

Development and Characterisation of a Topical Methyl Salicylate Patch: Effect of Solvents on Adhesion and Skin Permeation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Solvent Selection
2.2.1.1. Miscibility and Saturated Solubility Studies of MS
2.2.1.2. ATR-FTIR Spectroscopy
2.2.1.3. In Vitro Permeation and Mass Balance Studies
2.2.1.4. HPLC Analysis
2.3. Patch Formulation
2.3.1. Preparation of Patches
2.3.2. Characterisation of Patches
2.3.2.1. Weight of Patches
2.3.2.2. Thickness of Patches
2.3.2.3. Drug Content
2.3.2.4. ATR-FTIR Spectroscopy of Patch Formulation
2.3.2.5. Tack Force
2.3.2.6. Peel Strength
2.3.2.7. In Vitro Release Studies
2.3.2.8. In Vitro Permeation and Mass Balance Studies
2.3.2.9. Statistics
3. Results and Discussion
3.1. Solvent Selection
3.1.1. Miscibility and Saturated Solubility Studies of MS
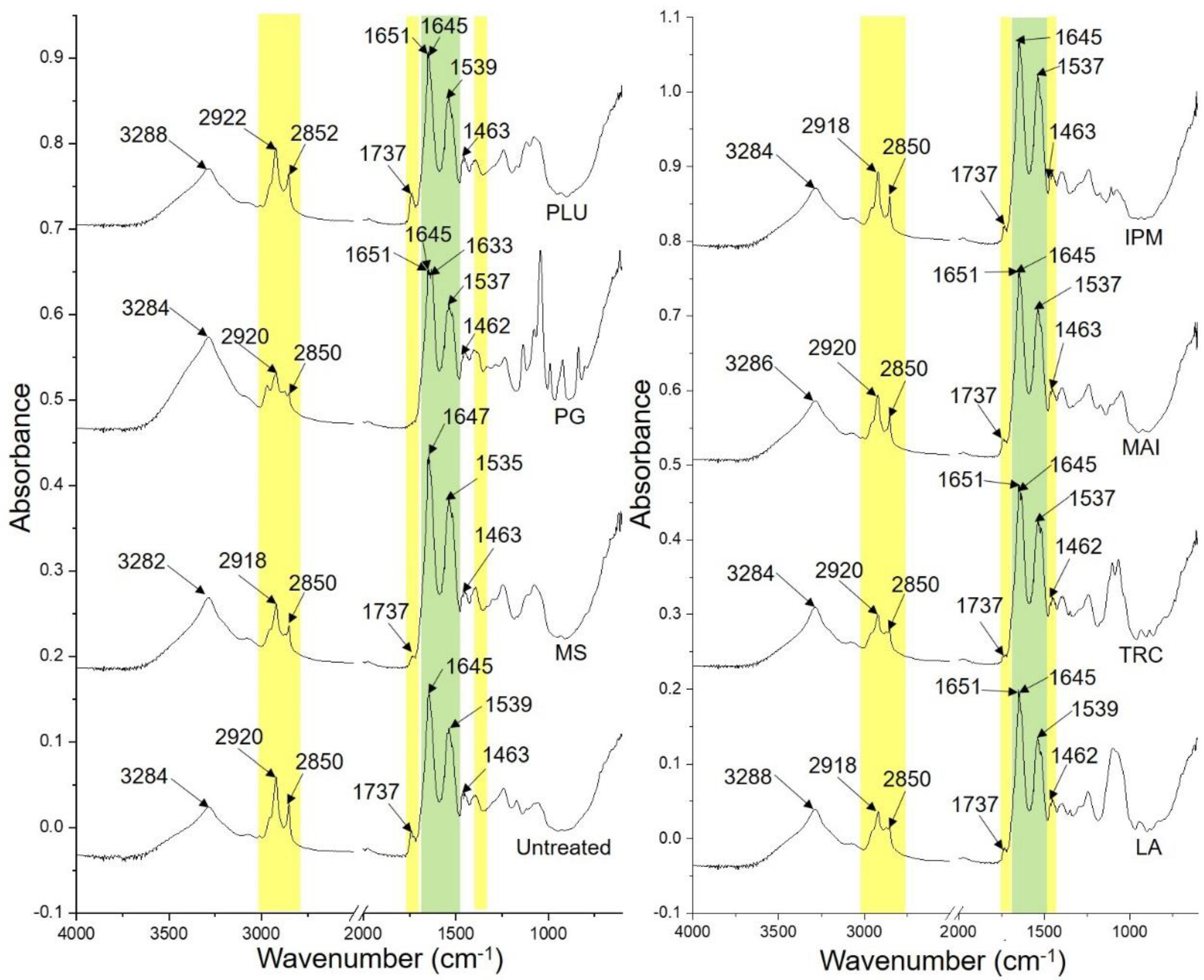
3.1.2. ATR-FTIR Spectroscopy
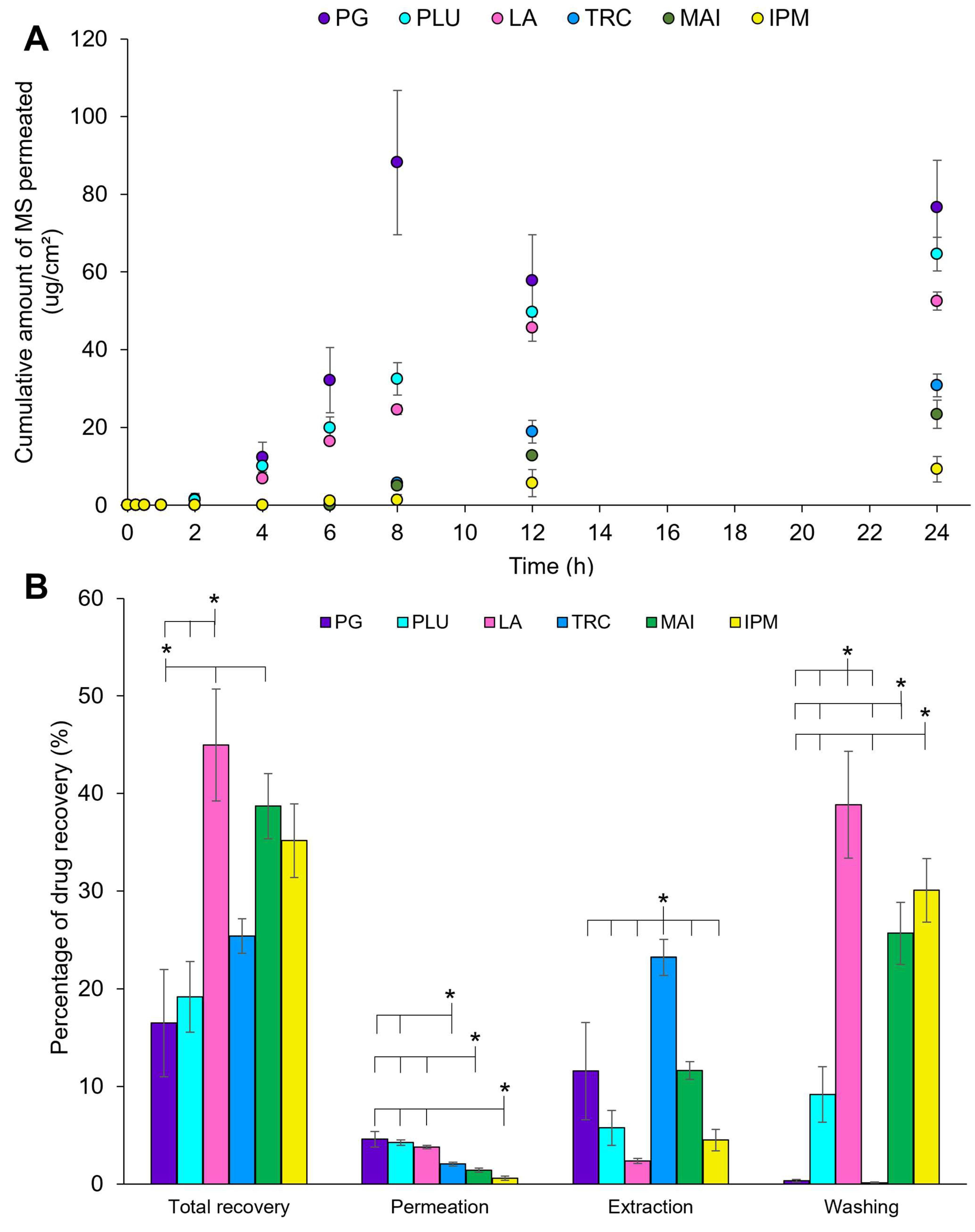
3.1.3. In Vitro Permeation and Mass Balance Studies
3.2. Patch Formulation
3.2.1. Characterisation of Patches
3.2.1.1. Patch Weight, Thickness and Drug Content
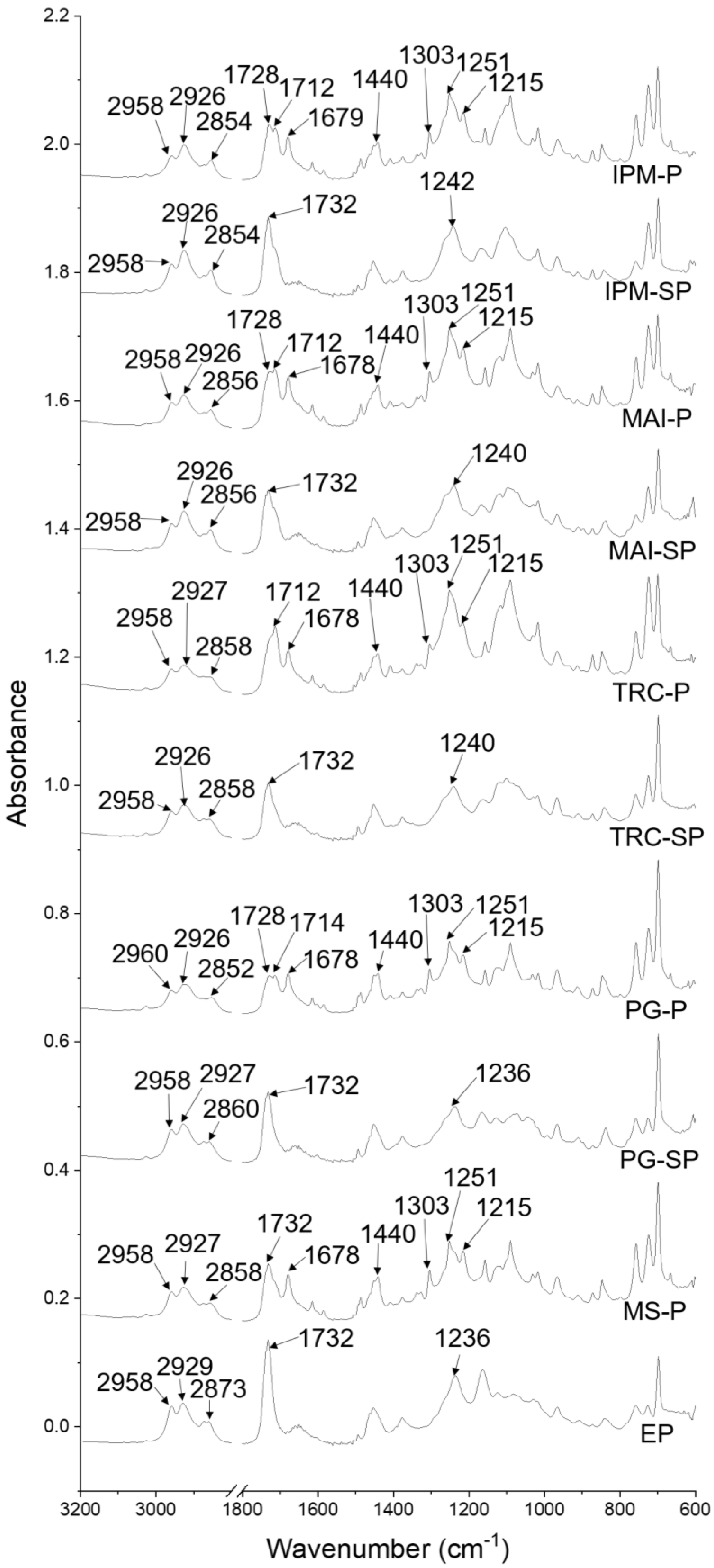
3.2.1.2. ATR-FTIR Spectroscopy of Patches
3.2.1.3. Tack Force
3.2.1.4. Peel Strength
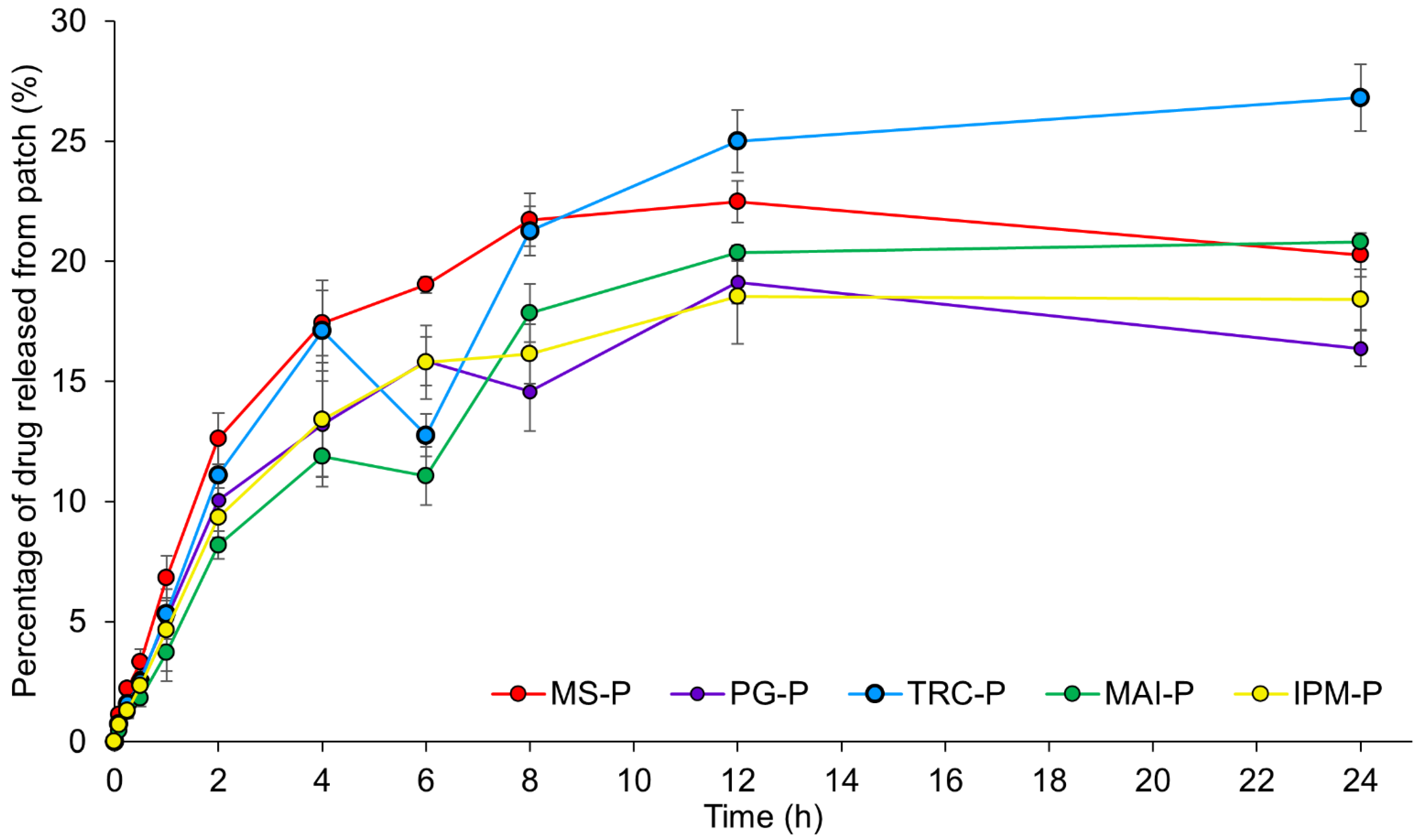
3.2.1.5. In Vitro Drug Release Studies
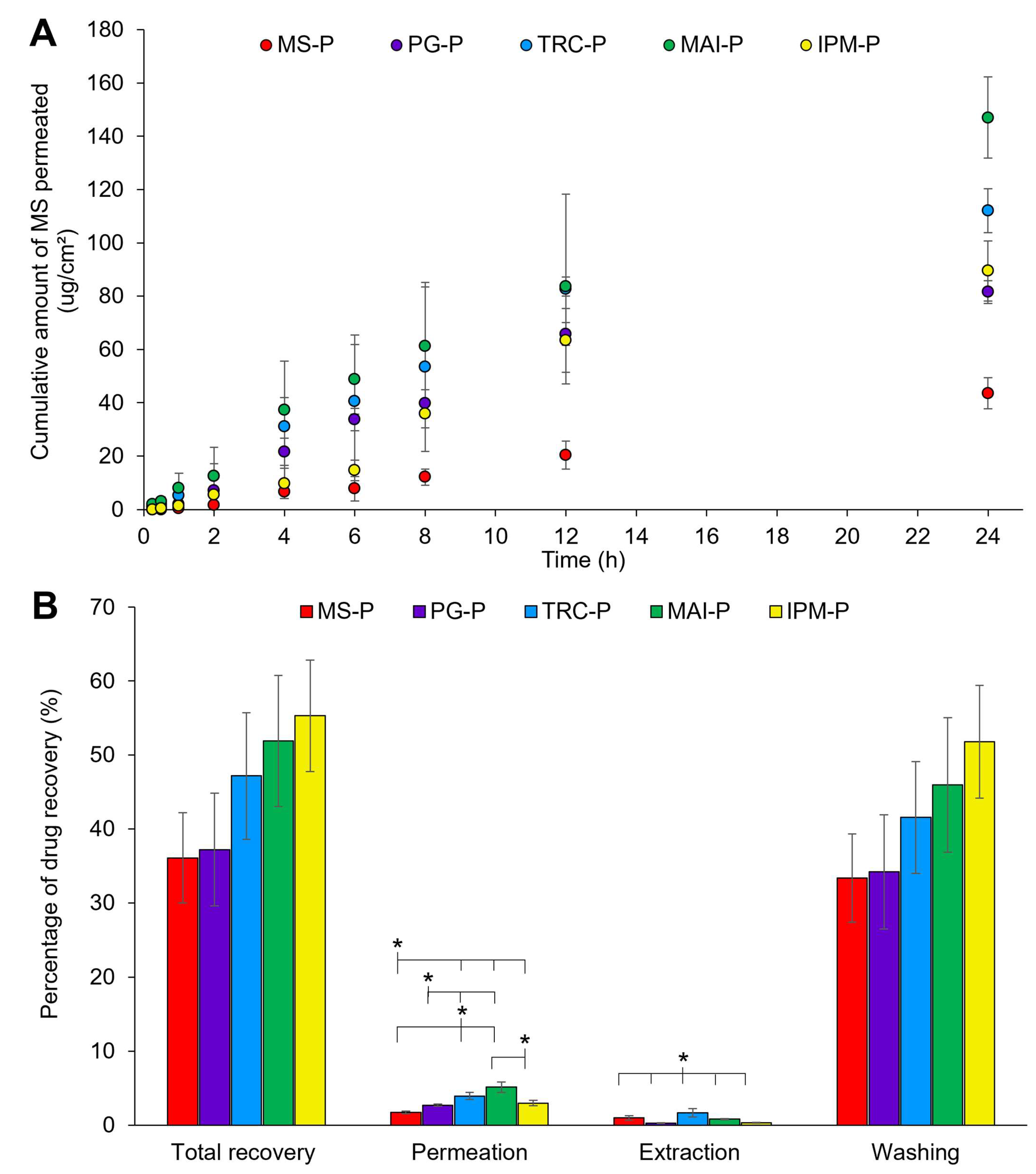
3.2.1.6. In Vitro Drug Permeation Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abd, E.; Benson, H.; Roberts, M.; Grice, J. Follicular penetration of caffeine from topically applied nanoemulsion formulations containing penetration enhancers: In vitro human skin studies. Ski. Pharmacol. Physiol. 2018, 31, 252–260. [Google Scholar] [CrossRef]
- Al-Akayleh, F.; Adwan, S.; Khanfer, M.; Idkaidek, N.; Al-Remawi, M. A novel eutectic-based transdermal delivery system for risperidone. AAPS PharmSciTech 2021, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Chadha, G.; Sathigari, S.; Parsons, D.; Jayachandra Babu, R. In Vitro percutaneous absorption of genistein from topical gels through human skin. Drug Dev. Ind. Pharm. 2011, 37, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Geevarghese, R.; Shirolkar, S. Formulation and evaluation of fluvastatin sodium drug-in-adhesive transdermal system. J. Res. Pharm. 2020, 24, 562–571. [Google Scholar] [CrossRef]
- Cilurzo, F.; Gennari, C.; Minghetti, P. Adhesive properties: A critical issue in transdermal patch development. Expert Opin. Drug Deliv. 2012, 9, 33–45. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, H.; Seong, M.; Lee, S.; Kang, M.; Yi, H.; Bae, W.; Kwak, M.; Jeong, H. Multifunctional smart skin adhesive patches for advanced health care. Adv. Healthc. Mater. 2018, 7, 1800275–1800295. [Google Scholar] [CrossRef]
- Cevc, G.; Blume, G.; Schätzlein, A.; Gebauer, D.; Paul, A. The skin: A pathway for systemic treatment with patches and lipid-based agent carriers. Adv. Drug Deliv. Rev. 1996, 18, 349–378. [Google Scholar] [CrossRef]
- Wohlrab, J.; Kreft, B.; Tamke, B. Skin tolerability of transdermal patches. Expert Opin. Drug Deliv. 2011, 8, 939–948. [Google Scholar] [CrossRef]
- Prabhakar, D.; Sreekanth, J.; Jayaveera, K. Transdermal drug delivery patches: A review. J. Drug Deliv. Ther. 2013, 3, 213–222. [Google Scholar] [CrossRef]
- Trommer, H.; Neubert, R. Overcoming the stratum corneum: The modulation of skin penetration. Ski. Pharmacol. Physiol. 2006, 19, 106–121. [Google Scholar] [CrossRef]
- Norn, S.; Permin, H.; Kruse, P.; Kruse, E. From willow bark to acetylsalicylic acid. Dan. Med. Arb. 2009, 37, 79–98. [Google Scholar]
- Levesque, H.; Lafont, O. Aspirin throughout the ages: A historical review. Rev. Med. Interne 2000, 21, 8–17. [Google Scholar]
