
Modification of the Tumor Microenvironment Enhances Anti-PD-1 Immunotherapy in Metastatic Melanoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Animals and Tumor Models
2.3. Tissue Preparation
2.4. Immunohistochemistry
2.5. Cytolytic Activity
2.6. ELISA
2.7. Flow Cytometry
2.8. Statistical Analysis
3. Results
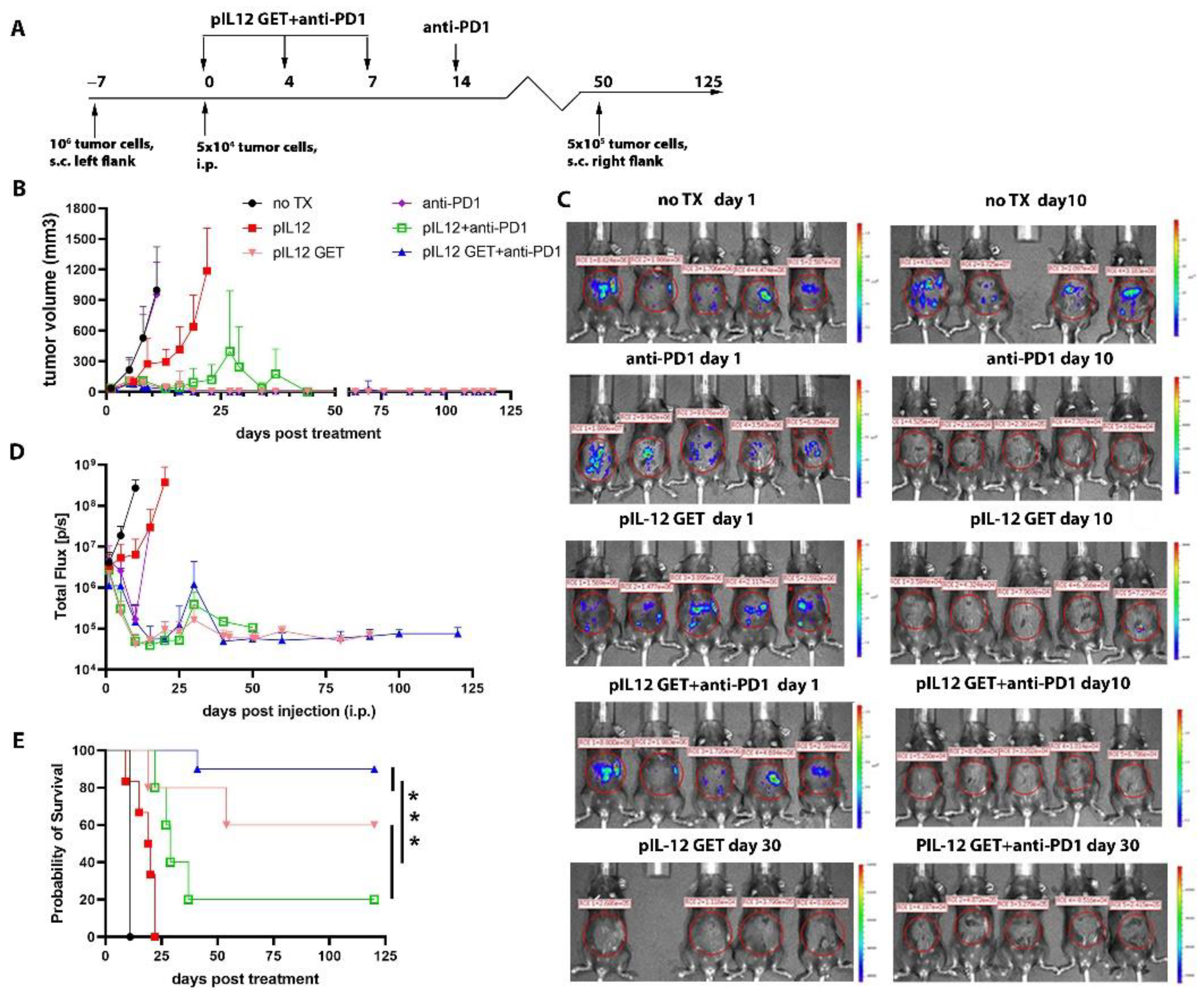
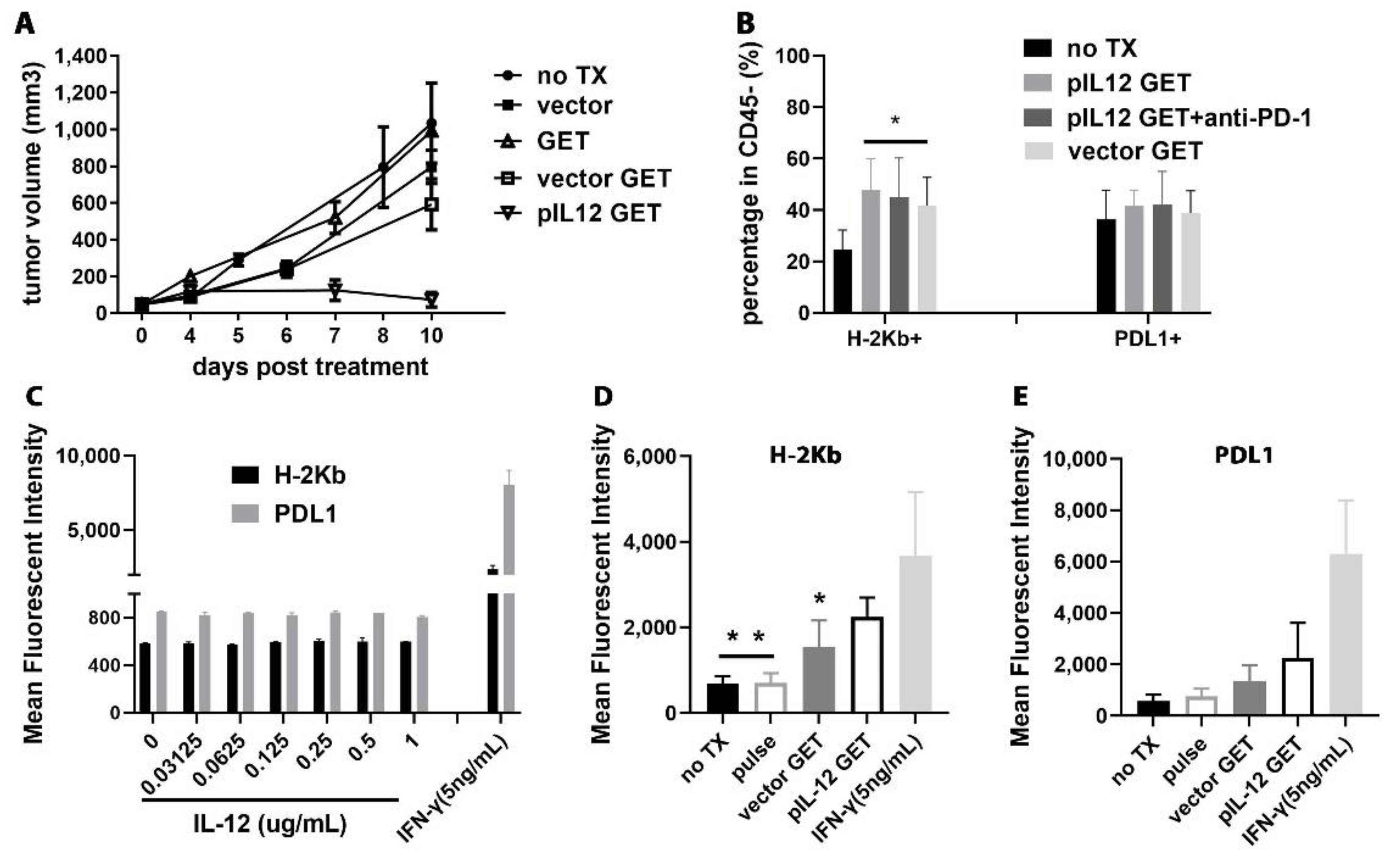
3.1. Tumor Regression by Combination Therapy
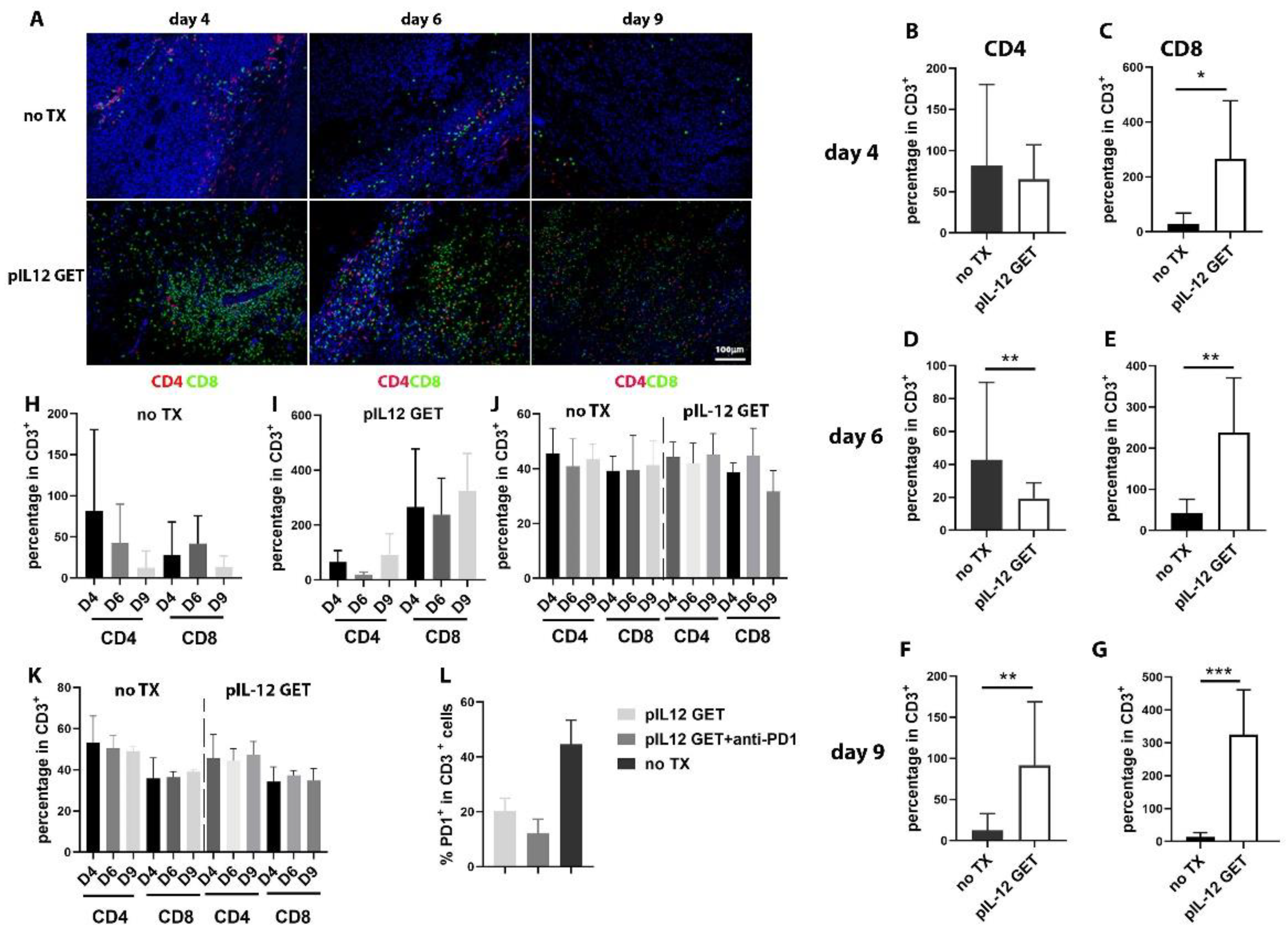
3.2. Dynamics of Immune Response in Tumor Microenvironment
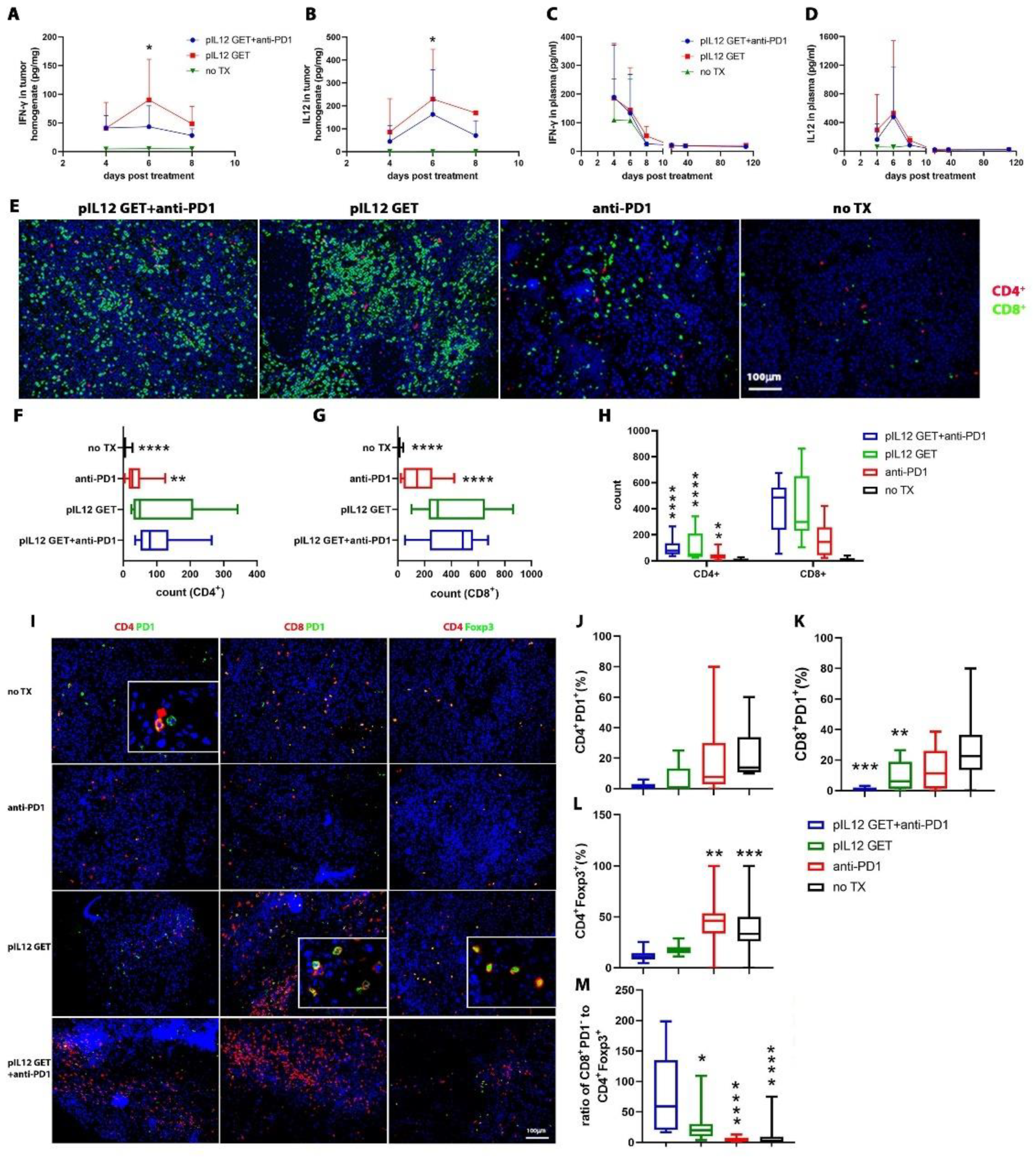
3.3. Endogenous IFN-γ Response within Tumors with pIL-12 GET Combination Treatment
3.4. Modifications of Immune Cell Infiltrate in Tumor Microenvironment
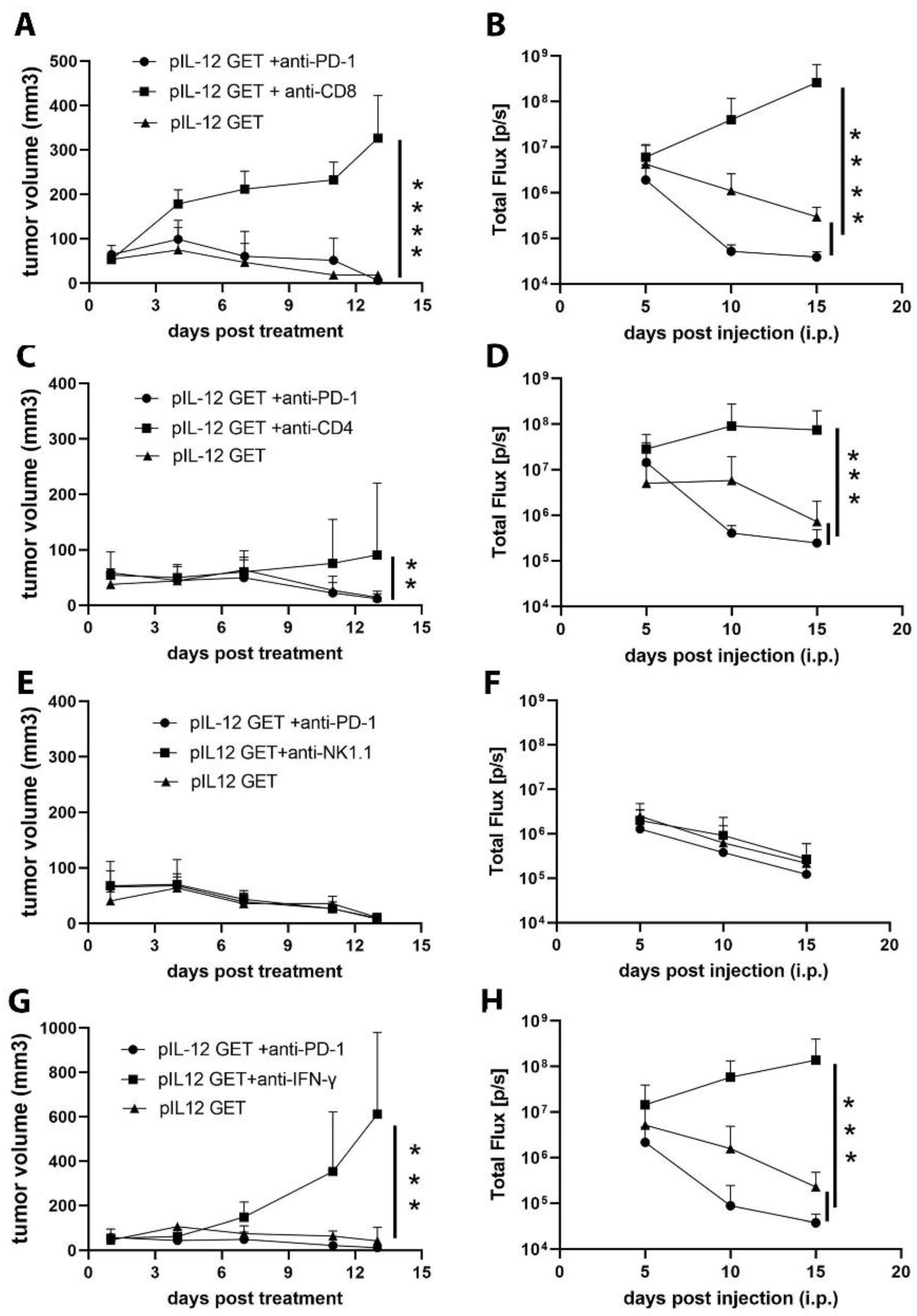
3.5. CD8–CD4- and IFN-γ-Dependent Manner of Anti-Tumor Efficacy
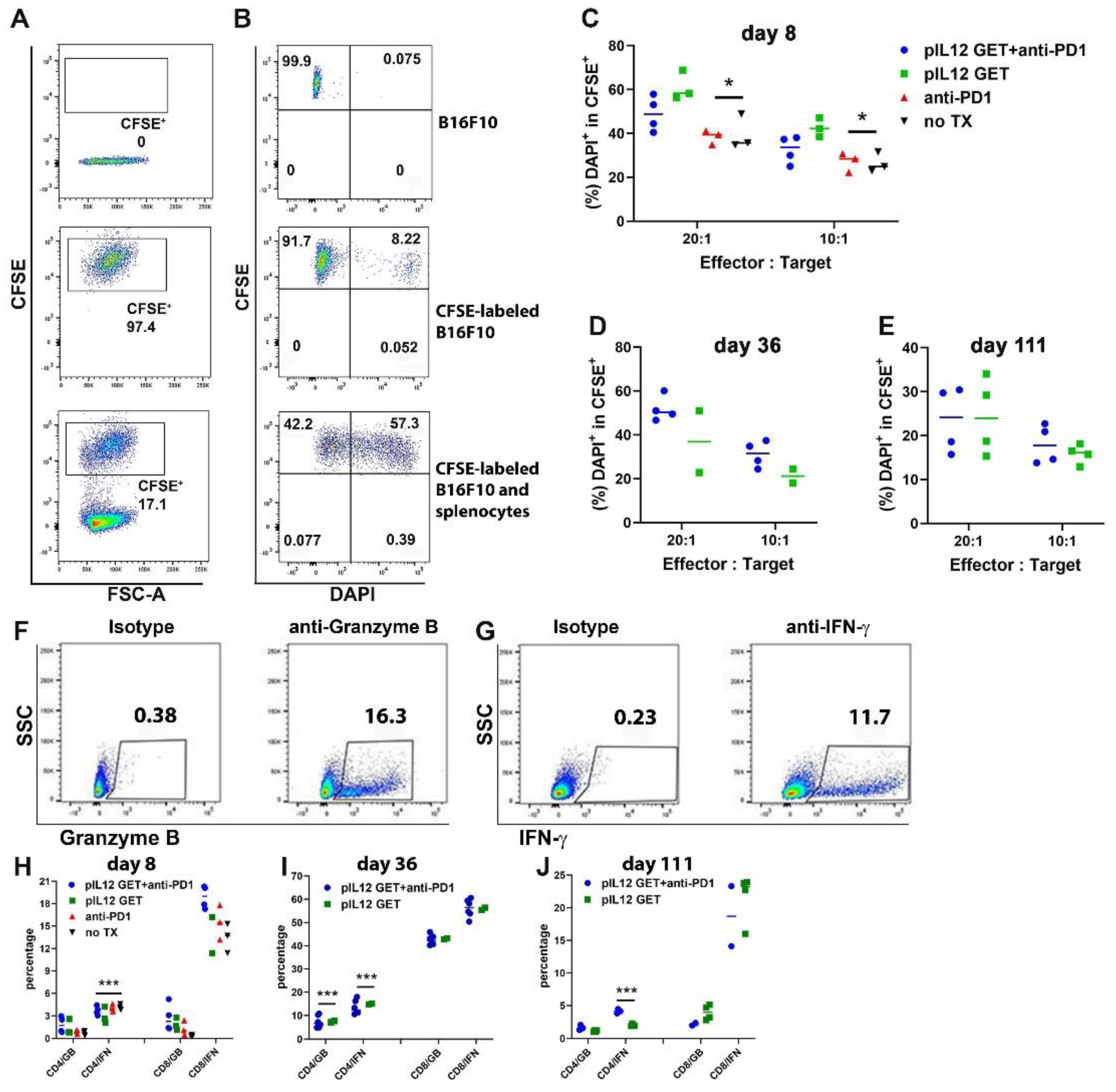
3.6. Augmentation of Killing Capability in T Cells
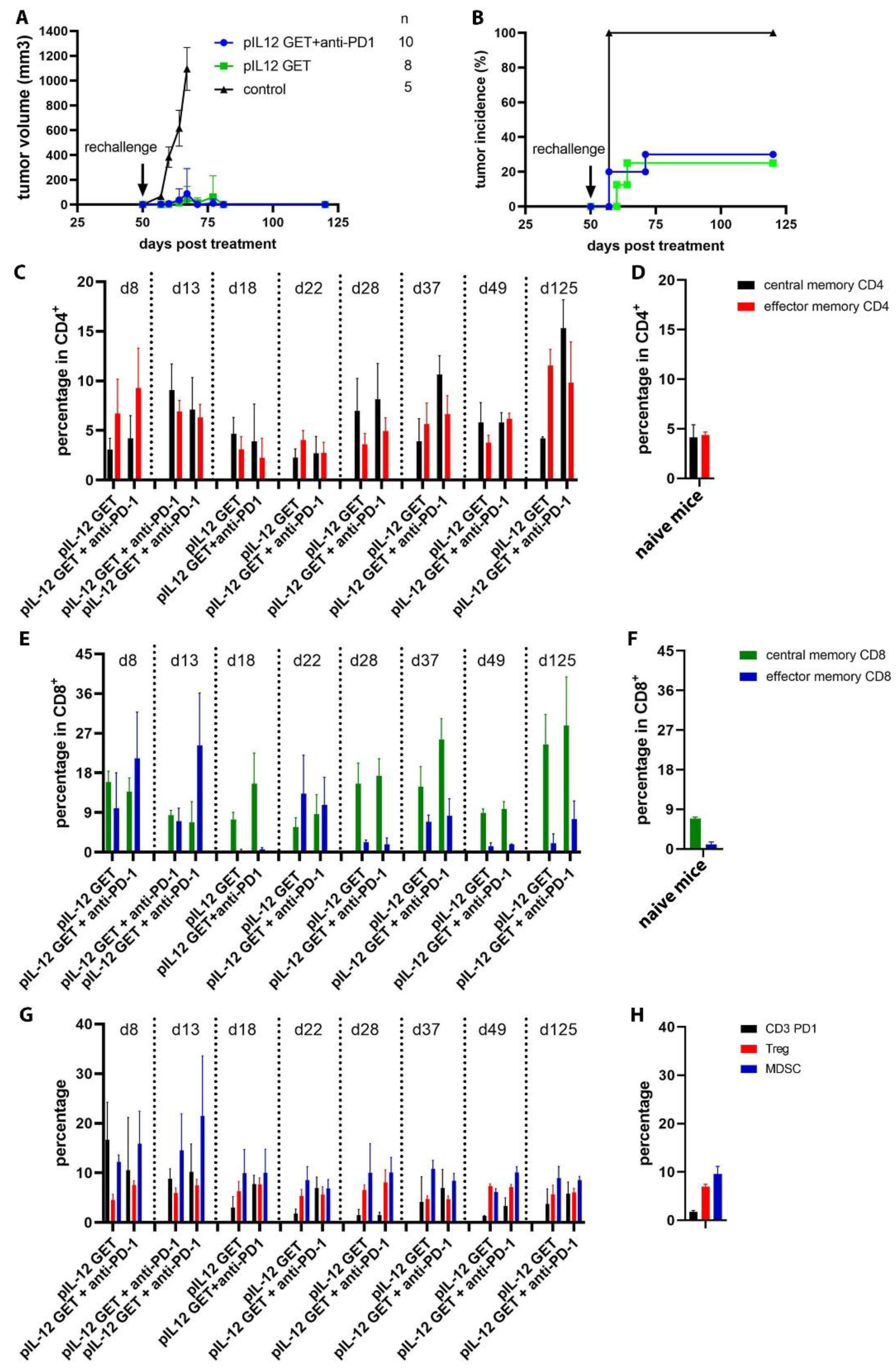
3.7. Mediating Long-Term Tumor Protection
3.8. pIL-12 GET Derived-Tumor Immune Modulatory Program
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Shi, G.; Edelblute, C.; Arpag, S.; Lundberg, C.; Heller, R. IL-12 Gene Electrotransfer Triggers a Change in Immune Response within Mouse Tumors. Cancers 2018, 10, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klevorn, L.E.; Teague, R.M. Adapting Cancer Immunotherapy Models for the Real World. Trends Immunol. 2016, 37, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, L.F.S.; Vardhana, S.A. Metabolic regulation of the cancer-immunity cycle. Trends Immunol. 2021, 42, 975–993. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Gaudreau, P.-O.; Wargo, J.A. Immune Checkpoint Blockade across the Cancer Care Continuum. Immunity 2018, 48, 1077–1080. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- de Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef]
