
Synthesis and Anticancer Activity of Indole-Functionalized Derivatives of Betulin

Abstract
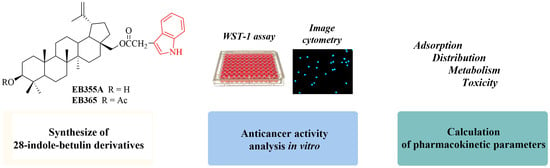
:
1. Introduction
2. Materials and Methods
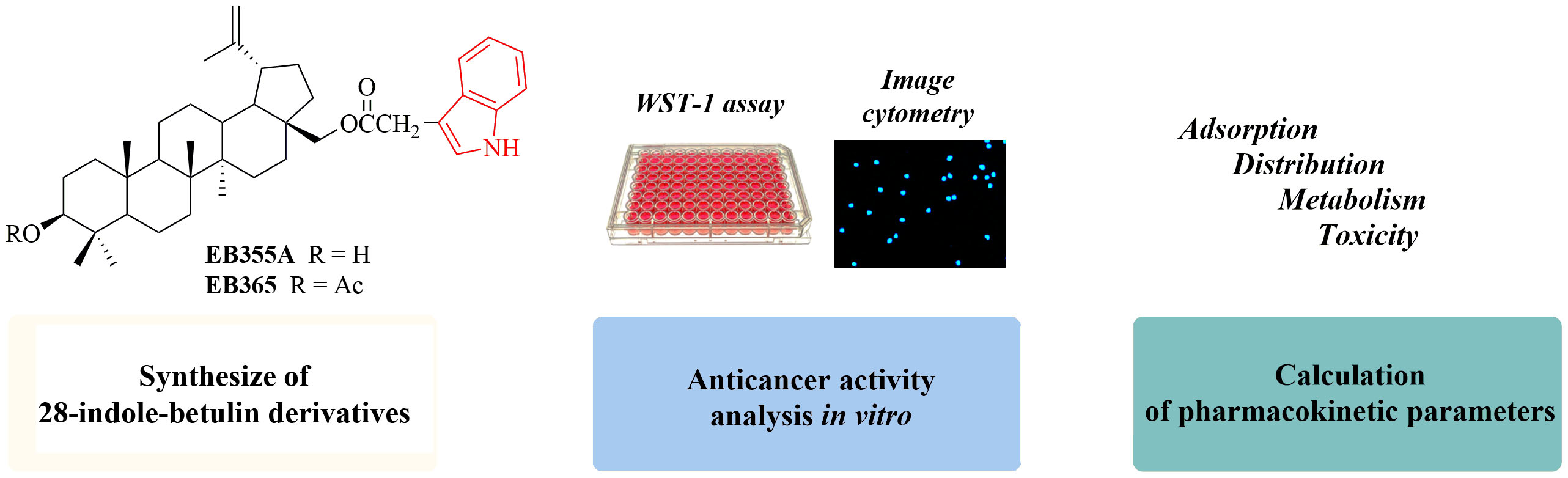
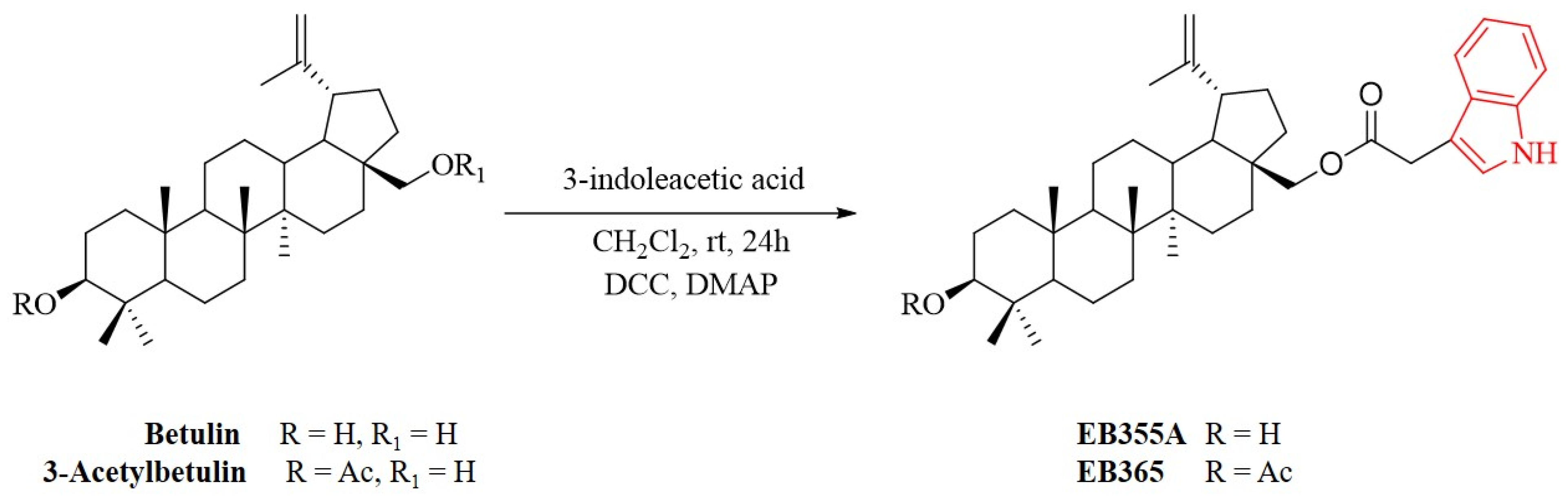
2.1. Chemistry
2.2. Synthesis of Indole Betulin Derivatives EB355A and EB365
2.3. Cell Culture and the Treatment
2.4. Cell Viability Assay
2.5. Cell Count Analysis
2.6. Cell Cycle and DNA Fragmentation Assay
2.7. Confocal Imaging
2.8. In Silico Study
2.9. Statistical Analysis
3. Results
3.1. Chemistry
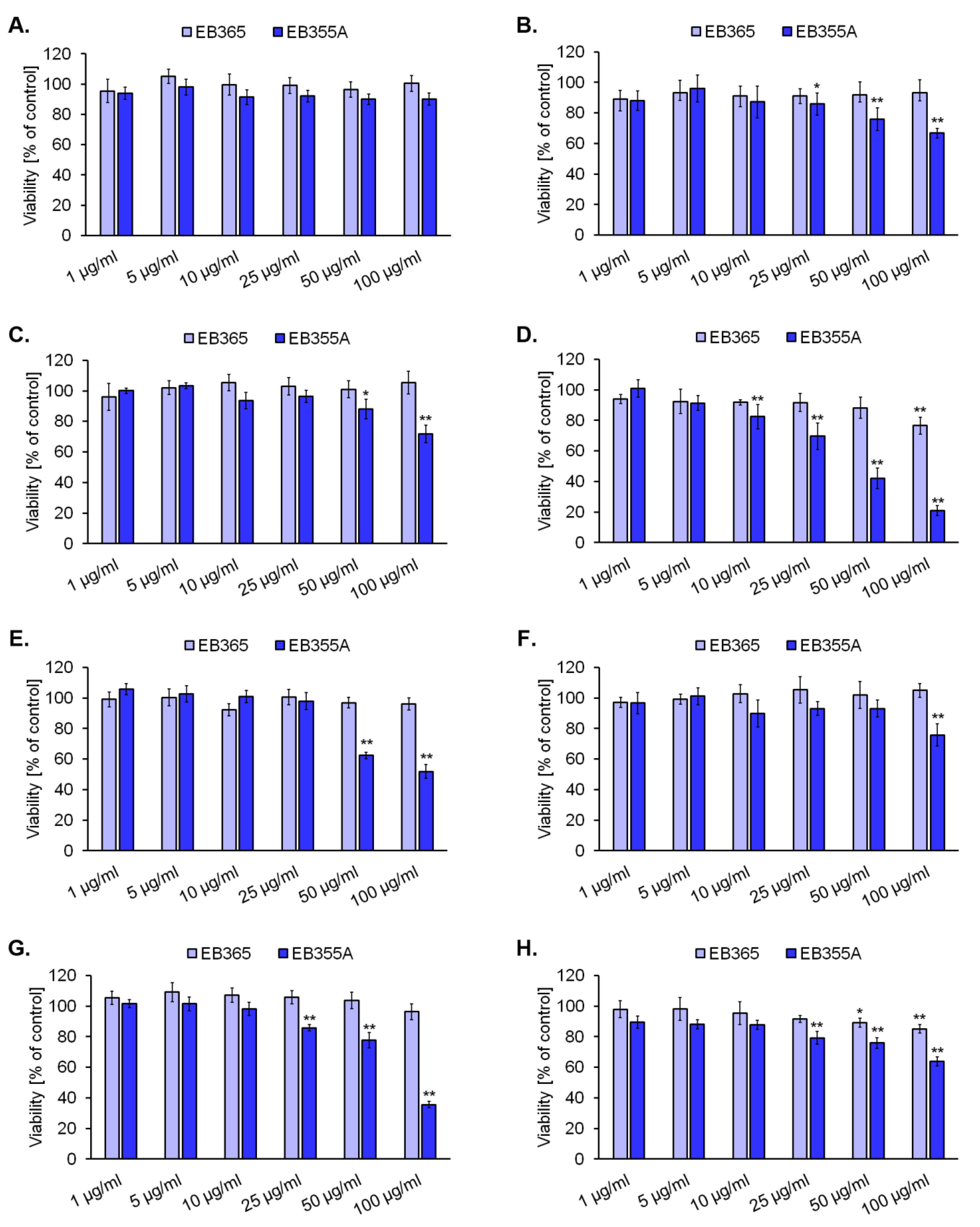
3.2. The Effect of Novel Betulin Derivatives on Cancer and Normal Cells Viability
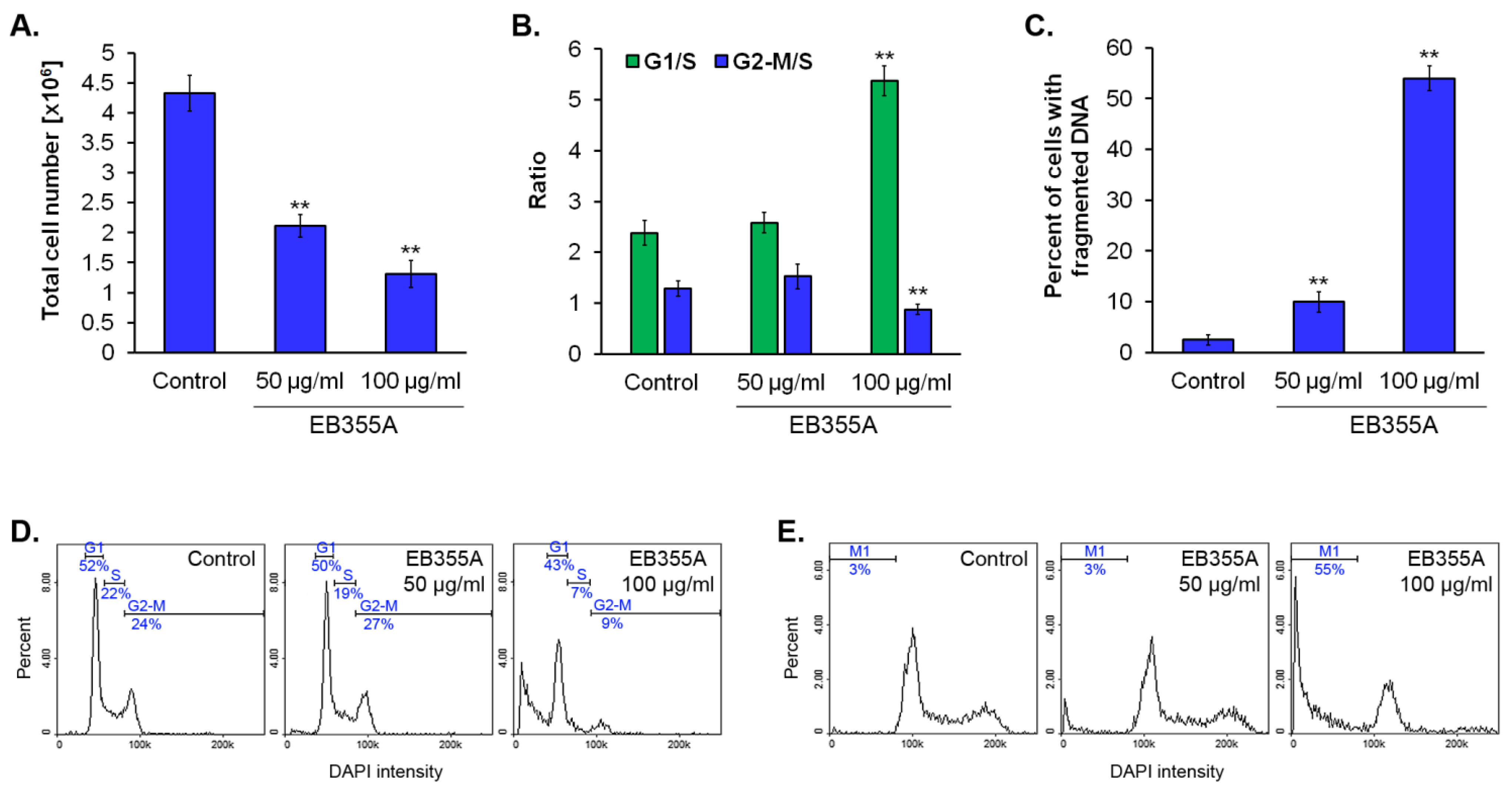
3.3. The Effect of EB355A on Proliferation, Cell Cycle, and DNA Fragmentation of MCF-7 Cells
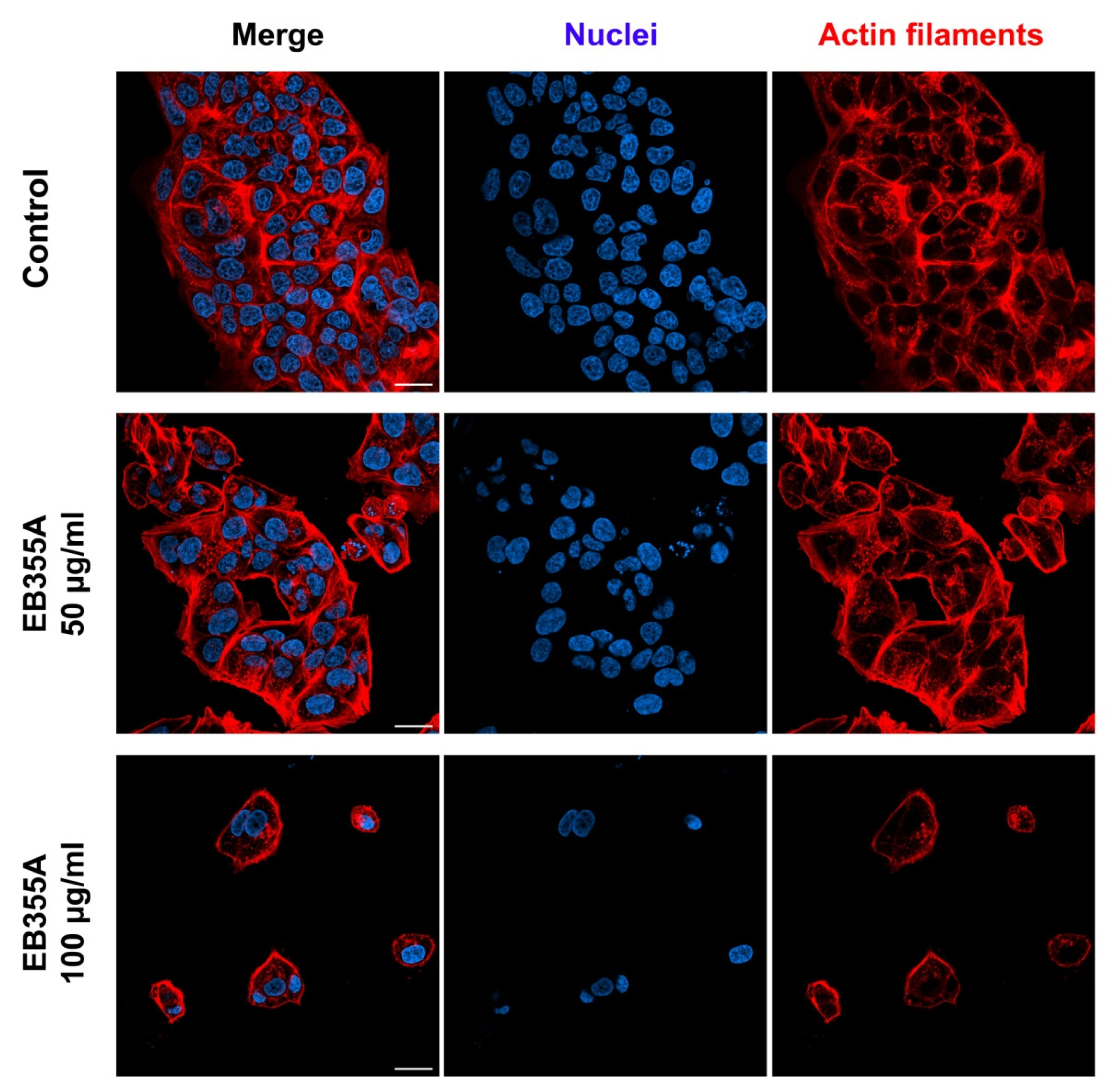
3.4. Confocal Imaging of Breast Cancer Cells Treated with EB355A
3.5. In Silico Analysis of Pharmacokinetic Profile of 28-Indolobetulin Derivatives
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Tilaoui, M.; Ait Mouse, H.; Zyad, A. Update and new insights on future cancer drug candidates from plant-based alkaloids. Front. Pharmacol. 2021, 12, 719694–719712. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409–107447. [Google Scholar] [CrossRef]
- Kaur, P.; Arora, S.; Singh, R. Isolation, characterization and biological activities of betulin from Acacia nilotica bark. Sci. Rep. 2022, 12, 9370–9380. [Google Scholar] [CrossRef]
- Cabaj, J.; Bąk, W.; Wróblewska-Łuczka, P. Anti-cancer effect of betulin and its derivatives, with particular emphasis on the treatment of melanoma. J. Pre-Clin. Clin. Res. 2021, 15, 73–79. [Google Scholar] [CrossRef]