- Martin, D.; Valdez, J.; Boren, J.; Mayersohn, M. Dermal absorption of camphor, menthol, and methyl salicylate in humans. J. Clin. Pharmacol. 2004, 44, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Reynaud, F.; Souza Picciani, P.; Silva, K.; Barradas, T. Chitosan-based films containing nanoemulsions of methyl salicylate: Formulation development, physical-chemical and in vitro drug release characterization. Int. J. Biol Macromol. 2020, 164, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, X.; Chen, J.; Li, X.; Hao, D.; Li, T.; Mei, K.; Lan, Y.; Wu, Q. Efficacy and mechanism of methyl salicylate in the enhancement of skin delivery of herbal medicines. J. Tradit. Chin. Med. Sci. 2021, 8, 336–342. [Google Scholar] [CrossRef]
- Yano, T.; Kanetake, T.; Saita, M.; Noda, K. Effects of 1-menthol and dl-camphor on the penetration and hydrolysis of methyl salicylate in hairless mouse skin. J. Pharm. Pharm. 1991, 14, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ma, J.; Liu, R.; Han, W.; Tang, X. A thermoplastic elastomer patch matrix for traditional Chinese medicine: Design and evaluation. Drug Dev. Ind. Pharm. 2014, 40, 211–221. [Google Scholar] [CrossRef]
- Yeoh, S.; Goh, C. Topical delivery of salicylates. Drug Deliv. Transl. Res. 2021, 12, 981–1001. [Google Scholar] [CrossRef]
- Spiandore, M.; Piram, A.; Lacoste, A.; Josse, D.; Doumenq, P. Hair analysis as a useful procedure for detection of vapour exposure to chemical warfare agents: Simulation of sulphur mustard with methyl salicylate. Drug Test. Anal. 2014, 6, 67–73. [Google Scholar] [CrossRef]
- Sharma, N. A brief review on transdermal patches. OMCIJ 2018, 7, 58–62. [Google Scholar]
- Pastore, M.; Kalia, Y.; Horstmann, M.; Roberts, M. Transdermal patches: History, development and pharmacology. Br. J. Pharmacol. 2015, 172, 2179–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otterbach, A.; Lamprecht, A. Enhanced skin permeation of estradiol by dimethyl sulfoxide containing transdermal patches. Pharmaceutics 2021, 13, 320. [Google Scholar] [CrossRef] [PubMed]
- Lane, M. The transdermal delivery of fentanyl. Eur. J. Pharm. Biopharm. Off. J. Arb. Fur. Pharm. Verfahr. e.V 2013, 84, 449–455. [Google Scholar] [CrossRef]
- Dhiman, S.; Singh, T.; Rehni, A. Transdermal patches: A recent approach to new drug delivery system. Int. J. Pharm. Pharm. Sci 2011, 3, 26–34. [Google Scholar]
- Benson, H.; Watkinson, A. Transdermal and Topical Drug Delivery; John Wiley & Sons, Inc.: New York, NY, USA, 2012; pp. 15–20. [Google Scholar]
- FDA. Salonpas Pain Relief New Drug Application Approval Letter (NDA 22-029/S-002); FDA: Rockville, MD, USA, 2008.
- Kováčik, A.; Kopečná, M.; Vávrová, K. Permeation enhancers in transdermal drug delivery: Benefits and limitations. Expert Opin. Drug Deliv. 2020, 17, 145–155. [Google Scholar] [CrossRef]
- Lane, M. Skin penetration enhancers. Int. J. Pharm. 2013, 447, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Karande, P.; Jain, A.; Ergun, K.; Kispersky, V.; Mitragotri, S. Design principles of chemical penetration enhancers for transdermal drug delivery. Proc. Natl. Acad. Sci. USA 2005, 102, 4688–4693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadzadeh, Y.; Adibkia, K.; Hamishekar, H. Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Modification of the Stratum Corneum; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1. [Google Scholar]
- Barry, B. Mode of action of penetration enhancers in human skin. J. Control. Release Off. J. Control. Release Soc. 1987, 6, 85–97. [Google Scholar] [CrossRef]
- Wokovich, A.; Prodduturi, S.; Doub, W.; Hussain, A.; Buhse, L. Transdermal drug delivery system (TDDS) adhesion as a critical safety, efficacy and quality attribute. Eur. J. Pharm. Biopharm. Off. J. Arb. Fur. Pharm. Verfahr. e.V 2006, 64, 1–8. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Baruch, N.; Sintov, A. Conjugates of unsaturated fatty acids with propylene glycol as potentially less-irritant skin penetration enhancers. Drug Dev. Ind. Pharm. 2007, 33, 1169–1175. [Google Scholar] [CrossRef]
- Trottet, L.; Merly, C.; Mirza, M.; Hadgraft, J.; Davis, A. Effect of finite doses of propylene glycol on enhancement of in vitro percutaneous permeation of loperamide hydrochloride. Int. J. Pharm. 2004, 274, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.; Heard, C. Film drying and complexation effects in the simultaneous skin permeation of ketoprofen and propylene glycol from simple gel formulations. Int. J. Pharm. 2006, 307, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Choi, H. Effect of ethanolamine salts and enhancers on the percutaneous absorption of piroxicam from a pressure sensitive adhesive matrix. Eur. J. Pharm. Sci. 2003, 18, 149–153. [Google Scholar] [CrossRef]
