
Selection of an Aptamer against the Enzyme 1-deoxy-D-xylulose-5-phosphate Reductoisomerase from Plasmodium falciparum
 , , , and
, , , and 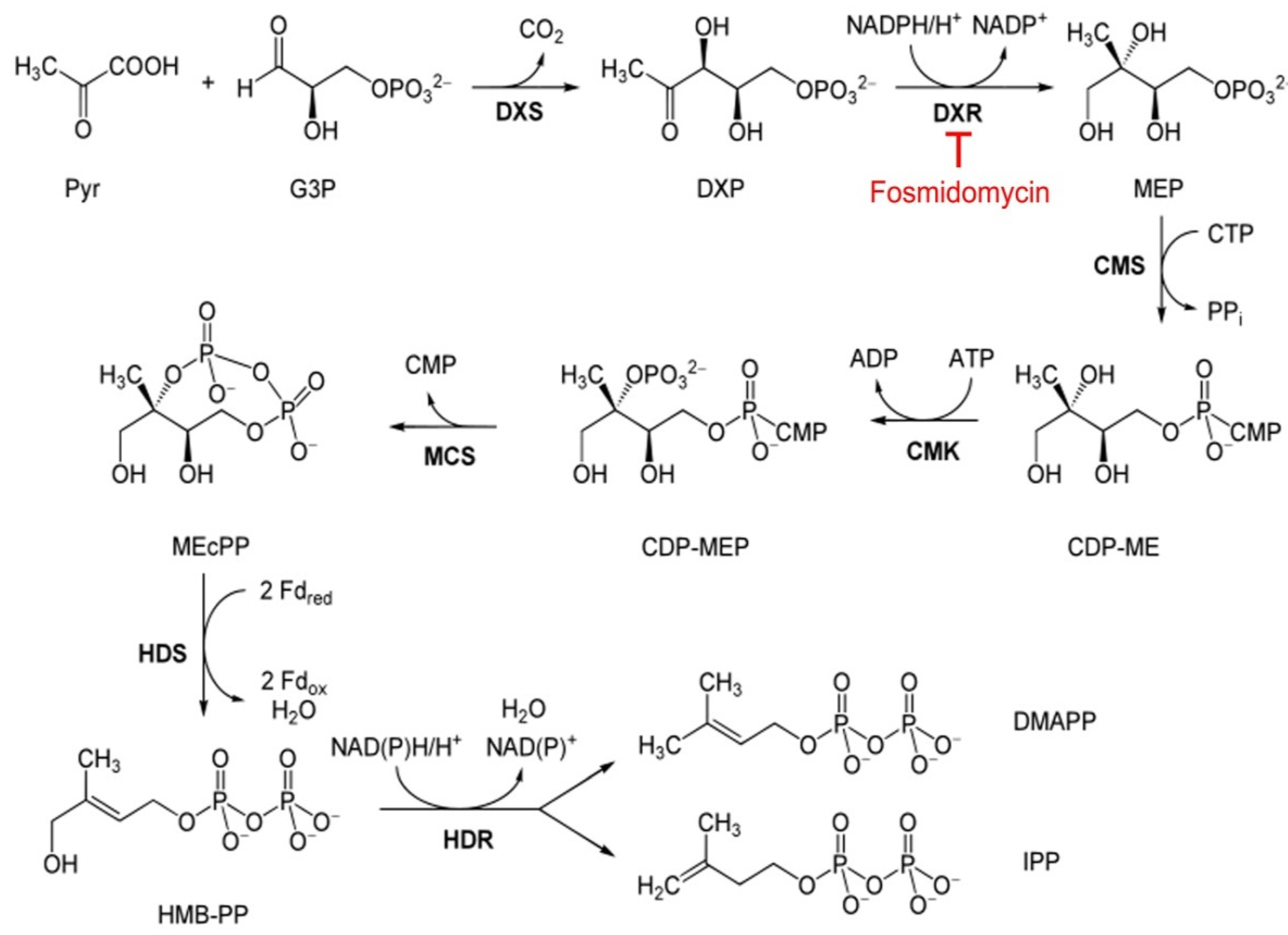{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Recombinant Enzymes
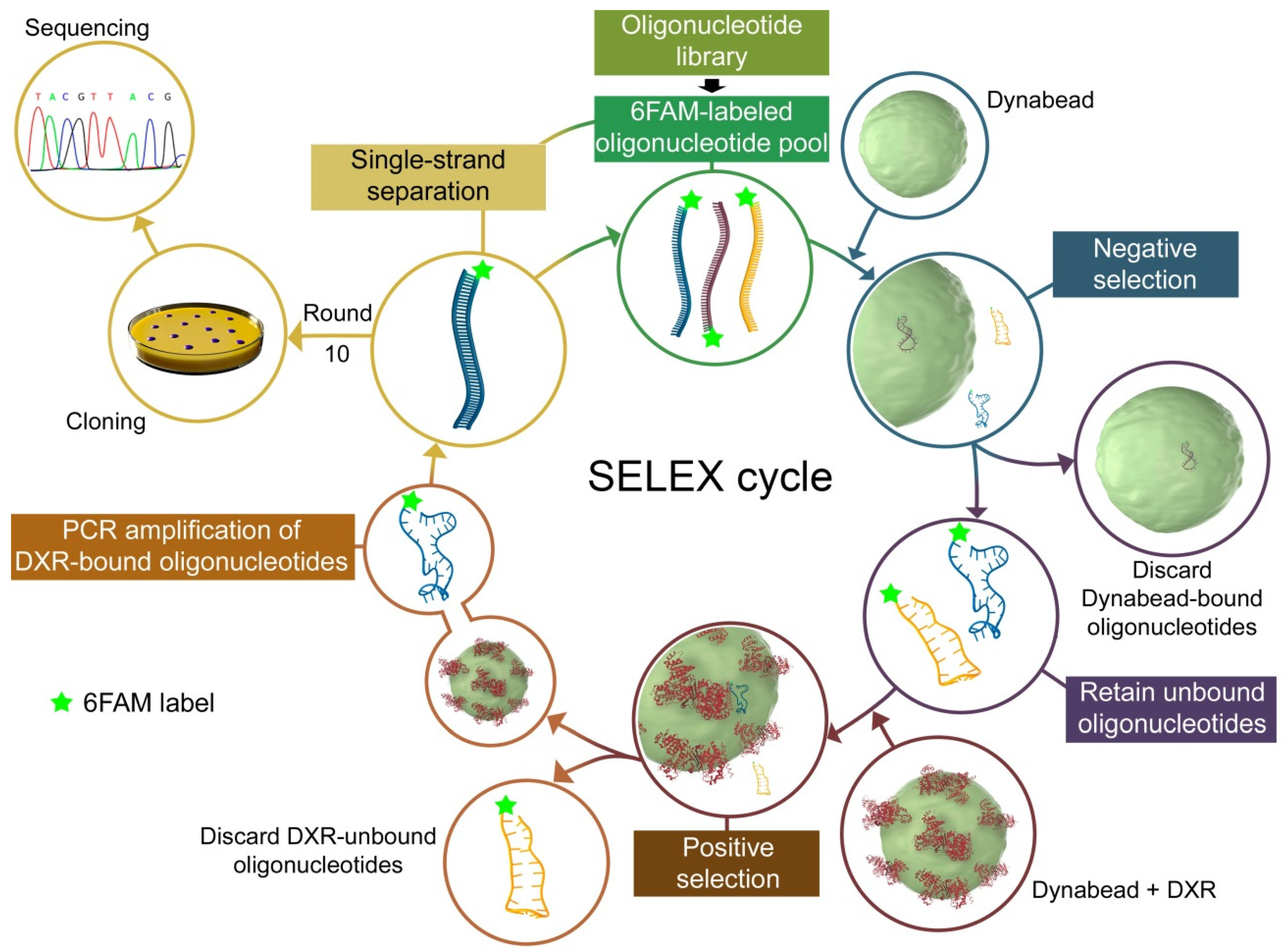