- Su, C.H.; Lin, C.Y.; Tsai, C.H.; Lee, H.P.; Lo, L.C.; Huang, W.C.; Wu, Y.C.; Hsieh, C.L.; Tang, C.H. Betulin suppresses TNF-α and IL-1β production in osteoarthritis synovial fibroblasts by inhibiting the MEK/ERK/NF-κB pathway. J. Funct. Foods 2021, 86, 104729–104739. [Google Scholar] [CrossRef]
- Oloyede, H.O.B.; Ajiboye, H.O.; Salawu, M.O.; Ajiboy, T.O. Influence of oxidative stress on the antibacterial activity of betulin, betulinic acid and ursolic acid. Microb. Pathog. 2017, 111, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Alhadrami, H.A.; Sayed, A.M.; Sharif, A.M.; Azhar, E.I.; Rateb, M.E. Olive-derived triterpenes suppress SARS COV-2 main protease: A promising scaffold for future therapeutics. Molecules 2021, 26, 2654. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, C.; Cai, Z.; Zhao, F.; He, L.; Lou, X.; Qi, X. Betulin induces cytochrome c release and apoptosis in colon cancer cells via NOXA. Oncol. Lett. 2018, 15, 7319–7327. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Jiang, D.; Lin, Y.; Wang, Y.; Li, Q.; Liu, L.; Jin, Y.H. Betulin induces reactive oxygen species-dependent apoptosis in human gastric cancer SGC7901 cells. Arch. Pharm. Res. 2016, 39, 1257–1265. [Google Scholar] [CrossRef]
- John, R.; Dalal, B.; Shankarkumar, A.; Devarajan, P.V. Innovative betulin nanosuspension exhibits enhanced anticancer activity in a triple negative breast cancer cell line and Zebrafish angiogenesis model. Int. J. Pharm. 2021, 600, 120511. [Google Scholar] [CrossRef] [PubMed]