- Shen, M.; Liu, C.; Wan, X.; Farah, N.; Fang, L. Development of a daphnetin transdermal patch using chemical enhancer strategy: Insights of the enhancement effect of Transcutol P and the assessment of pharmacodynamics. Drug Dev. Ind. Pharm. 2018, 44, 1642–1649. [Google Scholar] [CrossRef]
- Tuntiyasawasdikul, S.; Limpongsa, E.; Jaipakdee, N.; Sripanidkulchai, B. Development and evaluation of topical films containing phytoestrogenic diaryheptanoids from Curcuma comosa extract. Drug Dev. Ind. Pharm. 2018, 44, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Parhi, R.; Padilam, S. In vitro permeation and stability studies on developed drug-in-adhesive transdermal patch of simvastatin. Bull Fac. Pharm. Cairo. Univ. 2018, 56, 26–33. [Google Scholar] [CrossRef]
- Myoung, Y.; Choi, H. Permeation of ciclopirox across porcine hoof membrane: Effect of pressure sensitive adhesives and vehicles. Eur. J. Pharm. Sci. 2003, 20, 319–325. [Google Scholar] [CrossRef]
- Liu, C.; Farah, N.; Weng, W.; Jiao, B.; Shen, M.; Fang, L. Investigation of the permeation enhancer strategy on benzoylaconitine transdermal patch: The relationship between transdermal enhancement strength and physicochemical properties of permeation enhancer. Eur. J. Pharm. Sci. 2019, 138, 105009–105041. [Google Scholar] [CrossRef]
- Yang, D.; Liu, C.; Quan, P.; Fang, L. A systematic approach to determination of permeation enhancer action efficacy and sites: Molecular mechanism investigated by quantitative structure−activity relationship. J. Control. Release Off. J. Control. Release Soc. 2020, 322, 1–12. [Google Scholar] [CrossRef]
- Goh, C.; Hadgraft, J.; Lane, M. Thermal analysis of mammalian stratum corneum using differential scanning calorimetry for advancing skin research and drug delivery. Int. J. Pharm. 2022, 614, 121447–121465. [Google Scholar] [CrossRef]
- Tanojo, H.; Bouwstra, J.; Junginger, H.; Boddé, H. Thermal analysis studies on human skin and skin barrier modulation by fatty acids and propylene glycol. J. Therm. Anal. Calorim. 1999, 57, 313–322. [Google Scholar] [CrossRef]
- Zhang, Z.; Lunter, D. Confocal Raman microspectroscopy as an alternative to differential scanning calorimetry to detect the impact of emulsifiers and formulations on stratum corneum lipid conformation. Eur. J. Pharm. Sci. 2018, 121, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.; Moffat, J.; Craig, D.; Hadgraft, J.; Lane, M. Monitoring drug crystallization in percutaneous penetration using localized nanothermal analysis and photothermal microspectroscopy. Mol. Pharm. 2018, 16, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, F.; Goh, C.; Haque, T.; Rahma, A.; Lane, M. Dermal Delivery of Diclofenac Sodium—In vitro and in vivo Studies. Pharmaceutics 2022, 14, 2106. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.; Boyd, B.; Craig, D.; Lane, M. Profiling of drug crystallization in the skin. Expert Opin. Drug Deliv. 2020, 17, 1321–1334. [Google Scholar] [CrossRef]
- Musazzi, U.; Ortenzi, M.; Gennari, C.; Casiraghi, A.; Minghetti, P.; Cilurzo, F. Design of pressure-sensitive adhesive suitable for the preparation of transdermal patches by hot-melt printing. Int. J. Pharm. 2020, 586, 119607–119616. [Google Scholar] [CrossRef]
- Rezk, A.; Obiweluozor, F.; Choukrani, G.; Park, C.; Kim, C. Drug release and kinetic models of anticancer drug (BTZ) from a pH-responsive alginate polydopamine hydrogel: Towards cancer chemotherapy. Int. J. Biol. Macromol. 2019, 141, 388–400. [Google Scholar] [CrossRef]
- Rehman, Q.; Akash, M.; Rasool, M.; Rehman, K. Role of kinetic models in drug stability. In Drug Stability and Chemical Kinetics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 155–165. [Google Scholar]