2.2. In Vitro Selection of DNA Aptamers
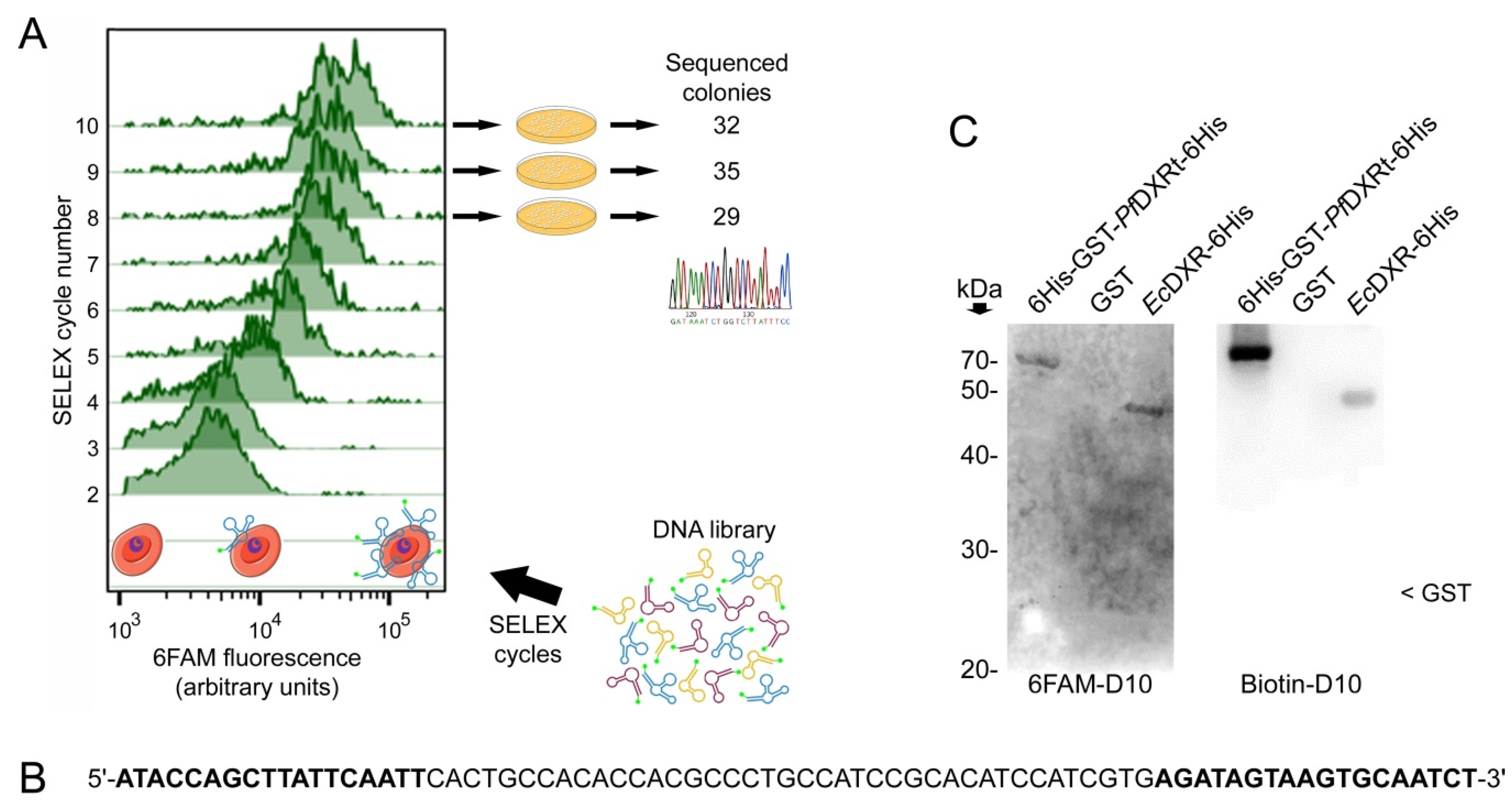
2.3. Subcloning and Sequencing of Candidate Oligonucleotides
2.4. Computational Analysis
2.5. Western Blots
2.6. Electrophoretic Mobility Shift Assay
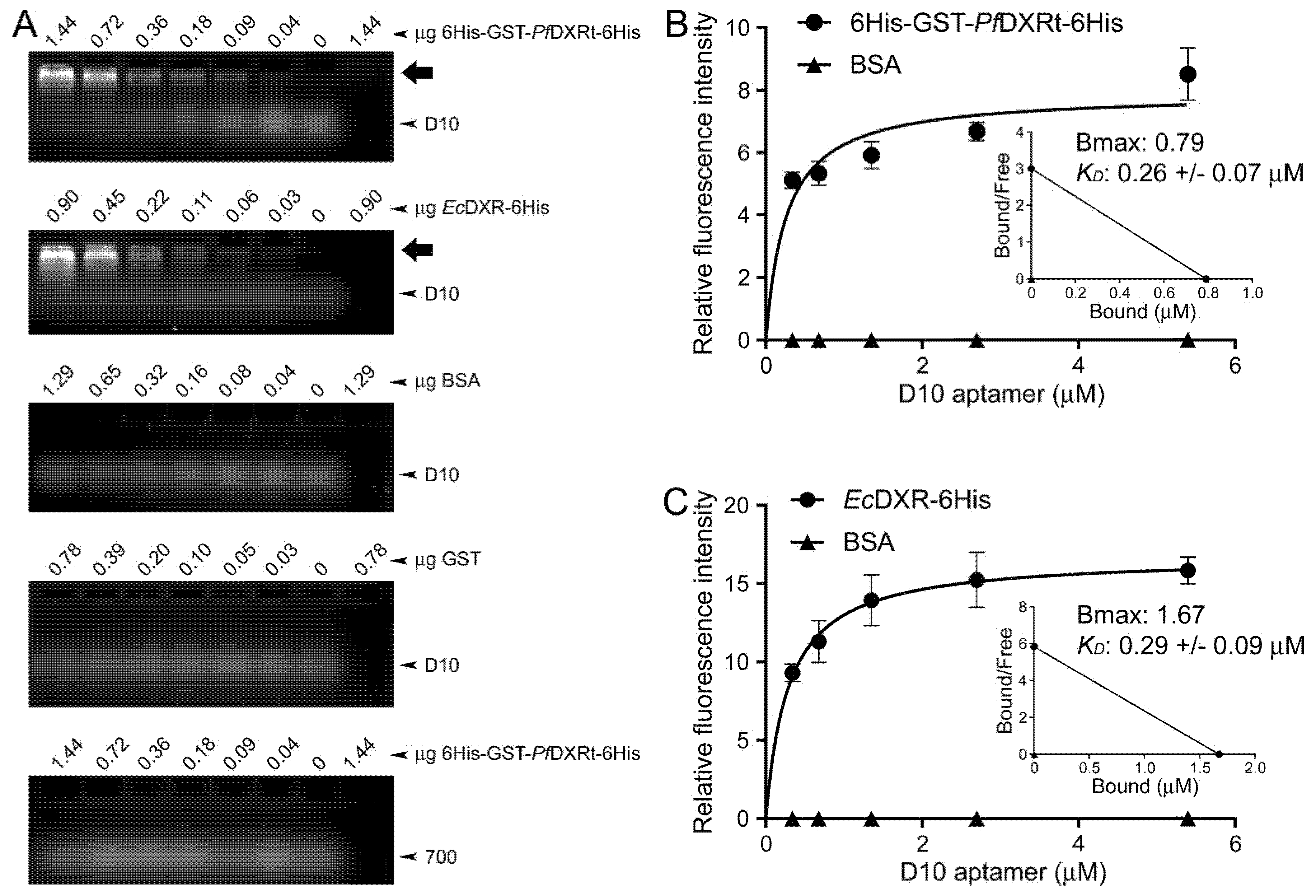