- Dehelean, C.A.; Feflea, S.; Molnár, J.; Zupko, I.; Soica, C. Betulin as an antitumor agent tested in vitro on A431, HeLa and MCF7, and as an angiogenic inhibitor in vivo in the CAM Assay. Nat. Prod. Commun. 2012, 7, 981–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Şoica, C.; Dehelean, C.; Danciu, C.; Wang, H.M.; Wenz, G.; Ambrus, R.; Bojin, F.; Anghel, M. Betulin complex in γ-cyclodextrin derivatives: Properties and antineoplasic activities in in vitro and in vivo tumor models. Int. J. Mol. Sci. 2012, 13, 14992–15011. [Google Scholar] [CrossRef]
- Dehelean, C.A.; Feflea, S.; Gheorgheosu, D.; Ganta, S.; Cimpean, A.M.; Muntean, D.; Amiji, M.M. Anti-angiogenic and anti-cancer evaluation of betulin nanoemulsion in chicken chorioallantoic membrane and skin carcinoma in Balb/c Mice. J. Biomed. Nanotechnol. 2013, 9, 577–589. [Google Scholar] [CrossRef]
- Bębenek, E.; Chrobak, E.; Marciniec, K.; Kadela-Tomanek, M.; Trynda, J.; Wietrzyk, J.; Boryczka, S. Biological activity and in silico study of 3-modified derivatives of betulin and betulinic aldehyde. Int. J. Mol. Sci. 2019, 20, 1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozubek, M.; Hoenke, S.; Schmidt, T.; Deigner, H.P.; Al-Harrasi, A.; Csuk, R. Synthesis and cytotoxicity of betulin and betulinic acid derived 30-oxo-amides. Steroids 2022, 182, 109014–109023. [Google Scholar] [CrossRef]
- Chrobak, E.; Jastrzębska, M.; Bębenek, E.; Kadela-Tomanek, M.; Marciniec, K.; Latocha, M.; Wrzalik, R.; Kusz, J.; Boryczka, S. Molecular structure, in vitro anticancer study and molecular docking of new phosphate derivatives of betulin. Molecules 2021, 26, 737–766. [Google Scholar]