- Wang, D.Y.; Eroglu, Z.; Ozgun, A.; Leger, P.D.; Zhao, S.; Ye, F.; Luke, J.J.; Joseph, R.W.; Haq, R.; Ott, P.A.; et al. Clinical Features of Acquired Resistance to Anti–PD-1 Therapy in Advanced Melanoma. Cancer Immunol. Res. 2017, 5, 357–362. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, N.; Della Corte, M.; Pelaia, C.; Piazzetta, G.; Lobello, N.; Del Duca, E.; Bennardo, L.; Nisticò, S.P. Primary Mucosal Melanoma Presenting with a Unilateral Nasal Obstruction of the Left Inferior Turbinate. Medicina 2021, 57, 359. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Woo, S.-R.; Zha, Y.; Spaapen, R.; Zheng, Y.; Corrales, L.; Spranger, S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 268–276. [Google Scholar] [CrossRef]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non–T-Cell–Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, L.; Harview, C.; Emerson, R.; Wang, X.; Mok, S.; Homet, B.; Comin-Anduix, B.; Koya, R.C.; Robins, H.; Tumeh, P.C.; et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. OncoImmunology 2014, 3, e29244. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Carmi, Y.; Spitzer, M.H.; Linde, I.L.; Burt, B.M.; Prestwood, T.R.; Perlman, N.; Davidson, M.G.; Kenkel, J.A.; Segal, E.; Pusapati, G.V.; et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 2015, 521, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef] [PubMed]
- Koşaloğlu-Yalçın, Z.; Lanka, M.; Frentzen, A.; Premlal, A.L.R.; Sidney, J.; Vaughan, K.; Greenbaum, J.; Robbins, P.; Gartner, J.; Sette, A.; et al. Predicting T cell recognition of MHC class I restricted neoepitopes. OncoImmunology 2018, 