- Yalkowsky, S.; Yan, H. Handbook of Aqueous Solubility Data; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Bronaugh, R.; Stewart, R. Methods for in vitro percutaneous absorption studies III: Hydrophobic compounds. J. Pharm. Sci. 1984, 73, 1255–1258. [Google Scholar] [CrossRef]
- Mansour, R.; Sallam, A.; Hamdan, I.; Khalil, E.; Yousef, I. Elucidation of penetration enhancement mechanism of Emu oil using FTIR microspectroscopy at EMIRA laboratory of SESAME synchrotron. Spectrochim. Acta. Mol. Biomol. Spectrosc. 2017, 185, 1–10. [Google Scholar] [CrossRef]
- Casiraghi, A.; Minghetti, P.; Cilurzo, F.; Selmin, F.; Gambaro, V.; Montanari, L. The effects of excipients for topical preparations on the human skin permeability of terpinen-4-ol contained in Tea tree oil: Infrared spectroscopic investigations. Pharm. Dev. Technol. 2010, 15, 545–552. [Google Scholar] [CrossRef]
- Obata, Y.; Utsumi, S.; Watanabe, H.; Suda, M.; Tokudome, Y.; Otsuka, M.; Takayama, K. Infrared spectroscopic study of lipid interaction in stratum corneum treated with transdermal absorption enhancers. Int. J. Pharm. 2010, 389, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, I.; Müller-Goymann, C. Role of isopropyl myristate, isopropyl alcohol and a combination of both in hydrocortisone permeation across the human stratum corneum. Ski. Pharmacol. Physiol. 2003, 16, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Leopold, C.; Lippold, B. An attempt to clarify the mechanism of the penetration enhancing effects of lipophilic vehicles with differential scanning calorimetry (DSC). J. Pharm. Pharmacol. 1995, 47, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Li, S. Chemical enhancer solubility in human stratum corneum lipids and enhancer mechanism of action on stratum corneum lipid domain. Int. J. Pharm. 2010, 383, 89–98. [Google Scholar] [CrossRef]
- Mitriaikina, S.; Muller-Goymann, C. Synergetic effects of isopropyl alcohol (IPA) and isopropyl myristate (IPM) on the permeation of betamethasone-17-valerate from semisolid Pharmacopoeia bases. J. Drug Deliv. Sci. Technol. 2007, 17, 1–8. [Google Scholar] [CrossRef]
- Salimi, A.; Moghimipour, E.; Rahmani, F. Effects of the various solvents on the in vitro permeability of indomethacin through whole abdominal rat skin. Annu. Res. Rev. Biol. 2015, 5, 335–346. [Google Scholar] [CrossRef]
- Jain, A.; Thomas, N.; Panchagnula, R. Transdermal drug delivery of imipramine hydrochloride.: I. Effect of terpenes. J. Control. Release Off. J. Control. Release Soc. 2002, 79, 93–101. [Google Scholar] [CrossRef]
- Boncheva, M.; Damien, F.; Normand, V. Molecular organization of the lipid matrix in intact stratum corneum using ATR-FTIR spectroscopy. Biochim. Biophys. Acta. Bioenerg. 2008, 1778, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Sugibayashi, K.; Morimoto, Y. Effect and mode of action of aliphatic esters on the in vitro skin permeation of nicorandil. Int. J. Pharm. 1988, 43, 31–40. [Google Scholar] [CrossRef]
- Eichner, A.; Stahlberg, S.; Sonnenberger, S.; Lange, S.; Dobner, B.; Ostermann, A.; Schrader, T.; Hauß, T.; Schroeter, A.; Huster, D. Influence of the penetration enhancer isopropyl myristate on stratum corneum lipid model membranes revealed by neutron diffraction and 2H NMR experiments. Biochim. Et Biophys. Acta Biomembr. 2017, 1859, 745–755. [Google Scholar] [CrossRef]
- Engelbrecht, T.; Demé, B.; Dobner, B.; Neubert, R. Study of the influence of the penetration enhancer isopropyl myristate on the nanostructure of stratum corneum lipid model membranes using neutron diffraction and deuterium labelling. Ski. Pharmacol. Physiol. 2012, 25, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Torii, H. Theoretical Analyses of the Amide I Infrared Bands of Globular Proteins; Wiley-Liss: New York, NY, USA, 1996; pp. 1–18. [Google Scholar]
- He, W.; Guo, X.; Xiao, L.; Feng, M. Study on the mechanisms of chitosan and its derivatives used as transdermal penetration enhancers. Int. J. Pharm. 2009, 382, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Covi-Schwarz, J.; Klang, V.; Valenta, C. ATR-FTIR spectroscopy and the skin barrier: Evaluation of penetration-enhancement effects. In Percutaneous Penetration Enhancers Drug Penetration into/through the Skin; Springer: Berlin/Heidelberg, Germany, 2017; pp. 247–254. [Google Scholar]