2.7. Determination of Aptamer Dissociation Constants
2.8. DXR Activity Assay
2.9. P. falciparum Cultures
2.10. Confocal Fluorescence Microscopy and Flow Cytometry Analysis
2.11. Bacterial Cultures
2.12. Ethics Statement
3. Results and Discussion
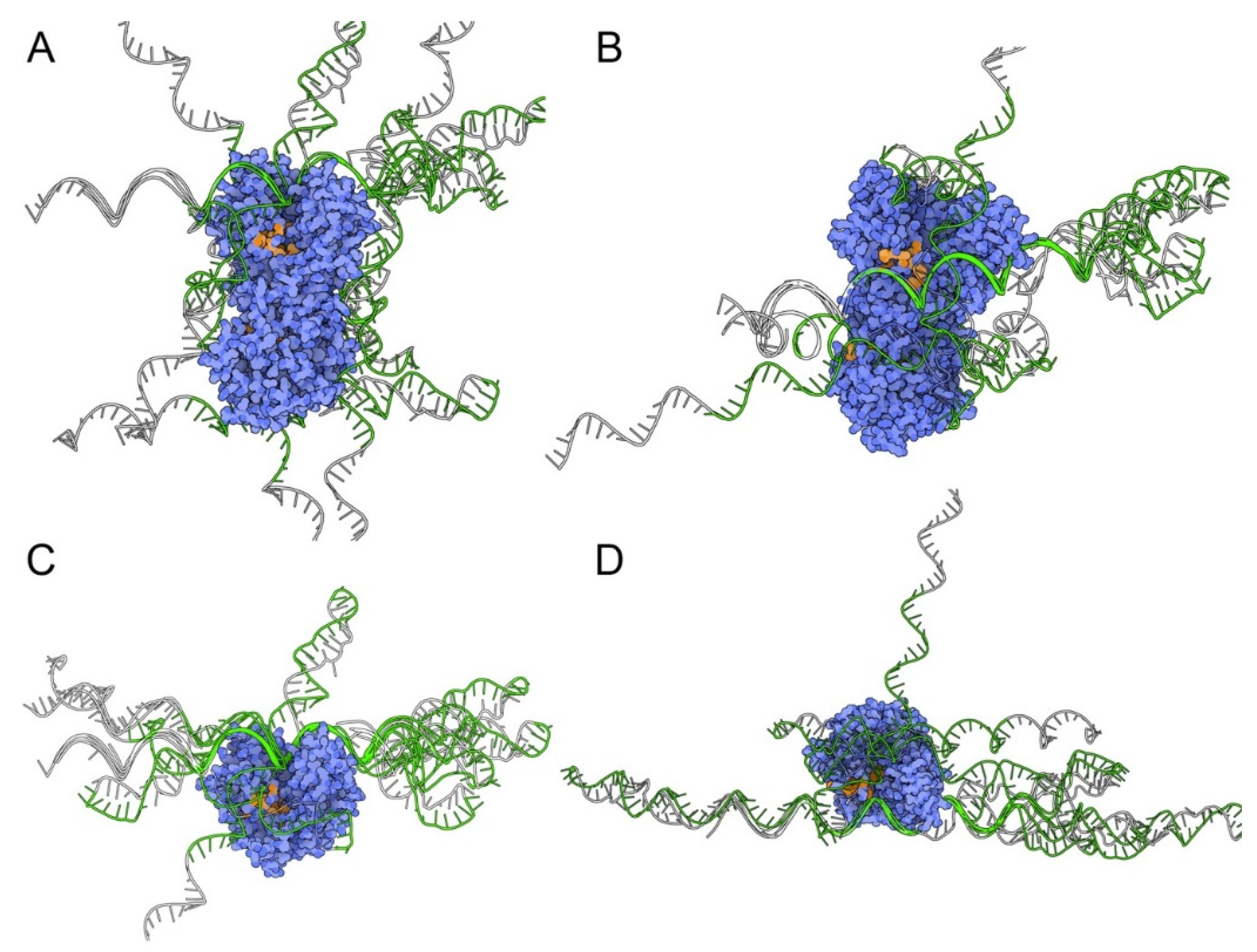
3.1. Aptamer Selection
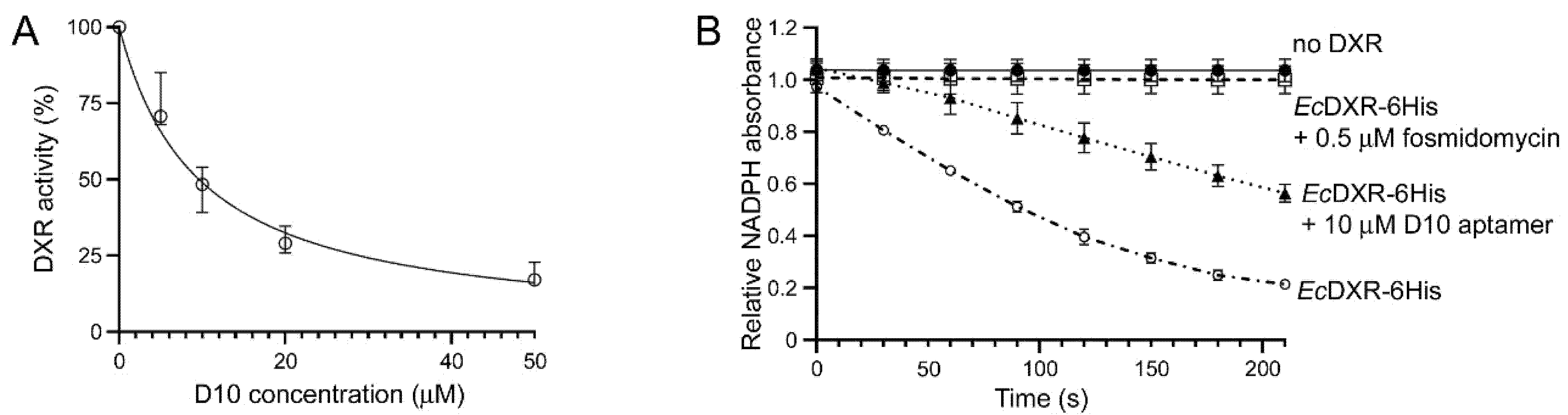
3.2. Characterization of Aptamer Binding to DXR In Vitro
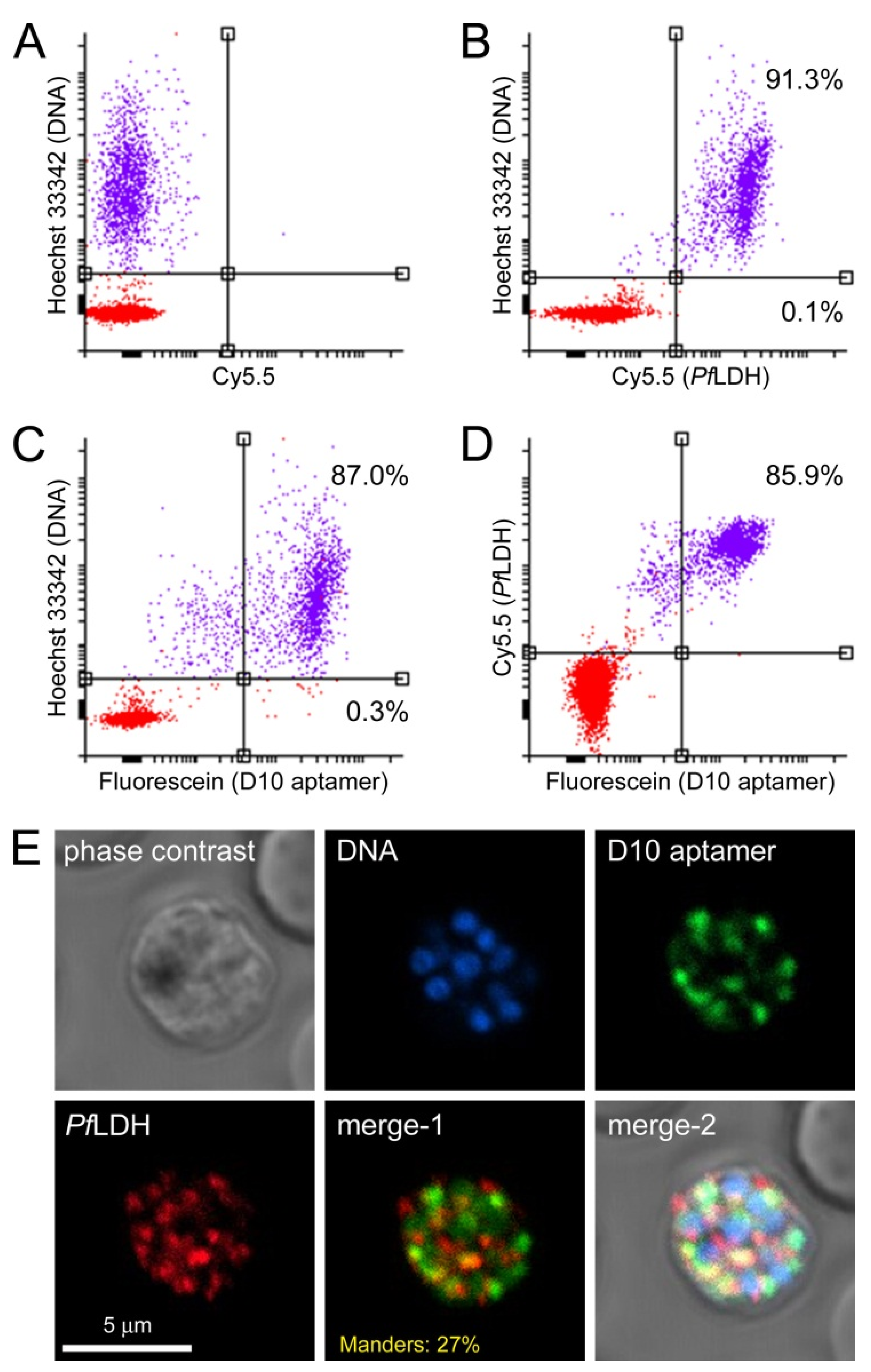
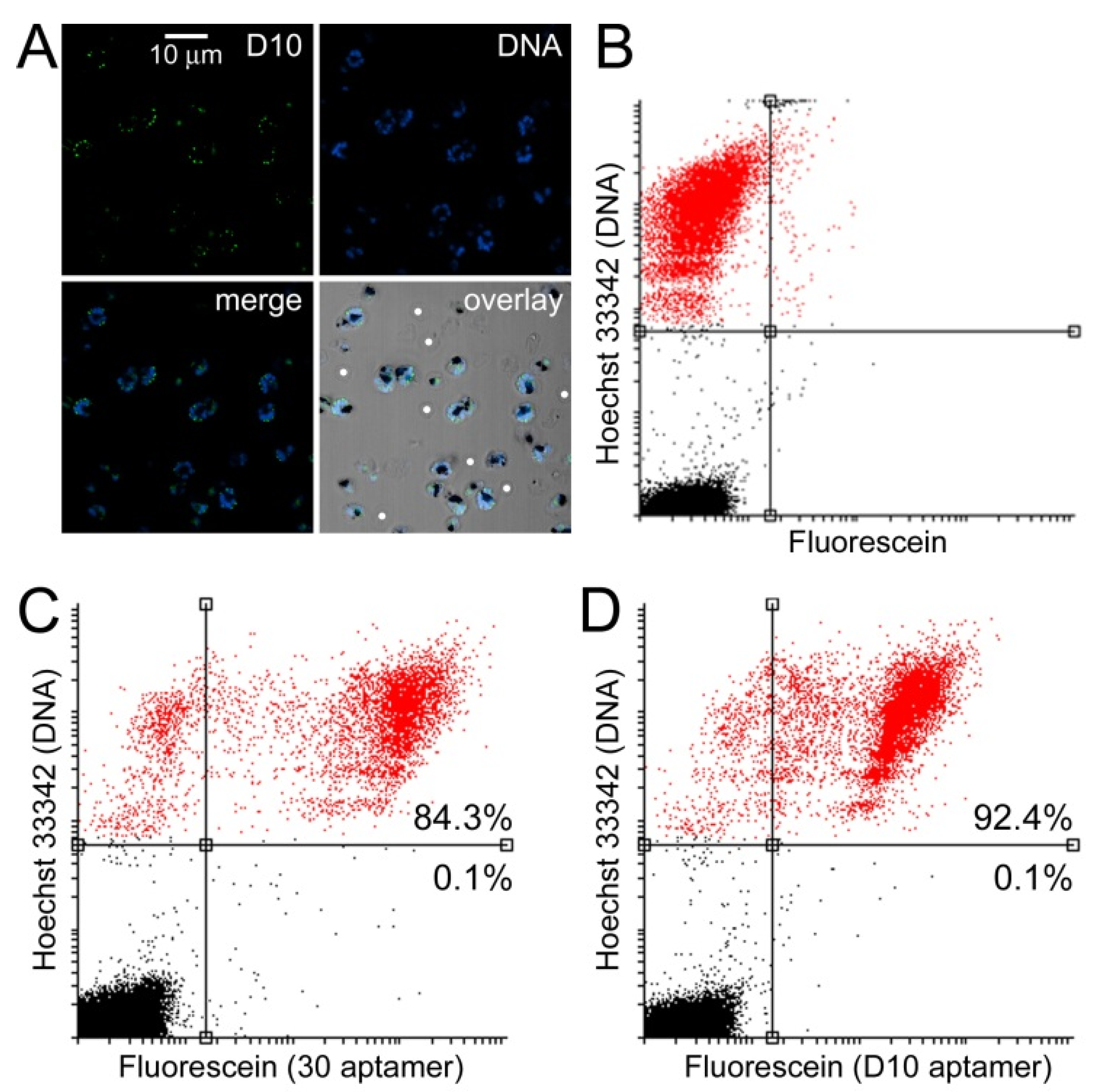
3.3. Characterization of Aptamer Binding to Plasmodium Cells