- Yang, S.J.; Liu, M.C.; Xiang, H.M.; Zhao, Q.; Xue, W.; Yang, S. Synthesis and in vitro antitumor evaluation of betulin acid ester derivatives as novel apoptosis inducers. Eur. J. Med. Chem. 2015, 102, 249–255. [Google Scholar] [CrossRef]
- Ali-Seyed, M.; Jantan, I.; Vijayaraghavan, K.; Bukhari, S.N.A. Betulinic acid: Recent advances in chemical modifications, effective delivery, and molecular mechanisms of a promising anticancer therapy. Chem. Biol. Drug. Des. 2016, 87, 517–536. [Google Scholar] [CrossRef]
- Lombrea, A.; Scurtu, A.D.; Avram, S.; Pavel, I.Z.; Turks, M.; Lugiņina, J.; Peipinš, U.; Dehelean, C.A.; Soica, C.; Danciu, C. Anticancer potential of betulonic acid derivatives. Int. J. Mol. Sci. 2021, 22, 3676. [Google Scholar] [CrossRef]
- Dvořák, Z.; Poulíková, K.; Mani, S. Indole scaffolds as a promising class of the aryl hydrocarbon receptor ligands. Eur. J. Med. Chem. 2021, 215, 113231–113240. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wen, X.; Gong, Y.; Wang, X. Current scenario of indole derivatives with potential anti-drug resistant cancer activity. Eur. J. Med. Chem. 2020, 200, 112359–112379. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.K.; Im, S.A.; Min, A.; Kim, H.P.; Hur, H.S.; Lee, K.H.; Han, S.W.; Song, S.H.; Oh, D.Y.; Kim, T.Y.; et al. Sunitinib synergizes the antitumor effect of cisplatin via modulation of ERCC1 expression in models of gastric cancer. Cancer Lett. 2012, 321, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.Z.; Ullrich, A.; Häring, H.U.; Ullrich, S.; Gerst, F. Sunitinib specifically augments glucose-induced insulin secretion. Cell Signal. 2017, 36, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bhakhar, K.A.; Sureja, D.K.; Dhameliya, T.M. Synthetic account of indoles in search of potential anti-mycobacterial agents: A review and future insights. J. Mol. Struct. 2022, 1248, 131522–131548. [Google Scholar] [CrossRef]