- Goh, C.; Moffat, J.; Craig, D.; Hadgraft, J.; Lane, M. Nano-thermal imaging of the stratum corneum and its potential use for understanding of the mechanism of skin penetration enhancer. Thermochim. Acta 2017, 655, 278–283. [Google Scholar] [CrossRef]
- Shah, D.; Khandavilli, S.; Panchagnula, R. Alteration of skin hydration and its barrier function by vehicle and permeation enhancers: A study using TGA, FTIR, TEWL and drug permeation as markers. Methods Find Exp. Clin. Pharmacol. 2008, 30, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.; Collett, J.; Garcia, S.; Thomazini, J.; Bentley, M. Glycerol monooleate/solvents systems for progesterone transdermal delivery: In vitro permeation and microscopic studies. Rev. Bras. De Ciências Farm. 2002, 38, 55–62. [Google Scholar] [CrossRef]
- Björklund, S.; Pham, Q.; Jensen, L.; Knudsen, N.; Nielsen, L.; Ekelund, K.; Ruzgas, T.; Engblom, J.; Sparr, E. The effects of polar excipients Transcutol and dexpanthenol on molecular mobility, permeability, and electrical impedance of the skin barrier. J. Colloid Interface Sci. 2016, 479, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Hathout, R.; Mansour, S.; Mortada, N.; Geneidi, A.; Guy, R. Uptake of microemulsion components into the stratum corneum and their molecular effects on skin barrier function. Mol. Pharm. 2010, 7, 1266–1273. [Google Scholar] [CrossRef]
- Kong, R.; Bhargava, R. Characterization of porcine skin as a model for human skin studies using infrared spectroscopic imaging. Analyst 2011, 136, 2359–2366. [Google Scholar] [CrossRef]
- Jacobi, U.; Kaiser, M.; Toll, R.; Mangelsdorf, S.; Audring, H.; Otberg, N.; Sterry, W.; Lademann, J. Porcine ear skin: An in vitro model for human skin. Skin Res. Technol. 2007, 13, 19–24. [Google Scholar] [CrossRef]
- Tsai, J.; Flynn, G.; Weiner, N.; Cappel, M.; Kreuter, J.; Ferry, J. Drug and vehicle deposition from topical applications: Use of in vitro mass balance technique with minoxidil solutions. J. Pharm. Sci. 1992, 81, 736–743. [Google Scholar] [CrossRef]
- Intarakumhaeng, R.; Li, S. Effects of solvent on percutaneous absorption of nonvolatile lipophilic solute. Int. J. Pharm. 2014, 476, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, F.; Sil, B.; Al Hossain, A.; Moore, D.; Lucas, R.; Lane, M. Topical delivery of niacinamide: Influence of neat solvents. Int. J. Pharm. 2020, 579, 119137–119145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, K.; Kusawake, Y.; Ito, Y.; Sugioka, N.; Shibata, N.; Takada, K. Enhancing mechanism of Labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. Eur. J. Pharm. Biopharm. Off. J. Arb. Fur Pharm. Verfahr. e.V 2006, 64, 82–91. [Google Scholar] [CrossRef]
- Williams, A.; Barry, B. Penetration enhancers. Adv. Drug Deliv. Rev. 2012, 64, 128–137. [Google Scholar] [CrossRef]
- Zidan, S.; Kamal, N.; Alayoubi, A.; Seggel, M.; Ibrahim, S.; Rahman, Z.; Cruz, C.N.; Ashraf, M. Effect of isopropyl myristate on transdermal permeation of testosterone from carbopol gel. J. Pharm. Sci. 2017, 106, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Chávez, J.; Quintanar-Guerrero, D.; Ganem-Quintanar, A. In vivo skin permeation of sodium naproxen formulated in Pluronic F-127 gels: Effect of Azone® and Transcutol®. Drug Dev. Ind. Pharm. 2005, 31, 447–454. [Google Scholar] [CrossRef]
- Osborne, D.; Musakhanian, J. Skin penetration and permeation properties of Transcutol®—Neat or diluted mixtures. AAPS PharmSciTech 2018, 19, 3512–3533. [Google Scholar] [CrossRef]
- Shah, P.; Desai, P.; Patlolla, R.; Klevans, L.; Singh, M. Effect of combination of hydrophilic and lipophilic permeation enhancers on the skin permeation of kahalalide F. J. Pharm. Pharmacol. 2014, 66, 760–768. [Google Scholar] [CrossRef]