7, e1492508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MNoblejas-López, D.M.; Nieto-Jiménez, C.; García, S.M.; Pérez-Peña, J.; Nuncia-Cantarero, M.; Andrés-Pretel, F.; Galán-Moya, E.M.; Amir, E.; Pandiella, A.; Győrffy, B.; et al. Expression of MHC class I, HLA-A and HLA-B identifies immune-activated breast tumors with favorable outcome. Oncoimmunology 2019, 8, e1629780. [Google Scholar] [CrossRef] [PubMed]
- Shklovskaya, E.; Rizos, H. MHC Class I Deficiency in Solid Tumors and Therapeutic Strategies to Overcome It. Int. J. Mol. Sci. 2021, 22, 6741. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.; Yshii, L.; Junius, S.; Geukens, N.; Liston, A.; Hollevoet, K.; Declerck, P. Intratumoral DNA-based delivery of checkpoint-inhibiting antibodies and interleukin 12 triggers T cell infiltration and anti-tumor response. Cancer Gene Ther. 2022, 29, 984–992. [Google Scholar] [CrossRef]
- Telli, M.L.; Nagata, H.; Wapnir, I.; Acharya, C.R.; Zablotsky, K.; Fox, B.A.; Bifulco, C.B.; Jensen, S.M.; Ballesteros-Merino, C.; Le, M.H.; et al. Intratumoral Plasmid IL12 Expands CD8+ T Cells and Induces a CXCR3 Gene Signature in Triple-negative Breast Tumors that Sensitizes Patients to Anti–PD-1 Therapy. Clin. Cancer Res. 2021, 27, 2481–2493. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Labiano, S.; Aznar, M.; Bolaños, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sánchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PDL1Blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Nishi, T.; Kobayashi, O.; Tamura, T.; Dev, S.B.; Takeshima, H.; Kochi, M.; Kuratsu, J.-I.; Sakata, T.; Ushio, Y. Combination electro-gene therapy using herpes virus thymidine kinase and interleukin-12 expression plasmids is highly efficient against murine carcinomas in vivo. Mol. Ther. 2004, 10, 929–937. [Google Scholar] [CrossRef]