- Yang, L.; Chen, X.; Ni, K.; Li, Y.; Wu, J.; Chen, W.; Ji, Y.; Feng, L.; Li, F.; Chen, D. Proton-exchanged montmorillonite-mediated reactions of hetero-benzyl acetates: Application to the synthesis of Zafirlukast. Tetrahedron Lett. 2020, 61, 152123–152129. [Google Scholar] [CrossRef]
- Lange, J.H.M.; de Jong, J.C.; Sanders, H.J.; Visser, G.M.; Kruse, C.G. A straightforward synthetic approach for roxindole. Bioorg. Med. Chem. Lett. 1999, 9, 1055–1056. [Google Scholar] [CrossRef]
- Sasmal, P.K.; Ramachandran, G.; Zhang, Y.; Liu, Z. First total synthesis of 3a-hydroxy-1,1-dimethyl-5-((N-methylsulfamoyl) methyl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-1-ium 2,2,2-trifluoroacetate by mimicking the oxidative degradation pathway of sumatriptan. Results Chem. 2021, 3, 100173–100176. [Google Scholar] [CrossRef]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef]
- Kumar, V.; Rani, N.; Aggarwal, P.; Sanna, V.K.; Singh, A.T.; Jaggi, M.; Joshi, N.; Sharma, P.K.; Irchhaiya, R.; Burman, A.C. Synthesis and cytotoxic activity of heterocyclic ring-substituted betulinic acid derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 5058–5062. [Google Scholar] [CrossRef]
- Li, A.L.; Hao, Y.; Wang, W.Y.; Liu, Q.S.; Sun, Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel indole derivatives of ursolic acid as potential topoisomerase II inhibitors. Int. J. Mol. Sci. 2020, 21, 2876. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Hu, H.; Persoons, L.; Daelemans, D.; De Jonghe, S.; Luyten, W.; Krasniqi, B.; Dehaen, W. Antibacterial and antitumoral properties of 1,2,3-triazolo fused triterpenes and their mechanism of inhibiting the proliferation of HL-60 cells. Eur. J. Med. Chem. 2021, 224, 113727–113741. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.; Whymark, K.D.; Sydenham, J.; Sneddon, H.F. A solvent-reagent selection guide for Steglich-type esterification of carboxylic acids. Green Chem 2021, 23, 6405–6413. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, M. DNA fragmentation in apoptosis. Cell Res. 2000, 10, 205–211. [Google Scholar]
- Darzynkiewicz, Z.; Zhao, H. Detection of DNA strand breaks in apoptotic cells by flow- and image-cytometry. Methods Mol. Biol. 2011, 682, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhou, Y.; Li, L.; Shen, X.; Chen, G.; Wang, X.; Liang, X.; Tan, M.; Huang, Z. Computational approaches in preclinical studies on drug discovery and development. Front. Chem. 2020, 8, 726. [Google Scholar] [CrossRef]
- Li, X.; Chen, L.; Cheng, F.; Wu, Z.; Bian, H.; Xu, C.; Li, W.; Liu, G.; Shen, X.; Tang, Y. In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model 2014, 54, 1061–1069. [Google Scholar] [CrossRef]
- Rzeski, W.; Stepulak, A.; Szymański, M.; Juszczak, M.; Grabarska, A.; Sifringer, M.; Kaczor, J.; Kandefer-Szerszeń, M. Betulin elicits anti-cancer effects in tumour primary cultures and cell lines in vitro. Basic Clin. Pharmacol. Toxicol. 2009, 105, 425–432. [Google Scholar] [CrossRef]