- Haque, T.; Rahman, K.; Thurston, D.; Hadgraft, J.; Lane, M. Topical delivery of anthramycin I. Influence of neat solvents. Eur. J. Pharm. Sci. 2017, 104, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Intarakumhaeng, R.; Shi, Z.; Wanasathop, A.; Stella, Q.; Wei, K.; Styczynski, P.; Li, C.; Smith, E.; Li, S. In vitro skin penetration of petrolatum and soybean oil and effects of glyceryl monooleate. Int. J. Cosmet. Sci. 2018, 40, 367–376. [Google Scholar] [CrossRef]
- Güngör, S.; Erdal, M.; Özsoy, Y. Plasticizers in Transdermal Drug Delivery Systems; IntechOpen: London, UK, 2012; Volume 5, pp. 91–112. [Google Scholar]
- Snejdrova, E.; Dittrich, M.; Drastik, M. Plasticized branched aliphatic oligoesters as potential mucoadhesive drug carriers. Int. J. Pharm. 2013, 458, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, R.; Tang, X.; Han, W. A drug-in-adhesive matrix based on thermoplastic elastomer: Evaluation of percutaneous absorption, adhesion, and skin irritation. AAPS PharmSciTech 2012, 13, 1179–1189. [Google Scholar] [CrossRef] [Green Version]
- Can, A.; Erdal, M.; Güngör, S.; Özsoy, Y. Optimization and characterization of chitosan films for transdermal delivery of ondansetron. Molecules 2013, 18, 5455–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuntiyasawasdikul, S.; Limpongsa, E.; Jaipakdee, N.; Sripanidkulchai, B. A monolithic drug-in-adhesive patch of methoxyflavones from Kaempferia parviflora: In vitro and in vivo evaluation. Int. J. Pharm. 2015, 478, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, S.; Soroushnia, A.; Mohamadnia, F. Preparation and in vitro evaluation of a new fentanyl patch based on functional and non-functional pressure sensitive adhesives. AAPS PharmSciTech 2010, 11, 278–284. [Google Scholar] [CrossRef]
- Taghizadeh, S.; Lahootifard, F. Effect of different skin permeation enhancers on peel strength of an acrylic PSA. J. Appl. Polym. Sci. 2003, 90, 2987–2991. [Google Scholar] [CrossRef]
- Chamarthy, S.; Pinal, R. Plasticizer concentration and the performance of a diffusion-controlled polymeric drug delivery system. Colloids Surf. Physicochem. Eng. Asp. 2008, 331, 25–30. [Google Scholar] [CrossRef]
- Jennings, C.; Dziubla, T.; Puleo, D. Combined effects of drugs and plasticizers on the properties of drug delivery films. J. Bioact. Compat. Polym. 2016, 31, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Jackson Jr, W.; Caldwell, J. Antiplasticization. II. Characteristics of antiplasticizers. J. Appl. Polym. Sci. 1967, 11, 211–226. [Google Scholar] [CrossRef]
- Alrimawi, B.; Chan, M.; Ooi, X.; Chan, S.; Goh, C. The interplay between drug and sorbitol contents determines the mechanical and swelling properties of potential rice starch films for buccal drug delivery. Polymers 2021, 13, 578. [Google Scholar] [CrossRef]
- Taghizadeh, S.; Mirzadeh, H.; Barikani, M.; Yousefi, M. Miscibility and tack of blends of poly (vinylpyrrolidone)/acrylic pressure-sensitive adhesive. Int. J. Adhes. Adhes. 2009, 29, 302–308. [Google Scholar] [CrossRef]
- Lir, I.; Haber, M.; Dodiuk-Kenig, H. Skin surface model material as a substrate for adhesion-to-skin testing. J. Adhes. Sci. Technol. 2007, 21, 1497–1512. [Google Scholar] [CrossRef]
- Manly, R. Part I-Principles of adhesion in biological systems. In Adhesion in Biological Systems; Elsevier: Amsterdam, The Netherlands, 1970; Volume 1. [Google Scholar]
- Frederiksen, K.; Guy, R.; Petersson, K. The potential of polymeric film-forming systems as sustained delivery platforms for topical drugs. Expert Opin. Drug Deliv. 2016, 13, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, I.; Franke, P.; Schaefer, U.; Lehr, C. Development and characterization of film forming polymeric solutions for skin drug delivery. Eur. J. Pharm. Biopharm. Off. J. Arb. Fur. Pharm. Verfahr. e.V 2007, 65, 111–121. [Google Scholar] [CrossRef]
- Hai, N.; Kim, J.; Park, E.; Chi, S. Formulation and biopharmaceutical evaluation of transdermal patch containing benztropine. Int. J. Pharm. 2008, 357, 55–60. [Google Scholar] [CrossRef]
- Pichayakorn, W.; Suksaeree, J.; Boonme, P.; Amnuaikit, T.; Taweepreda, W.; Ritthidej, G. Nicotine transdermal patches using polymeric natural rubber as the matrix controlling system: Effect of polymer and plasticizer blends. J. Membr. Sci. 2012, 411, 81–90. [Google Scholar] [CrossRef]