- Lucas, M.L.; Heller, R. IL-12 Gene Therapy Using an Electrically Mediated Nonviral Approach Reduces Metastatic Growth of Melanoma. DNA Cell Biol. 2003, 22, 755–763. [Google Scholar] [CrossRef]
- Greaney, S.K.; Algazi, A.P.; Tsai, K.K.; Takamura, K.T.; Chen, L.; Twitty, C.G.; Zhang, L.; Paciorek, A.; Pierce, R.H.; Le, M.H.; et al. Intratumoral Plasmid IL12 Electroporation Therapy in Patients with Advanced Melanoma Induces Systemic and Intratumoral T-cell Responses. Cancer Immunol. Res. 2020, 8, 246–254. [Google Scholar] [CrossRef]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, E.; Benhar, I. Immune checkpoint inhibitor combinations: Current efforts and important aspects for success. Drug Resist. Updat. 2019, 45, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.M.; Subleski, J.J.; Wigginton, J.M.; Wiltrout, R.H. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin. Biol. Ther. 2007, 7, 1705–1721. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I.J.; Blasberg, R. Impact of the Tumor Microenvironment on Tumor-Infiltrating Lymphocytes: Focus on Breast Cancer. Breast Cancer 2017, 11, 1178223417731565. [Google Scholar] [CrossRef] [Green Version]
- Hicks, K.C.; Chariou, P.L.; Ozawa, Y.; Minnar, C.M.; Knudson, K.M.; Meyer, T.J.; Bian, J.; Cam, M.; Schlom, J.; Gameiro, S.R. Tumour-targeted interleukin-12 and entinostat combination therapy improves cancer survival by reprogramming the tumour immune cell landscape. Nat. Commun. 2021, 12, 5151. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef]
- Ahn, S.G.; Jeong, J.; Hong, S.; Jung, W.H. Current Issues and Clinical Evidence in Tumor-Infiltrating Lymphocytes in Breast Cancer. J. Pathol. Transl. Med. 2015, 49, 355–363. [Google Scholar] [CrossRef]