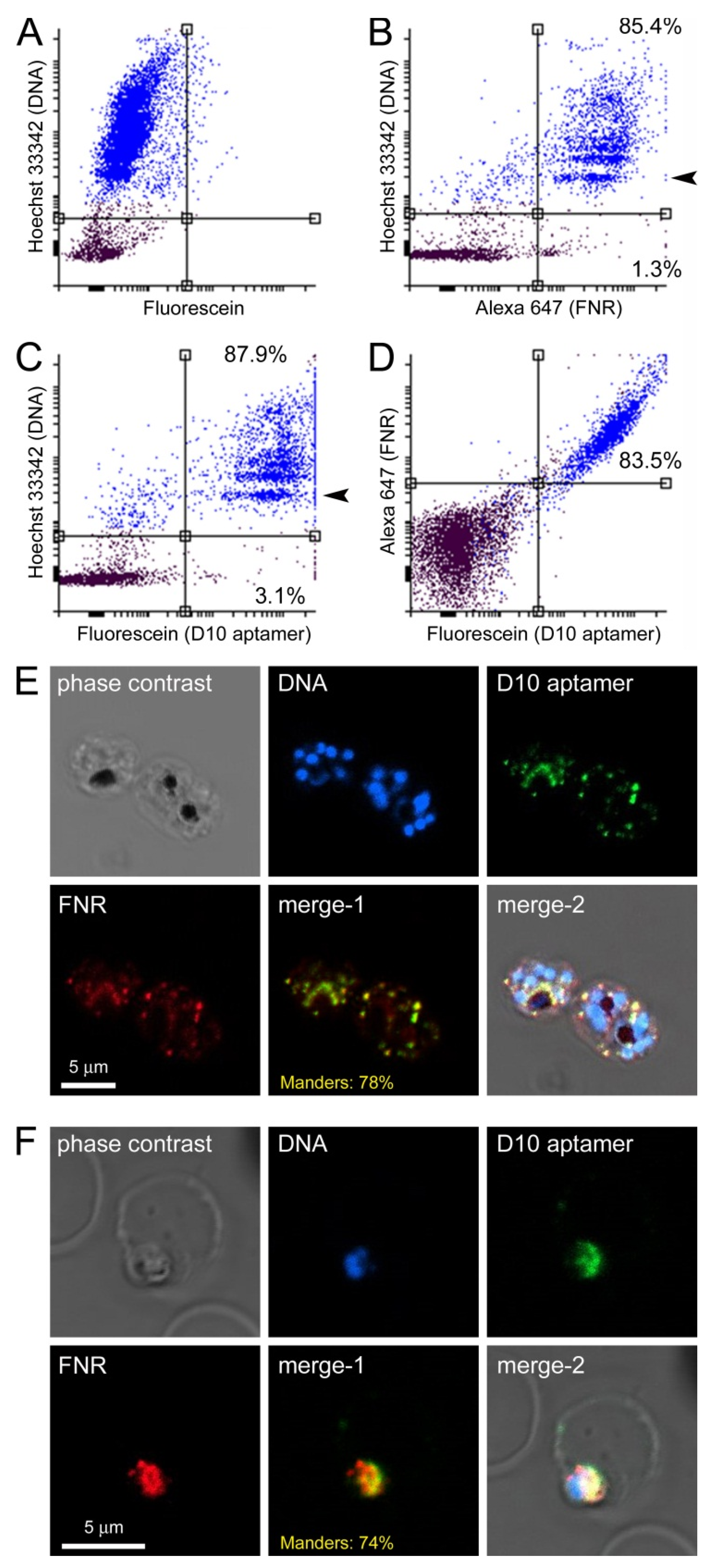
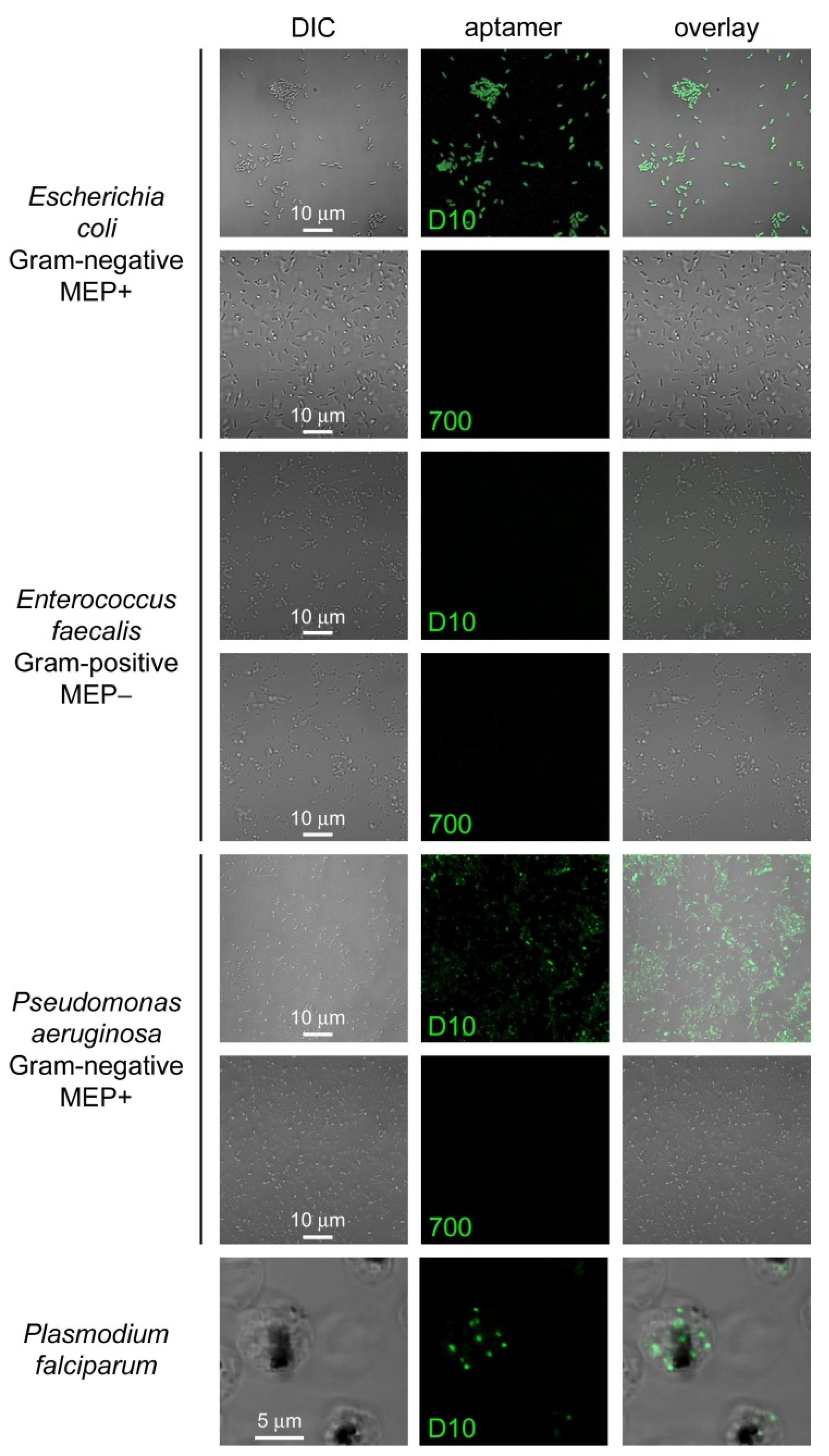
3.4. Characterization of Aptamer Binding to Bacterial Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. 2021. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed on 13 July 2022).
