J.; Arnold, S.R.; Ampofo, K.; Bramley, A.M.; Reed, C.; Stockmann, C.; Anderson, E.J.; Grijalva, C.G.; Self, W.H.; et al. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Children. N. Engl. J. Med. 2015, 372, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, H.C. Viral Bronchiolitis in Children. N. Engl. J. Med. 2016, 374, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Fedele, G.; Schiavoni, I.; Nenna, R.; Pierangeli, A.; Frassanito, A.; Leone, P.; Petrarca, L.; Scagnolari, C.; Midulla, F. Analysis of the immune response in infants hospitalized with viral bronchiolitis shows different Th1/Th2 profiles associated with respiratory syncytial virus and human rhinovirus. Pediatr. Allergy Immunol. 2018, 29, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E. The ABCs of rhinoviruses, wheezing, and asthma. J. Virol. 2010, 84, 7418–7426. [Google Scholar] [CrossRef] [Green Version]
- Merckx, J.; Ducharme, F.M.; Martineau, C.; Zemek, R.; Gravel, J.; Chalut, D.; Poonai, N.; Quach, C. Respiratory Viruses and Treatment Failure in Children with Asthma Exacerbation. Pediatrics 2018, 142, e20174105. [Google Scholar] [CrossRef] [Green Version]
- Seemungal, T.; Harper-Owen, R.; Bhowmik, A.; Moric, I.; Sanderson, G.; Message, S.; Maccallum, P.; Meade, T.W.; Jeffries, D.J.; Johnston, S.L.; et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1618–1623. [Google Scholar] [CrossRef]
- Coultas, J.A.; Cafferkey, J.; Mallia, P.; Johnston, S.L. Experimental Antiviral Therapeutic Studies for Human Rhinovirus Infections. J. Exp. Pharmacol. 2021, 13, 645–659. [Google Scholar] [CrossRef]
- Warner, S.M.; Wiehler, S.; Michi, A.N.; Proud, D. Rhinovirus replication and innate immunity in highly differentiated human airway epithelial cells. Respir. Res. 2019, 20, 150. [Google Scholar] [CrossRef]
- Arruda, E.; Boyle, T.R.; Winther, B.; Pevear, D.C.; Gwaltney, J.M., Jr.; Hayden, F.G. Localization of Human Rhinovirus Replication in the Upper Respiratory Tract by In Situ Hybridization. J. Infec. Dis. 1995, 171, 1329–1333. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Bates, P.J.; Bardin, P.G.; Papi, A.; Leir, S.H.; Fraenkel, D.J.; Meyer, J.; Lackie, P.M.; Sanderson, G.; Holgate, S.T.; et al. Rhinoviruses Infect the Lower Airways. J. Infec. Dis. 2000, 181, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Ganjian, H.; Rajput, C.; Elzoheiry, M.; Sajjan, U. Rhinovirus and Innate Immune Function of Airway Epithelium. Front. Cell. Infect. Microbiol. 2020, 10, 277. [Google Scholar] [CrossRef]
- Boland, H.; Adrian, E.; Schwarzbach, H.; Burger-Kentischer, A.; Jonigk, D.; Braubach, P.; Rohde, G.; Bellinghausen, C. Protective effect of interferon type I on barrier function of human airway epithelium during rhinovirus infections in vitro. ERJ Open Res. 2022, 8, 92. [Google Scholar] [CrossRef]
- Rao, R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 2008, 13, 7210–7226. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Kan, O.K.; Tatsuta, M.; Ishii, Y.; Ogawa, T.; Shinozaki, S.; Fukuyama, S.; Nakanishi, Y.; Matsumoto, K. Incense smoke-induced oxidative stress disrupts tight junctions and bronchial epithelial barrier integrity and induces airway hyperresponsiveness in mouse lungs. Sci. Rep. 2021, 11, 7222. [Google Scholar] [CrossRef]
- Biagioli, M.C.; Kaul, P.; Singh, I.; Turner, R.B. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic. Biol. Med. 1999, 26, 454–462. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.-Y.; Liu, G.-C.; Zhang, J.-X.; Wang, L.-H.; Xu, C.; Yan, Z.-A.; Wang, A.; Su, Y.-F.; Lee, J.-J.; Piao, G.-C.; et al. Pharmacological Properties of Ginsenoside Re. Front. Pharmacol. 2022, 13, 754191. [Google Scholar] [CrossRef]
- Huang, W.H.; Lee, A.R.; Yang, C.H. Antioxidative and anti-inflammatory activities of polyhydroxyflavonoids of Scutellaria baicalensis GEORGI. Biosci. Biotechnol. Biochem. 2006, 70, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Uyeki, T.M.; Hui, D.S.; Zambon, M.; Wentworth, D.E.; Monto, A.S. Influenza. Lancet 2022, 400, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.H.L. Transmissibility and transmission of respiratory viruses. Nat. Rev. Microbiol. 2021, 19, 528–545. [Google Scholar] [CrossRef]
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef] [PubMed]
- WHO. Influenza in Focus: Up to 650,000 People Die of Respiratory Diseases Linked to Seasonal Flu Each Year. Available online: https://www.who.int/news/item/13-12-2017-up-to-650-000-people-die-of-respiratory-diseases-linked-to-seasonal-flu-each-year (accessed on 3 November 2022).