- Zhang, N.; Cao, M.; Duan, Y.; Bai, H.; Li, X.; Wang, Y. Prognostic role of tumor-infiltrating lymphocytes in gastric cancer: A meta-analysis and experimental validation. Arch. Med Sci. 2020, 16, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Spector, M.E.; Bellile, E.; Amlani, L.; Zarins, K.; Smith, J.; Brenner, J.C.; Rozek, L.; Nguyen, A.; Thomas, D.; McHugh, J.B.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol. Head Neck Surg. 2019, 145, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.S.; Ohashi, P.S. The Roles of CD8+ T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2019, 20, 25–39. [Google Scholar] [CrossRef]
- Shirley, S.A.; Lundberg, C.G.; Li, F.; Burcus, N.; Heller, R. Controlled Gene Delivery Can Enhance Therapeutic Outcome for Cancer Immune Therapy for Melanoma. Curr. Gene Ther. 2015, 15, 32–43. [Google Scholar] [CrossRef]
- Bodles-Brakhop, A.M.; Heller, R.; Draghia-Akli, R. Electroporation for the Delivery of DNA-based Vaccines and Immunotherapeutics: Current Clinical Developments. Mol. Ther. 2009, 17, 585–592. [Google Scholar] [CrossRef]
- Heller, L.C.; Heller, R. Electroporation Gene Therapy Preclinical and Clinical Trials for Melanoma. Curr. Gene Ther. 2010, 10, 312–317. [Google Scholar] [CrossRef]
- Frelin, L.; Brab, A.; Ahlen, G.; Brenndörfer, E.D.; Chen, M.; Sällberg, M. Electroporation: A promising method for the nonviral delivery of DNA vaccines in humans? Drug News Perspect. 2010, 23, 647–653. [Google Scholar] [CrossRef]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef] [PubMed]
- de Vry, J.; Martínez-Martínez, P.; Losen, M.; Temel, Y.; Steckler, T.; Steinbusch, H.W.; de Baets, M.H.; Prickaerts, J. Invivo electroporation of the central nervous system: A non-viral approach for targeted gene delivery. Prog. Neurobiol. 2010, 92, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiu, S.; Zhang, X.; Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I Trial of Interleukin-12 Plasmid Electroporation in Patients with Metastatic Melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef] [Green Version]