- Sato, S. Plasmodium—A brief introduction to the parasites causing human malaria and their basic biology. J. Physiol. Anthropol. 2021, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Ghosh, S.; Zhang, L.; Lal, A.; Jang, S.W.; Ju, A.; Zhang, S.; Wang, X.; Ralph, S.A.; Zou, J.; et al. Integrative proteomics and bioinformatic prediction enable a high-confidence apicoplast proteome in malaria parasites. PLoS Biol. 2018, 16, e2005895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, E.; de Risi, J.L. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011, 9, e1001138. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.D.; McFadden, G.I. Targeting apicoplasts in malaria parasites. Expert Opin. Ther. Targets 2013, 17, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Dahl, E.L.; Rosenthal, P.J. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 2007, 51, 3485–3490. [Google Scholar] [CrossRef] [Green Version]
- Dahl, E.L.; Rosenthal, P.J. Apicoplast translation, transcription and genome replication: Targets for antimalarial antibiotics. Trends Parasitol. 2008, 24, 279–284. [Google Scholar] [CrossRef]
- Kennedy, K.; Cobbold, S.A.; Hanssen, E.; Birnbaum, J.; Spillman, N.J.; McHugh, E.; Brown, H.; Tilley, L.; Spielmann, T.; McConville, M.J.; et al. Delayed death in the malaria parasite Plasmodium falciparum is caused by disruption of prenylation-dependent intracellular trafficking. PLoS Biol. 2019, 17, e3000376. [Google Scholar] [CrossRef] [Green Version]
- Biosca, A.; Ramírez, M.; Gomez-Gomez, A.; Lafuente, A.; Iglesias, V.; Pozo, O.J.; Imperial, S.; Fernàndez-Busquets, X. Characterization of domiphen bromide as a new fast-acting antiplasmodial agent inhibiting the apicoplastidic methyl erythritol phosphate pathway. Pharmaceutics 2022, 14, 1320. [Google Scholar] [CrossRef]
- Chakraborty, A. Understanding the biology of the Plasmodium falciparum apicoplast; an excellent target for antimalarial drug development. Life Sci. 2016, 158, 104–110. [Google Scholar] [CrossRef]
- Ramya, T.N.; Mishra, S.; Karmodiya, K.; Surolia, N.; Surolia, A. Inhibitors of nonhousekeeping functions of the apicoplast defy delayed death in Plasmodium falciparum. Antimicrob. Agents Chemother. 2007, 51, 307–316. [Google Scholar] [CrossRef]
- Waller, R.F.; Ralph, S.A.; Reed, M.B.; Su, V.; Douglas, J.D.; Minnikin, D.E.; Cowman, A.F.; Besra, G.S.; McFadden, G.I. A type II pathway for fatty acid biosynthesis presents drug targets in Plasmodium falciparum. Antimicrob. Agents Chemother. 2003, 47, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Naik, R.S.; Davidson, E.A.; Gowda, D.C. Developmental stage-specific biosynthesis of glycosylphosphatidylinositol anchors in intraerythrocytic Plasmodium falciparum and its inhibition in a novel manner by mannosamine. J. Biol. Chem. 2000, 275, 24506–24511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meer, J.Y.; Hirsch, A.K. The isoprenoid-precursor dependence of Plasmodium spp. Nat. Prod. Rep. 2012, 29, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Qidwai, T.; Priya, A.; Khan, N.A.; Tripathi, H.; Khan, F.; Darokar, M.P.; Pal, A.; Bawankule, D.U.; Shukla, R.K.; Bhakuni, R.S. Antimalarial drug targets and drugs targeting dolichol metabolic pathway of Plasmodium falciparum. Curr. Drug Targets 2014, 15, 374–409. [Google Scholar] [CrossRef]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggu, G.S.; Pala, Z.R.; Garg, S.; Saxena, V. New insight into isoprenoids biosynthesis process and future prospects for drug designing in Plasmodium. Front. Microbiol. 2016, 7, 1421. [Google Scholar] [CrossRef] [Green Version]
- Goble, J.L.; Adendorff, M.R.; de Beer, T.A.; Stephens, L.L.; Blatch, G.L. The malarial drug target Plasmodium falciparum 1-deoxy-D-xylulose-5-phosphate reductoisomerase (PfDXR): Development of a 3-D model for identification of novel, structural and functional features and for inhibitor screening. Protein Pept. Lett. 2010, 17, 109–120. [Google Scholar] [CrossRef]
- Kholodar, S.A.; Allen, C.L.; Gulick, A.M.; Murkin, A.S. The role of phosphate in a multistep enzymatic reaction: Reactions of the substrate and intermediate in pieces. J. Am. Chem. Soc. 2015, 137, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; McFadden, G.I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Giménez-Oya, V.; Villacañas, O.; Obiol-Pardo, C.; Antolin-Llovera, M.; Rubio-Martinez, J.; Imperial, S. Design of novel ligands of CDP-methylerythritol kinase by mimicking direct protein-protein and solvent-mediated interactions. J. Mol. Recognit. 2011, 24, 71–80. [Google Scholar] [CrossRef]
- da Silva, M.F.; Saito, A.Y.; Peres, V.J.; Oliveira, A.C.; Katzin, A.M. In vitro antimalarial activity of different inhibitors of the plasmodial isoprenoid synthesis pathway. Antimicrob. Agents Chemother. 2015, 59, 5084–5087. [Google Scholar] [CrossRef] [Green Version]
- Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M.; Turbachova, I.; Eberl, M.; Zeidler, J.; Lichtenthaler, H.K.; et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, J.; Ziemann, C.; Hintz, M.; Reichenberg, A.; Ortmann, R.; Schlitzer, M.; Fuhst, R.; Timmesfeld, N.; Vilcinskas, A.; Jomaa, H. FR-900098, an antimalarial development candidate that inhibits the non-mevalonate isoprenoid biosynthesis pathway, shows no evidence of acute toxicity and genotoxicity. Virulence 2016, 7, 718–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization WHO Global Technical Strategy for Malaria 2016–2030. 2015. Available online: http://www.who.int/malaria/areas/global_technical_strategy/en/ (accessed on 13 July 2022).