- CDC. Estimated Flu-Related Illnesses, Medical Visits, Hospitalizations, and Deaths in the United States—2019–2020 Flu Season. Available online: https://www.cdc.gov/flu/about/burden/2019-2020.html (accessed on 3 November 2022).
- Huang, Q.J.; Song, K.; Xu, C.; Bolon, D.N.A.; Wang, J.P.; Finberg, R.W.; Schiffer, C.A.; Somasundaran, M. Quantitative structural analysis of influenza virus by cryo-electron tomography and convolutional neural networks. Structure 2022, 30, 777–786.e3. [Google Scholar] [CrossRef] [PubMed]
- Denney, L.; Ho, L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Mauad, T.; Hajjar, L.A.; Callegari, G.D.; da Silva, L.F.; Schout, D.; Galas, F.R.; Alves, V.A.; Malheiros, D.M.; Auler, J.O., Jr.; Ferreira, A.F.; et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am. J. Respir. Crit. Care Med. 2010, 181, 72–79. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The induction and consequences of Influenza A virus-induced cell death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Branche, A.R.; Falsey, A.R. Parainfluenza Virus Infection. Semin. Respir. Crit. Care Med. 2016, 37, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Henrickson, K.J. Parainfluenza viruses. Clin. Microbiol. Rev. 2003, 16, 242–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z. Parainfluenza virus 5-vectored vaccines against human and animal infectious diseases. Rev. Med. Virol. 2018, 28, e1965. [Google Scholar] [CrossRef]
- Burke, C.W.; Bridges, O.; Brown, S.; Rahija, R.; Russell, C.J. Mode of parainfluenza virus transmission determines the dynamics of primary infection and protection from reinfection. PLoS Pathog. 2013, 9, e1003786. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Gordon, B.; Fielding, B.C. Coronaviruses. In Encyclopedia of Infection and Immunity; Rezaei, N., Ed.; Elsevier: Oxford, UK, 2022; pp. 241–258. [Google Scholar]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef]
- Groot, R.J.d.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.M.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Commentary: Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Chafekar, A.; Fielding, B.C. MERS-CoV: Understanding the Latest Human Coronavirus Threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 3 November 2022).