- Zaharoff, D.A.; Barr, R.C.; Li, C.-Y.; Yuan, F. Electromobility of plasmid DNA in tumor tissues during electric field-mediated gene delivery. Gene Ther. 2002, 9, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Komel, T.; Bosnjak, M.; Brezar, S.K.; De Robertis, M.; Mastrodonato, M.; Scillitani, G.; Pesole, G.; Signori, E.; Sersa, G.; Cemazar, M. Gene electrotransfer of IL-2 and IL-12 plasmids effectively eradicated murine B16.F10 melanoma. Bioelectrochemistry 2021, 141, 107843. [Google Scholar] [CrossRef]
- Lucas, M.; Heller, L.; Coppola, D.; Heller, R. IL-12 Plasmid Delivery by in Vivo Electroporation for the Successful Treatment of Established Subcutaneous B16.F10 Melanoma. Mol. Ther. 2002, 5, 668–675. [Google Scholar] [CrossRef]
- Hanna, E.; Zhang, X.; Woodlis, J.; Breau, R.; Suen, J.; Li, S. Intramuscular electroporation delivery of IL-12 gene for treatment of squamous cell carcinoma located at distant site. Cancer Gene Ther. 2001, 8, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Nguyen, B.; Lee, J.Y.; Browning, E.; Zhang, J.; Mukhopadhyay, A.; Gujar, R.; Salazar, J.; Hermiz, R.; Svenson, L.; et al. Intratumoral Electroporation of Plasmid Encoded IL12 and Membrane-Anchored Anti-CD3 Increases Systemic Tumor Immunity. Mol. Cancer Res. 2022, 20, 983–995. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Kraynyak, K.A.; Shields, A.F.; McRee, A.J.; Johnson, J.M.; Sun, W.; Chintakuntlawar, A.V.; Pawlicki, J.; Sylvester, A.J.; McMullan, T.; et al. Phase 1 study of safety, tolerability and immunogenicity of the human telomerase (hTERT)-encoded DNA plasmids INO-1400 and INO-1401 with or without IL-12 DNA plasmid INO-9012 in adult patients with solid tumors. J. Immunother. Cancer 2021, 9, e003019. [Google Scholar] [CrossRef]