- Menard, D.; Dondorp, A. Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619. [Google Scholar] [CrossRef] [Green Version]
- Sola, M.; Menon, A.P.; Moreno, B.; Meraviglia-Crivelli, D.; Soldevilla, M.M.; Cartón-García, F.; Pastor, F. Aptamers against live targets: Is in vivo SELEX finally coming to the edge? Mol. Ther. Nucleic Acids 2020, 21, 192–204. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ospina-Villa, J.D.; López-Camarillo, C.; Castañón-Sánchez, C.A.; Soto-Sánchez, J.; Ramírez-Moreno, E.; Marchat, L.A. Advances on aptamers against protozoan parasites. Genes 2018, 9, 584. [Google Scholar] [CrossRef] [Green Version]
- Parashar, A. Aptamers in therapeutics. J. Clin. Diagn. Res. 2016, 10, BE01–BE06. [Google Scholar] [CrossRef]
- Barfod, A.; Persson, T.; Lindh, J. In vitro selection of RNA aptamers against a conserved region of the Plasmodium falciparum erythrocyte membrane protein 1. Parasitol. Res. 2009, 105, 1557–1566. [Google Scholar] [CrossRef] [Green Version]
- Niles, J.C.; de Risi, J.L.; Marletta, M.A. Inhibiting Plasmodium falciparum growth and heme detoxification pathway using heme-binding DNA aptamers. Proc. Natl. Acad. Sci. USA 2009, 106, 13266–13271. [Google Scholar] [CrossRef]
- Cheung, Y.W.; Dirkzwager, R.M.; Wong, W.C.; Cardoso, J.; D’Arc Neves, C.J.; Tanner, J.A. Aptamer-mediated Plasmodium-specific diagnosis of malaria. Biochimie 2018, 145, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Lantero, E.; Belavilas-Trovas, A.; Biosca, A.; Recolons, P.; Moles, E.; Sulleiro, E.; Zarzuela, F.; Avalos-Padilla, Y.; Ramírez, M.; Fernàndez-Busquets, X. Development of DNA aptamers against Plasmodium falciparum blood stages using cell-systematic evolution of ligands by exponential enrichment. J. Biomed. Nanotechnol. 2020, 16, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. FluMag-SELEX as an advantageous method for DNA aptamer selection. Anal. Bioanal. Chem. 2005, 383, 83–91. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX-a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef]
- Roos, D.S.; Crawford, M.J.; Donald, R.G.; Fraunholz, M.; Harb, O.S.; He, C.Y.; Kissinger, J.C.; Shaw, M.K.; Striepen, B. Mining the Plasmodium genome database to define organellar function: What does the apicoplast do? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2002, 357, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, T.; Tanaka, N.; Kusakabe, Y.; Nakanishi, M.; Kitade, Y.; Nakamura, K.T. Crystallization and preliminary X-ray crystallographic study of 1-deoxy-D-xylulose 5-phosphate reductoisomerase from Plasmodium falciparum. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2010, 66 Pt 3, 330–332. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.M.; Jarmusik, N.A.; Endrizzi, E.J.; Leipzig, N.D. Expression, isolation, and purification of soluble and insoluble biotinylated proteins for nerve tissue regeneration. J. Vis. Exp. 2014, 83, e51295. [Google Scholar]
- Stoltenburg, R.; Schubert, T.; Strehlitz, B. In vitro selection and interaction studies of a DNA aptamer targeting Protein A. PLoS ONE 2015, 10, e0134403. [Google Scholar] [CrossRef] [Green Version]
- Cline, J.; Braman, J.C.; Hogrefe, H.H. PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996, 24, 3546–3551. [Google Scholar] [CrossRef] [Green Version]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Li, S.; Olson, W.K.; Lu, X.J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res. 2019, 47, W26–W34. [Google Scholar] [CrossRef] [Green Version]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the FoldX forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. S9), 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Tomasello, G.; Armenia, I.; Molla, G. The Protein Imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, H.; Sefah, K.; Liu, B.; Pu, Y.; Van, S.D.; Tan, W. Selection of aptamers specific for adipose tissue. PLoS ONE 2012, 7, e37789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cranmer, S.L.; Magowan, C.; Liang, J.; Coppel, R.L.; Cooke, B.M. An alternative to serum for cultivation of Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 363–365. [Google Scholar] [CrossRef]
- Cheung, Y.W.; Kwok, J.; Law, A.W.L.; Watt, R.M.; Kotaka, M.; Tanner, J.A. Structural basis for discriminatory recognition of Plasmodium lactate dehydrogenase by a DNA aptamer. Proc. Natl. Acad. Sci. USA. 2013, 110, 15967–15972. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224 Pt 3, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Froger, A.; Hall, J.E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 2007, 6, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.K.; Chakma, B.; Jain, P.; Goswami, P. Protein-induced fluorescence enhancement based detection of Plasmodium falciparum glutamate dehydrogenase using carbon dot coupled specific aptamer. ACS Comb. Sci. 2018, 20, 350–357. [Google Scholar] [CrossRef]
- Giessmann, D.; Heidler, P.; Haemers, T.; Van Calenbergh, S.; Reichenberg, A.; Jomaa, H.; Weidemeyer, C.; Sanderbrand, S.; Wiesner, J.; Link, A. Towards new antimalarial drugs: Synthesis of non-hydrolyzable phosphate mimics as feed for a predictive QSAR study on 1-deoxy-D-xylulose-5-phosphate reductoisomerase inhibitors. Chem. Biodivers. 2008, 5, 643–656. [Google Scholar] [CrossRef]
- Joseph, D.F.; Nakamoto, J.A.; Garcia Ruiz, O.A.; Peñaranda, K.; Sanchez-Castro, A.E.; Castillo, P.S.; Milón, P. DNA aptamers for the recognition of HMGB1 from Plasmodium falciparum. PLoS ONE 2019, 14, e0211756. [Google Scholar] [CrossRef] [Green Version]
- Frith, K.A.; Fogel, R.; Goldring, J.P.D.; Krause, R.G.E.; Khati, M.; Hoppe, H.; Cromhout, M.E.; Jiwaji, M.; Limson, J.L. Towards development of aptamers that specifically bind to lactate dehydrogenase of Plasmodium falciparum through epitopic targeting. Malar. J. 2018, 17, 191. [Google Scholar] [CrossRef] [Green Version]
- Mitkevich, O.V.; Kochneva-Pervukhova, N.V.; Surina, E.R.; Benevolensky, S.V.; Kushnirov, V.V.; Ter-Avanesyan, M.D. DNA aptamers detecting generic amyloid epitopes. Prion 2012, 6, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Godonoga, M.; Lin, T.Y.; Oshima, A.; Sumitomo, K.; Tang, M.S.L.; Cheung, Y.W.; Kinghorn, A.B.; Dirkzwager, R.M.; Zhou, C.; Kuzuya, A.; et al. A DNA aptamer recognising a malaria protein biomarker can function as part of a DNA origami assembly. Sci. Rep. 2016, 6, 21266. [Google Scholar] [CrossRef] [Green Version]
- Sooriyaarachchi, S.; Chofor, R.; Risseeuw, M.D.; Bergfors, T.; Pouyez, J.; Dowd, C.S.; Maes, L.; Wouters, J.; Jones, T.A.; Van Calenbergh, S.; et al. Targeting an aromatic hotspot in Plasmodium falciparum 1-deoxy-D-xylulose-5-phosphate reductoisomerase with b-arylpropyl analogues of fosmidomycin. ChemMedChem 2016, 11, 2024–2036. [Google Scholar] [CrossRef]
- Chofor, R.; Sooriyaarachchi, S.; Risseeuw, M.D.; Bergfors, T.; Pouyez, J.; Johny, C.; Haymond, A.; Everaert, A.; Dowd, C.S.; Maes, L.; et al. Synthesis and bioactivity of beta-substituted fosmidomycin analogues targeting 1-deoxy-D-xylulose-5-phosphate reductoisomerase. J. Med. Chem. 2015, 58, 2988–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, T.; Tanaka, N.; Kusakabe, Y.; Nakanishi, M.; Kitade, Y.; Nakamura, K.T. Molecular basis of fosmidomycin’s action on the human malaria parasite Plasmodium falciparum. Sci. Rep. 2011, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Mac Sweeney, A.; Lange, R.; Fernandes, R.P.; Schulz, H.; Dale, G.E.; Douangamath, A.; Proteau, P.J.; Oefner, C. The crystal structure of E.coli 1-deoxy-D-xylulose-5-phosphate reductoisomerase in a ternary complex with the antimalarial compound fosmidomycin and NADPH reveals a tight-binding closed enzyme conformation. J. Mol. Biol. 2005, 345, 115–127. [Google Scholar] [CrossRef]