- Sharif-Yakan, A.; Kanj, S.S. Emergence of MERS-CoV in the Middle East: Origins, transmission, treatment, and perspectives. PLoS Pathog. 2014, 10, e1004457. [Google Scholar] [CrossRef] [Green Version]
- Van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef]
- Meselson, M. Droplets and Aerosols in the Transmission of SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2063. [Google Scholar] [CrossRef]
- Mann, R.; Perisetti, A.; Gajendran, M.; Gandhi, Z.; Umapathy, C.; Goyal, H. Clinical Characteristics, Diagnosis, and Treatment of Major Coronavirus Outbreaks. Front. Med. 2020, 7, 581521. [Google Scholar] [CrossRef]
- Robinson, P.C.; Liew, D.F.L.; Tanner, H.L.; Grainger, J.R.; Dwek, R.A.; Reisler, R.B.; Steinman, L.; Feldmann, M.; Ho, L.-P.; Hussell, T.; et al. COVID-19 therapeutics: Challenges and directions for the future. Proc. Natl. Acad. Sci. USA 2022, 119, e2119893119. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Song, L.-G.; Xie, Q.-X.; Lao, H.-L.; Lv, Z.-Y. Human coronaviruses and therapeutic drug discovery. Infect. Dis. Poverty 2021, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Travanty, E.A.; Oko, L.; Edeen, K.; Berglund, A.; Wang, J.; Ito, Y.; Holmes, K.V.; Mason, R.J. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am. J. Respir. Cell Mol. Biol. 2013, 48, 742–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Bakhrebah, M.A.; Abdulaal, W.H.; Zamzami, M.A.; Baothman, O.A.; Hassan, M.A.; Zeyadi, M.; Helmi, N.; Alzahrani, F.; Ali, A.; et al. Middle East respiratory syndrome: Pathogenesis and therapeutic developments. Future Virol. 2019, 14, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.E8. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, R.; Huo, S.; Zhou, Y.; Jiang, S.; Wang, Q.; Yu, F. Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Signal Transduct. Target. Ther. 2021, 6, 233. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- D’Agnillo, F.; Walters, K.-A.; Xiao, Y.; Sheng, Z.-M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790. [Google Scholar] [CrossRef]
- Rouaud, F.; Méan, I.; Citi, S. The ACE2 Receptor for Coronavirus Entry Is Localized at Apical Cell—Cell Junctions of Epithelial Cells. Cells 2022, 11, 627. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, X.-W.; Margolis, B. PALS1 Regulates E-Cadherin Trafficking in Mammalian Epithelial Cells. Mol. Biol. Cell 2007, 18, 874–885. [Google Scholar] [CrossRef] [Green Version]
- De Maio, F.; Lo Cascio, E.; Babini, G.; Sali, M.; Della Longa, S.; Tilocca, B.; Roncada, P.; Arcovito, A.; Sanguinetti, M.; Scambia, G.; et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect. 2020, 22, 592–597. [Google Scholar] [CrossRef]
- Shepley-McTaggart, A.; Sagum, C.A.; Oliva, I.; Rybakovsky, E.; DiGuilio, K.; Liang, J.; Bedford, M.T.; Cassel, J.; Sudol, M.; Mullin, J.M.; et al. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PLoS ONE 2021, 16, e0251955. [Google Scholar] [CrossRef]
- Hekman, R.M.; Hume, A.J.; Goel, R.K.; Abo, K.M.; Huang, J.; Blum, B.C.; Werder, R.B.; Suder, E.L.; Paul, I.; Phanse, S.; et al. Actionable Cytopathogenic Host Responses of Human Alveolar Type 2 Cells to SARS-CoV-2. Mol. Cell 2020, 80, 1104–1122.E9. [Google Scholar] [CrossRef]
- Adil, M.S.; Khulood, D.; Narayanan, S.P.; Somanath, P.R. Bioinformatics analyses reveal cell-barrier junction modulations in lung epithelial cells on SARS-CoV-2 infection. Tissue Barriers 2022, 10, 2000300. [Google Scholar] [CrossRef]