- Saidi, M.; Dabbaghi, A.; Rahmani, S. Swelling and drug delivery kinetics of click-synthesized hydrogels based on various combinations of PEG and star-shaped PCL: Influence of network parameters on swelling and release behavior. Polym. Bull. 2020, 77, 3989–4010. [Google Scholar] [CrossRef]
- Koprda, V.; Falson-Rieg, F.; Hadgraft, J. Skin penetration studies of Transcutol using radiotracer technique. Mamm Skin Barrier Funct. 1995, 31, 1–10. [Google Scholar]
- Panchagnula, R.; Ritschel, W. Development and evaluation of an intracutaneous depot formulation of corticosteroids using Transcutol as a cosolvent: In-vitro, ex-vivo and in-vivo rat studies. J. Pharm. Pharmacol. 1991, 43, 609–614. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition (% w/w) | |||
|---|---|---|---|---|
| EP | MS-P | Solvent-SP | Solvent-P | |
| MS | - | 10 | - | 10 |
| Solvents | - | - | 5 | 5 |
| Durabond | 8 | |||
| Nikasol | 7 | |||
| Tartaric acid | 3 | |||
| D-sorbitol solution | 5 | |||
| Tween 80 | 0.4 | |||
| CMC | 3.5 | |||
| PVP K-90 | 3 | |||
| Distilled water | Add up to 100 | |||
| SC Components | Tentative Assignment | Wavenumber (cm−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Untreated | MS | PG | PLU | LA | TRC | MAI | IPM | ||
| Protein | Amide I | 1645 | 1647 | 1651, 1645, 1633 | 1651, 1645 | 1651, 1645 | 1651, 1645 | 1651, 1645 | 1645 |
| Amide II | 1539 | 1535 | 1537 | 1539 | 1539 | 1537 | 1537 | 1537 | |
| Lipids | CH2 asym str | 2920 | 2918 | 2920 | 2922 | 2918 | 2920 | 2920 | 2918 |
| CH2 sym str | 2850 | 2850 | 2850 | 2852 | 2850 | 2850 | 2850 | 2850 | |
| C=O str | 1737 | 1737 | - | 1737 | 1737 | 1737 | 1737 | 1737 | |
| CH2 scissoring | 1463 | 1463 | 1462 | 1463 | 1462 | 1462 | 1463 | 1463 | |
| Type of Patches | Weight * (mg/cm2) | Thickness * (mm) | Amount of MS (mg/cm2) |
|---|---|---|---|
| EP | 6.3 ± 0.3 | 0.087 ± 0.002 | - |
| PG-SP | 6.9 ± 0.7 | 0.085 ± 0.003 | - |
| TRC-SP | 6.2 ± 0.8 | 0.087 ± 0.002 | - |
| MAI-SP | 6.8 ± 0.5 | 0.087 ± 0.004 | - |
| IPM-SP | 6.5 ± 0.6 | 0.086 ± 0.003 | - |
| MS-P | 7.0 ± 0.5 | 0.087 ± 0.004 | 2.5 ± 0.1 |
| PG-P | 6.8 ± 0.6 | 0.084 ± 0.003 | 2.3 ± 0.1 |
| TRC-P | 6.4 ± 0.7 | 0.086 ± 0.003 | 2.4 ± 0.1 |
| MAI-P | 6.7 ± 0.6 | 0.087 ± 0.003 | 2.4 ± 0.2 |
| IPM-P | 6.4 ± 0.6 | 0.086 ± 0.004 | 2.4 ± 0.1 |
| Tentative Assignment | Materials | Wavenumber (cm−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EP | MS-P | PG-SP | PG-P | TRC-SP | TRC-P | MAI-SP | MAI-P | IPM-SP | IPM-P | ||
| CH3 asym str | MS, polymeric matrix and PSA | 2958 | 2958 | 2958 | 2960 | 2958 | 2958 | 2958 | 2958 | 2958 | 2958 |
| CH2 asym str | Polymeric matrix and PSA | 2929 | 2927 | 2927 | 2926 | 2926 | 2927 | 2926 | 2926 | 2926 | 2926 |
| CH2 sym str | PSA | 2873 | 2858 | 2860 | 2852 | 2858 | 2858 | 2856 | 2854 | 2854 | 2854 |
| C=O str | PSA | 1732 | 1732 | 1732 | 1728, 1714 | 1732 | 1712 | 1732 | 1728, 1712 | 1732 | 1728, 1712 |
| C=O str | MS | - | 1678 | - | 1678 | - | 1678 | - | 1678 | - | 1679 |
| O-H bending | MS | - | 1440 | - | 1440 | - | 1440 | - | 1440 | - | 1440 |
| C-O str | MS | - | 1303 | - | 1303 | - | 1303 | - | 1303 | - | 1303 |
| C-O str | Polymeric matrix | - | 1251 | - | 1251 | - | 1251 | - | 1251 | - | 1251 |
| C-N str | Polymeric matrix | 1236 | 1215 | 1236 | 1215 | 1240 | 1215 | 1240 | 1215 | 1242 | 1215 |
| C-O str | PSA | 1163, 1124, 1082 | 1157, 1118, 1089 | 1165, 1128, 1072 | 1157, 1126, 1118, 1089 | 1161, 1116, 1101, 1083 | 1157, 1116, 1097, 1089 | 1166, 1118, 1097, 1085, 1072 | 1157, 1116, 1099, 1089 | 1166, 1103, 1085 | 1157, 1121, 1101, 1089 |
| C-N str | PSA | 1068, 1029, 1018 | 1031, 1016 | 1043, 1031, 1018 | 1043, 1031, 1016 | 1068, 1029, 1018 | 1031, 1016 | 1029, 1016 | 1029, 1016 | 1029, 1016 | 1031, 1016 |
| Formulation | Ingredient | Concentration (% w/w, Otherwise Stated) | Tack Force (N/cm2, Otherwise Stated) | Peel Strength (N/cm, Otherwise Stated) | Test & Substrate Used | Reference |
|---|---|---|---|---|---|---|