- Birch, C.M.; Hou, H.W.; Han, J.; Niles, J.C. Identification of malaria parasite-infected red blood cell surface aptamers by inertial microfluidic SELEX (I-SELEX). Sci. Rep. 2015, 5, 11347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra-Pérez, N.; Ramos, E.; García-Hernández, M.; Pinto, C.; Soto, M.; Martín, M.E.; González, V.M. Molecular and functional characterization of ssDNA aptamers that specifically bind Leishmania infantum PABP. PLoS ONE 2015, 10, e0140048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lijuan, C.; Xing, Y.; Minxi, W.; Wenkai, L.; Le, D. Development of an aptamer-ampicillin conjugate for treating biofilms. Biochem. Biophys. Res. Commun. 2017, 483, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.; Vahed, S.Z.; Ahmadian, E.; Dizaj, S.M.; Eftekhari, A.; Khalilov, R.; Ahmadi, M.; Hamidi-Asl, E.; Labib, M. Detection of pathogenic bacteria via nanomaterials-modified aptasensors. Biosens. Bioelectron. 2020, 150, 111933. [Google Scholar] [CrossRef]
- Heuston, S.; Begley, M.; Gahan, C.G.M.; Hill, C. Isoprenoid biosynthesis in bacterial pathogens. Microbiology 2012, 158 Pt 6, 1389–1401. [Google Scholar] [CrossRef]
- Zacco, E.; Kantelberg, O.; Milanetti, E.; Armaos, A.; Panei, F.P.; Gregory, J.; Jeacock, K.; Clarke, D.J.; Chandran, S.; Ruocco, G.; et al. Probing TDP-43 condensation using an in silico designed aptamer. Nat. Commun. 2022, 13, 3306. [Google Scholar] [CrossRef] [PubMed]
- Berzosa, P.; de Lucio, A.; Romay-Barja, M.; Herrador, Z.; González, V.; García, L.; Fernández-Martínez, A.; Santana-Morales, M.; Ncogo, P.; Valladares, B.; et al. Comparison of three diagnostic methods (microscopy, RDT, and PCR) for the detection of malaria parasites in representative samples from Equatorial Guinea. Malar. J. 2018, 17, 333. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Pande, V.; Bhatt, R.M.; Shah, N.K.; Mishra, N.; Srivastava, B.; Valecha, N.; Anvikar, A.R. Genetic deletion of HRP2 and HRP3 in Indian Plasmodium falciparum population and false negative malaria rapid diagnostic test. Acta Trop. 2013, 125, 119–121. [Google Scholar] [CrossRef]
- Koita, O.A.; Doumbo, O.K.; Ouattara, A.; Tall, L.K.; Konaré, A.; Diakité, M.; Diallo, M.; Sagara, I.; Masinde, G.L.; Doumbo, S.N.; et al. False-negative rapid diagnostic tests for malaria and deletion of the histidine-rich repeat region of the hrp2 gene. Am. J. Trop. Med. Hyg. 2012, 86, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, A.M.; Marques, R.F.; Regiart, M.; Bargieri, D.Y. Diagnostic methods for non-falciparum malaria. Front. Cell. Infect. Microbiol. 2021, 11, 681063. [Google Scholar] [CrossRef]
- Mayor, A.; Bassat, Q. “Resistance” to diagnostics: A serious biological challenge for malaria control and elimination. EBioMedicine 2019, 50, 9–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Manjunatha, D.H.; Jeon, W.; Ban, C. Cationic surfactant-based colorimetric detection of Plasmodium lactate dehydrogenase, a biomarker for malaria, using the specific DNA aptamer. PLoS ONE 2014, 9, e100847. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.J.; Ban, C. Crystal structure of a DNA aptamer bound to PvLDH elucidates novel single-stranded DNA structural elements for folding and recognition. Sci. Rep. 2016, 6, 34998. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Chakma, B.; Singh, N.K.; Patra, S.; Goswami, P. Aromatic surfactant as aggregating agent for aptamer-gold nanoparticle-based detection of Plasmodium lactate dehydrogenase. Mol. Biotechnol. 2016, 58, 497–508. [Google Scholar] [CrossRef]
- Fraser, L.A.; Kinghorn, A.B.; Dirkzwager, R.M.; Liang, S.; Cheung, Y.W.; Lim, B.; Shiu, S.C.; Tang, M.S.L.; Andrew, D.; Manitta, J.; et al. A portable microfluidic Aptamer-Tethered Enzyme Capture (APTEC) biosensor for malaria diagnosis. Biosens. Bioelectron. 2018, 100, 591–596. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; 2015. Available online: http://apps.who.int/iris/bitstream/10665/162441/1/9789241549127_eng.pdf (accessed on 13 July 2022).
- Figueroa-Miranda, G.; Chen, S.; Neis, M.; Zhou, L.; Zhang, Y.; Lo, Y.; Tanner, J.A.; Kreidenweiss, A.; Offenhäusser, A.; Mayer, D. Multi-target electrochemical malaria aptasensor on flexible multielectrode arrays for detection in malaria parasite blood samples. Sens. Actuators B Chem. 2021, 349, 130812. [Google Scholar] [CrossRef]
- Linzke, M.; Yan, S.L.R.; Tárnok, A.; Ulrich, H.; Groves, M.R.; Wrenger, C. Live and let dye: Visualizing the cellular compartments of the malaria parasite Plasmodium falciparum. Cytom. A 2020, 97, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Kimata-Ariga, Y.; Morihisa, R. Effect of artemisinin on the redox system of NADPH/FNR/ferredoxin from malaria parasites. Antioxidants 2022, 11, 273. [Google Scholar] [CrossRef]
- Swift, R.P.; Rajaram, K.; Elahi, R.; Liu, H.B.; Prigge, S.T. Roles of ferredoxin-dependent proteins in the apicoplast of Plasmodium falciparum parasites. mBio 2022, 13, e0302321. [Google Scholar] [CrossRef]
- Gallagher, J.R.; Prigge, S.T. Plasmodium falciparum acyl carrier protein crystal structures in disulfide-linked and reduced states and their prevalence during blood stage growth. Proteins 2010, 78, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Kehr, S.; Sturm, N.; Rahlfs, S.; Przyborski, J.M.; Becker, K. Compartmentation of redox metabolism in malaria parasites. PLoS Pathog. 2010, 6, e1001242. [Google Scholar] [CrossRef] [PubMed]
- Walczak, M.; Ganesan, S.M.; Niles, J.C.; Yeh, E. ATG8 Is essential specifically for an autophagy-independent function in apicoplast biogenesis in blood-stage malaria parasites. mBio 2018, 9, e02021-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutsiopoulou, A.; Broyles, D.; Dikici, E.; Daunert, S.; Deo, S.K. Molecular aptamer beacons and their applications in sensing, imaging, and diagnostics. Small 2019, 15, e1902248. [Google Scholar] [CrossRef]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-SELEX. Nat. Protoc. 2010, 5, 1169. [Google Scholar] [CrossRef]
- Zeng, Y.; Qi, P.; Wang, Y.; Chen, C.; Zhang, D. DNA pom-pom nanostructure as a multifunctional platform for pathogenic bacteria determination and inactivation. Biosens. Bioelectron. 2021, 177, 112982. [Google Scholar] [CrossRef]
- McConnell, E.M.; Nguyen, J.; Li, Y. Aptamer-based biosensors for environmental monitoring. Front. Chem. 2020, 8, 434. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roca, C.; Avalos-Padilla, Y.; Prieto-Simón, B.; Iglesias, V.; Ramírez, M.; Imperial, S.; Fernàndez-Busquets, X. Selection of an Aptamer against the Enzyme 1-deoxy-D-xylulose-5-phosphate Reductoisomerase from Plasmodium falciparum. Pharmaceutics 2022, 14, 2515. https://doi.org/10.3390/pharmaceutics14112515
Roca C, Avalos-Padilla Y, Prieto-Simón B, Iglesias V, Ramírez M, Imperial S, Fernàndez-Busquets X. Selection of an Aptamer against the Enzyme 1-deoxy-D-xylulose-5-phosphate Reductoisomerase from Plasmodium falciparum. Pharmaceutics. 2022; 14(11):2515. https://doi.org/10.3390/pharmaceutics14112515
Chicago/Turabian StyleRoca, Carlota, Yunuen Avalos-Padilla, Beatriz Prieto-Simón, Valentín Iglesias, Miriam Ramírez, Santiago Imperial, and Xavier Fernàndez-Busquets. 2022. "Selection of an Aptamer against the Enzyme 1-deoxy-D-xylulose-5-phosphate Reductoisomerase from Plasmodium falciparum" Pharmaceutics 14, no. 11: 2515. https://doi.org/10.3390/pharmaceutics14112515