- Tugizov, S. Virus-associated disruption of mucosal epithelial tight junctions and its role in viral transmission and spread. Tissue Barriers 2021, 9, 1943274. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Microorganism | Model | Impacts on Airway Epithelial Barrier Function | Impacts on Apical Junctional Complex (AJC) | Mechanisms | Potential Therapeutics | Ref |
|---|---|---|---|---|---|---|
| Staphylococcus aureus (S. aureus) | Primary epithelial cells from CRSwNP patients-ALI; S. aureus enterotoxin B | ↓ TEER, ↑ permeability | ↓ ZO-1 (IF), ↓ occludin (IF), ↓ phospho-occludin (WB), ↓ claudin-1(WB) | TLR2 pathway | TLR2 blocking antibody | [21] |
| C57BL/6J mice; S. aureus enterotoxin B | ↑ Permeability | ↓ ZO-1 (mRNA), ↓ occludin (mRNA) | ||||
| Primary HNECs-ALI; S. aureus conditioned media | ↓ TEER | TJ separation (EM), discontinuous staining of ZO-1 | - | - | [26] | |
| Primary HNECs-ALI; S. aureus extracellular proteases | ↓ TEER, ↑ permeability | Discontinuous staining of ZO-1 | - | - | [27] | |
| Human airway epithelial cells (H441) | ↑ Permeability | ↓ ZO-1 (IF), ↓ occludin (IF) | AMPK-PKCζ pathway | Metformin | [28] | |
| C57BL/6J mice; S. aureus α-toxin | ↑ Permeability | ↑ E-cadherin cleavage (WB) | ADAM10 activity | ADAM10 inhibition | [29] | |
| A549; S. aureus α-toxin | ↓ Focal adhesion | - | FAK/Src/F-actin reorganization | - | [30] | |
| 16HBE; S. aureus α-toxin | ↑ Paracellular gaps | - | PAK/LIMK/cofilin/F-actin reorganization | - | [31] | |
| Streptococcus pneumoniae (S. pneumoniae) | A549, 16HBE, C57BL/6J mice | Bacterial transmigration, ↑ permeability | - | - | IFN-β | [20] |
| A549 | - | ↑ E-cadherin cleavage/degradation (WB) | ADAM10 activation by pneumolysin | ADAM10 inhibition | [29] | |
| 16HBE, C57BL/6J mice | ↓ TEER, ↑ permeability | ↓ Claudin-7,10 (mRNA) | TLR/p38/TGF-β/Snail1 | - | [32] | |
| Human respiratory tissues | - | TJ separation (EM) | - | - | [33] | |
| H292, BALB/c mice | - | ↓ E-cadherin (IF) | - | - | [34] | |
| human lung tissue | - | (IF, WB) ↓ ZO-1, ↓ occludin, ↓ claudin-5, ↓ VE-cadherin | - | - | [35] | |
| A549 | Bacterial transmigration | ↑VE-cadherin cleavage/degradation (WB) | bacteria-bound plasmin | - | [36] | |
| Pseudomonas aeruginosa (P. aeruginosa) | Calu3 | ↓ TEER, ↑ permeability | ↓ occludin and claudin-1, ↑ cleavage of occludin (WB) | hyperglycemia | Metformin | [37] |
| 16HBE | ↓ TEER, ↑ permeability | ↓ ZO-1 (IF) | - | - | [38] | |
| C57BL/6 mice | ↑ Permeability | - | - | - | ||
| Primary HNECs; P. aeruginosa elastase (PE) | ↓ TEER (transient) | Transient ↓ claudin-1 and -4, occludin, and tricellulin (WB) | Decreased PAR-2; activation of PKC, MAPK, PI3K, p38 MAPK, JNK, COX-1 and -2, and NF-κB pathways | PAR-2 agonist; inhibitors for PKC, MAPK, PI3K, p38 MAPK, JNK, COX-1 and -2, and NF-κB pathways | [39] | |
| Calu3; P. aeruginosa elastase (PE) | ↓ TEER | ↓ localization of ZO-1 and occludin on membrane (IF, WB) | PKC signaling/ F-actin reorganization | PKC inhibitor | [40] | |
| Primary HNECs-ALI | ↓ TEER, ↑ permeability | - | endotoxin rhamnolipids | - | [41] | |
| 16HBE | ↑ Permeability | Altered distribution of ZO-1 (IF), ezrin (IF), and occludin (IF, WB) | Type III toxins (ExoS, -T, and -Y. ExoS) | - | [42] | |