| Benztropine patch (DURO-TAK® 2525) | Benztropine | 5 | 1358 ± 35 g | 980 ± 36 12.5 mm of patch length | Tack force (stainless-steel probe) & peel strength (stainless-steel) | Hai, et al. [104] |
| 10 | Decrease (1310 ± 29 g) | Increase (1214 ± 79 12.5 mm of patch length) | ||||
| 15 | Decrease (1140 ± 48 g) | Increase (1809 ± 63 12.5 mm of patch length) | ||||
| Patch (Gelva 737 acrylic/Silicone Adhesive 7-4503) | Lauryl alcohol | 0 | 7 N/mm2 | 30 N/25 mm width of patch | Tack force (stainless-steel probe) & peel strength (stainless-steel) | Taghizadeh, Soroushnia and Mohamadnia [93] |
| 6 | Increase (70 N/mm2) | Increase (65 N/25 mm width of patch) | ||||
| 12 | Decrease (8 N/mm2) | Increase (13 N/25 mm width of patch) | ||||
| Patch (Gelva 737 acrylic/Silicone Adhesive 7-4503/ PVP 27/23) | 0 | 1 N/mm2 | 65 N/25 mm width of patch | |||
| 6 | Increase (67 N/mm2) | Same (65 N/25 mm width of patch) | ||||
| 12 | Decrease (7 N/25 mm width of patch) | Decrease (43 N/25 mm width of patch) | ||||
| Patch (styrene-isoprene-styrene block copolymer 3620) | Liquid paraffin | 26 | 0.31 ± 0.09 | 0.53 ± 0.11 | Tack force (stainless-steel probe) & peel strength (bakelite plate) | Wang, Liu, Tang and Han [90] |
| 32.6 | Increase (0.79 ± 0.2) | Increase (0.77 ± 0.08) | ||||
| 39 | Increase (2.76 ± 0.14) | Increase (1.59 ± 0.14) | ||||
| 45.5 | Increase (3.31 ± 0.23) | Decrease (1.34 ± 0.12) | ||||
| Patch (deproteinised natural rubber latex) | - | Control | 1.51 ± 0.51 | 0.10 ± 0.01 N/cm2 | Tack force (stainless-steel) & peel strength (polyvinyl chloride surface) | Pichayakorn, et al. [105] |
| Sodium CMC | 15 parts per hundred rubber | Increase (1.86 ± 0.05) | Decrease (0.07 ± 0.01 N/cm2) | |||
| Sodium CMC & dibutylphthalate | 15:10 parts per hundred rubber | Increase (1.79 ± 0.15) | Decrease (0.08 ± 0.01 N/cm2) | |||
| Patch (styrene-isoprene-styrene block copolymer 3620) | Liquid paraffin | 0.2 | 1.72 ± 0.14 | 2.81 ± 0.05 | Tack force (stainless-steel probe) & peel strength (bakelite plate) | Wang, Ma, Liu, Han and Tang [17] |
| 0.3 | Increase (3.21 ± 0.27) | Increase (4.71 ± 0.27) | ||||
| 0.35 | Increase (1.79 ± 0.16) | Decrease (2.11 ± 0.17) | ||||
| 0.5 | Decrease (0.93 ± 0.09) | Decrease (0.61 ± 0.07) | ||||
| Methoxyflavones (7% w/w) patch (DURO-TAK® 2853) | - | Control | 2.75 ± 0.07 kg | 8.50 ± 0.05 N | Tack force (stainless-steel probe) & peel strength (stainless-steel) | Tuntiyasawasdikul, Limpongsa, Jaipakdee and Sripanidkulchai [92] |
| IPM | 3 | Increase (3.06 ± 0.13 kg) | Decrease (8.32 ± 0.16 N) | |||
| Menthol and N-methyl-2-pyrrolidone | 3:3 | Increase (2.95 ± 0.19 kg) | Decrease (8.30 ± 0.37 N) | |||
| Menthol and PG | Increase (3.30 ± 0.03 kg) | Decrease (7.81 ± 0.10 N) | ||||
| Oleic acid, menthol and N-methyl-2-pyrrolidone | 3:3:3 | Increase (2.99 ± 0.05 kg) | Decrease (6.82 ± 0.20 N) |
| Formulation | Zero Order Model | First Order Model | Higuchi Model | Korsmeyer-Peppas Model | |
|---|---|---|---|---|---|
| R2 | R2 | R2 | R2 | n | |
| MS-P | 0.713 | 0.723 | 0.878 | 0.932 | 0.579 |
| PG-P | 0.732 | 0.755 | 0.889 | 0.931 | 0.615 |
| TRC-P | 0.858 | 0.891 | 0.954 | 0.959 | 0.680 |
| MAI-P | 0.855 | 0.854 | 0.955 | 0.966 | 0.716 |
| IPM-P | 0.783 | 0.809 | 0.923 | 0.947 | 0.647 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeoh, S.C.; Loh, P.L.; Murugaiyah, V.; Goh, C.F. Development and Characterisation of a Topical Methyl Salicylate Patch: Effect of Solvents on Adhesion and Skin Permeation. Pharmaceutics 2022, 14, 2491. https://doi.org/10.3390/pharmaceutics14112491
Yeoh SC, Loh PL, Murugaiyah V, Goh CF. Development and Characterisation of a Topical Methyl Salicylate Patch: Effect of Solvents on Adhesion and Skin Permeation. Pharmaceutics. 2022; 14(11):2491. https://doi.org/10.3390/pharmaceutics14112491
Chicago/Turabian StyleYeoh, Soo Chin, Poh Lee Loh, Vikneswaran Murugaiyah, and Choon Fu Goh. 2022. "Development and Characterisation of a Topical Methyl Salicylate Patch: Effect of Solvents on Adhesion and Skin Permeation" Pharmaceutics 14, no. 11: 2491. https://doi.org/10.3390/pharmaceutics14112491