- Atkins, M.B.; Robertson, M.J.; Gordon, M.; Lotze, M.T.; Decoste, M.; Dubois, J.S.; Ritz, J.; Sandler, A.B.; Edington, H.D.; Garzone, P.D.; et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin. Cancer Res. 1997, 3, 409–471. [Google Scholar] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farber, D.L. Differential TCR signaling and the generation of memory T cells. J. Immunol. 1998, 160, 535–539. [Google Scholar] [PubMed]
- Ahmed, R.; Gray, D. Immunological Memory and Protective Immunity: Understanding Their Relation. Science 1996, 272, 54–60. [Google Scholar] [CrossRef]
- Opferman, J.T.; Ober, B.T.; Ashton-Rickardt, P.G. Linear Differentiation of Cytotoxic Effectors into Memory T Lymphocytes. Science 1999, 283, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Dobrzanski, M.J.; Reome, J.B.; Dutton, R.W. Type 1 and Type 2 CD8+ Effector T Cell Subpopulations Promote Long-Term Tumor Immunity and Protection to Progressively Growing Tumor. J. Immunol. 2000, 164, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Lora, A.M.G.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Früh, K.; Yang, Y. Antigen presentation by MHC class I and its regulation by interferon gamma. Curr. Opin. Immunol. 1999, 11, 76–81. [Google Scholar] [CrossRef]
- Fusi, A.; Festino, L.; Botti, G.; Masucci, G.; Melero, I.; Lorigan, P.; A Ascierto, P. PD-L1 expression as a potential predictive biomarker. Lancet Oncol. 2015, 16, 1285–1287. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Santarpia, M.; Karachaliou, N. Tumor immune microenvironment characterization and response to anti-PD-1 therapy. Cancer Biol. Med. 2015, 12, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Thiem, A.; Hesbacher, S.; Kneitz, H.; Di Primio, T.; Heppt, M.V.; Hermanns, H.M.; Goebeler, M.; Meierjohann, S.; Houben, R.; Schrama, D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J. Exp. Clin. Cancer Res. 2019, 38, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, K.; Teh, J.L.; Okayama, H.; Shiraishi, K.; Kua, L.-F.; Koh, V.; Smoot, D.T.; Ashktorab, H.; Oike, T.; Suzuki, Y.; et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018, 109, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-γ/PDL1axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PDL1therapy. J. Neuroinflamm. 2018, 15, 290. [Google Scholar] [CrossRef] [Green Version]
- Yam, A.O.; Chtanova, T. The Ins and Outs of Chemokine-Mediated Immune Cell Trafficking in Skin Cancer. Front. Immunol. 2019, 10, 386. [Google Scholar] [CrossRef]
- Jacquelot, N.; Duong, C.P.M.; Belz, G.T.; Zitvogel, L. Targeting Chemokines and Chemokine Receptors in Melanoma and Other Cancers. Front. Immunol. 2018, 9, 2480. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, G.; Scott, M.; Mangiamele, C.G.; Heller, R. Modification of the Tumor Microenvironment Enhances Anti-PD-1 Immunotherapy in Metastatic Melanoma. Pharmaceutics 2022, 14, 2429. https://doi.org/10.3390/pharmaceutics14112429
Shi G, Scott M, Mangiamele CG, Heller R. Modification of the Tumor Microenvironment Enhances Anti-PD-1 Immunotherapy in Metastatic Melanoma. Pharmaceutics. 2022; 14(11):2429. https://doi.org/10.3390/pharmaceutics14112429
Chicago/Turabian StyleShi, Guilan, Megan Scott, Cathryn G. Mangiamele, and Richard Heller. 2022. "Modification of the Tumor Microenvironment Enhances Anti-PD-1 Immunotherapy in Metastatic Melanoma" Pharmaceutics 14, no. 11: 2429. https://doi.org/10.3390/pharmaceutics14112429