| Burkholderia | 16HBE | ↓ TEER, ↑ permeability | ↓ occludin (IF) | occludin dephosphorylation | - | [43] |
| Human lung explant | ↑ Permeability | - | - | - | [44] | |
| Primary Human AECs-ALI; B. cepacia BC-7 | Invasion and destruction of epithelial cells (EM) | - | Biofilm-dependent, rearrangements of the actin cytoskeleton | - | [45] | |
| Primary Human AECs-ALI; B. cepacia HI2258 | Invasion and destruction of epithelial cells (EM) | - | Biofilm-independent | - | ||
| Primary Human AECs-ALI; B. cepacia J-1 | Invasion and destruction of epithelial cells (EM) | - | Biofilm-dependent and independent | - | ||
| Calu-3 | ↓ TEER | ↓ ZO-1 and E-cadherin (IF) | - | - | [46] | |
| Primary HBECs-ALI, 16HBE | ↓ TEER | Disrupted occludin (IF) | Increasing TNF-α cytokine | TNF-α neutralizing agent and steroids | [47] | |
| 16HBE, CF cell line (CFBE41o−) | ↓ TEER | ↓ ZO-1 (IF, WB) | - | - | [48] | |
| 16HBE, CF cell line (CFBE41o−) | ↓ TEER, ↑ permeability | ↓ ZO-1, occludin, and claudin-1 (WB) | - | - | [49] | |
| Haemophilus influenzae (H. influenzae) | Primary human alveolar epithelial cells type II, A549 | - | ↓ E-cadherin (IF, WB, mRNA), ↓ ZO-1 (IF) | FGF2 upregulation and activation of mTOR pathway | rapamycin | [50] |
| Microorganism | Model | Impacts on Airway Epithelial Barrier Function | Impacts on Apical Junctional Complex (AJC) | Mechanisms | Potential Therapeutics | Ref |
|---|---|---|---|---|---|---|
| Respiratory Syncytial Virus (RSV) | Primary mTECs -ALI | ↓ TEER, ↑ permeability | disassembly of ZO-1, occludin, β-catenin (IF), ↑ E-cadherin cleavage (WB) | - | - | [15] |
| C57BL/6J mice | ↑ Permeability | ↓ ZO-1 (IHC, WB), mislocalization of occludin (IHC), ↓ occludin (WB), ↓ claudin-1 (IHC, WB), ↑ claudin-2 (IHC, WB), ↑ E-cadherin cleavage (WB) | - | - | ||
| 16HBE | ↓ TEER, ↑ permeability | disassembly of ZO-1, occludin, E-cadherin, β-catenin (IF) | PKD activation/actin cytoskeletal remodeling | PKD inhibitors | [18] | |
| Primary NHBE cells-ALI, 16HBE | ↓ TEER, ↑ permeability | disassembly of ZO-1, occludin, E-cadherin, β-catenin (IF) | - | Forskolin, cAMP analog | [51] | |
| Primary HBECs, A549 | ↓TEER, ↑ paracellular gaps | - | p38 MAPK activation/actin cytoskeletal remodeling | p38 MAPK inhibitor | [52] | |
| 16HBE | ↓ TEER, ↑ permeability | disassembly of ZO-1, occludin, E-cadherin, β-catenin (IF) | cortactin decrease/Rap1 inhibition/actin cytoskeletal remodeling | F-actin stabilizer, Rap1 activator | [53] | |
| BALB/c mice | ↑ Permeability | ↓ occludin and claudin-1 (mRNA) | - | - | [54] | |
| Primary HNECs | ↑ TEER | ↑ claudin-4 and occludin (IF, WB, mRNA) | TGF-β1/PKCδ/HIF-1α/NF-κB | - | [55] | |
| Primary HBECs | ↓ TEER, ↑ paracellular gaps | - | inducing VEGF | VEGF antibody | [56] | |
| Human Rhinovirus (HRV) | Primary human AECs-ALI | ↓ TEER, ↑ permeability (transient) | ↓ occludin, Crumbs3, and E-cadherin (IF, WB, mRNA) | EGFR activation and Snail increase | Snail inhibition | [57] |
| Primary human AECs-ALI, 16HBE, calu-3, C57BL/6 mice | ↓ TEER, ↑ permeability | dissociation of ZO-1 from TJ (IF, IHC, WB) | - | - | [58] | |
| Primary HNECs-ALI | ↓ TEER | - | - | Betamethasone | [59] | |
| Primary NHBE cells-ALI | ↓ TEER, ↑ permeability | disruption of ZO-1 and occludin (IF) | PGC-1α decrease | PGC-1α activator | [60] | |
| Primary human AECs-ALI, NuLi-1 | ↑ Permeability | ↓ membrane ZO-1, occludin, and claudin-1 (In-Cell Western), ↓ ZO-1 (mRNA) | IL-15 | - | [61] | |
| Primary HNECs-ALI | ↓ TEER | ↓ ZO-1, occludin, claudin-1, and E-cadherin (IF, WB, mRNA) | - | - | [62] | |
| Primary HNECs-ALI | - | ↓ ZO-1, occludin, claudin-1, and E-cadherin (WB) | ROS-mediated phosphatases inhibition | NOX inhibitor | [63] | |
| Primary human AECs from asthmatic children-ALI | ↓ TEER, ↑ permeability | ↓ ZO-1 and occludin (In-Cell Western, IF), ↓ claudin-1 (In-Cell Western) | - | - | [64] | |
| Human precision-cut lung slices | - | ↓ claudin-8 (mRNA) | - | - | [65] | |
| 16HBE | ↓ TEER, ↑ bacterial transmigration | ↓ ZO-1, occludin (IF) | ROS generation | Rac1/NOX/NOX1 inhibitor, quercetin | [66] | |
| 16HBE | ↓ TEER, ↑ bacterial transmigration | ↓ occludin (IF, WB) | mitochondrial ROS generation | antioxidant targeted to mitochondria | [67] | |
| Primary HNECs-ALI | - | Disruption of ZO-1, occludin, claudin-1, and E-cadherin (WB) | ROS generation | Ginsenoside Re | [68] | |
| Primary HNECs-ALI, RPMI 2650 | - | Disruption of ZO-1, occludin, claudin-1, and E-cadherin (WB) | Akt/NF-κB and ERK1/2 activation | Wogonin | [69] | |
| Influenza A Virus (IAV) | Coculture of human alveolar epithelial and endothelial cells; H1N1 and H5N1 | ↓ TEER, ↑ permeability | ↓ AJC integrity (EM), ↓ JAM (IF), ↓ claudin-4 (IF) | - | - | [16] |
| Primary NHBE cells-ALI; H1N1 | ↓ TEER, ↑ permeability | Disassembly of ZO-1 (IF) | - | - | [17] | |
| Primary human alveolar epithelial cells type II; H1N1 | ↓ TEER, ↑ permeability | - | - | - | [70] | |
| A549, C57BL/6J mice; H1N1 | ↓ TEER ↑ permeability | ↓ ZO-1 (WB, IF) ↓ occludin (IF, WB) ↓ E-cadherin (WB) | MAPK/PI3K/Gli1/Snail activation | Gli1 Inhibitor | [71] | |
| A549, NL20, C57BL/6J mice; H5N1 | ↓ TEER | ↓ ZO-1(WB), ↓ occludin (IF, WB), ↓ claudin-1 (IF, WB), ↓ E-cadherin (WB) | TAK1/Itch- mediated protein degradation | TAK1-Itch inhibition | [72] | |
| Human Parainfluenza Virus (HPIV) | Primary human AECs-ALI; HPIV3 and HPIV5 | ↓ TEER, ↑ permeability | - | - | - | [73] |
| A549; HPIV2 | Cell-to-cell spread of HPIV2 | ↑ claudin-1 mRNA | HPIV2 V protein | Increasing claudin-1 | [74] | |
| Severe Acute Respiratory Syndrome (SARS-CoV) | MDCKII; SARS-E protein overexpression | ↓ TEER | ↓ E-cadherin and ZO-1(IF), delayed the formation of TJs (IF) | PALS1 Binding | - | [75] |
| Severe Acute Respiratory Syndrome 2 (SARS-CoV-2) | Organotypic human airway epithelial cultures-ALI | ↓ TEER | - | - | - | [76] |
| Reconstructed human bronchial epithelium-ALI | ↓ TEER, ↑ permeability (transient) | Disrupted ZO-1 (IF) | - | - | [77] | |
| Chip model of human epithelial, endothelial, and mononuclear cells | ↑ Permeability | Disrupted cell–cell contact (SEM), Disrupted E-cadherin (IF) | - | - | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, N.; Rezaee, F. Airway Epithelial Cell Junctions as Targets for Pathogens and Antimicrobial Therapy. Pharmaceutics 2022, 14, 2619. https://doi.org/10.3390/pharmaceutics14122619
Gao N, Rezaee F. Airway Epithelial Cell Junctions as Targets for Pathogens and Antimicrobial Therapy. Pharmaceutics. 2022; 14(12):2619. https://doi.org/10.3390/pharmaceutics14122619
Chicago/Turabian StyleGao, Nannan, and Fariba Rezaee. 2022. "Airway Epithelial Cell Junctions as Targets for Pathogens and Antimicrobial Therapy" Pharmaceutics 14, no. 12: 2619. https://doi.org/10.3390/pharmaceutics14122619