- Han, Y.H.; Mun, J.G.; Jeon, H.D.; Kee, J.Y.; Hong, S.H. Betulin inhibits lung metastasis by inducing cell cycle arrest, autophagy, and apoptosis of metastatic colorectal cancer cells. Nutrients 2019, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fei, Z.; Huang, C. Betulin terpenoid targets OVCAR-3 human ovarian carcinoma cells by inducing mitochondrial mediated apoptosis, G2/M phase cell cycle arrest, inhibition of cell migration and invasion and modulating mTOR/PI3K/AKT signalling pathway. Cell Mol. Biol. 2021, 67, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Latocha, M.; Boryczka, S. Novel triazoles of 3-acetylbetulin and betulone as anticancer agents. Med. Chem. Res. 2018, 27, 2051–2061. [Google Scholar] [CrossRef] [Green Version]
- Kommera, H.; Kaluderović, G.N.; Kalbitz, J.; Paschke, R. Synthesis and anticancer activity of novel betulinic acid and betulin derivatives. Arch. Pharm. 2010, 343, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Güttler, A.; Eiselt, Y.; Funtan, A.; Thiel, A.; Petrenko, M.; Keßler, J.; Thondorf, I.; Paschke, R.; Vordermark, D.; Bache, M. Betulin sulfonamides as carbonic anhydrase inhibitors and anticancer agents in breast cancer cells. Int. J. Mol. Sci. 2021, 22, 8808. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Singh, S.; Skvortsova, I.; Kumar, V. Promising targets in anti-cancer drug development: Recent updates. Curr. Med. Chem. 2017, 24, 4729–4752. [Google Scholar] [CrossRef]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel triazole hybrids of betulin: Synthesis and biological activity profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [Green Version]
- Kadela, M.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Książek, M.; Boryczka, S. Synthesis, Structure and Cytotoxic Activity of Mono- and Dialkoxy Derivatives of 5,8-Quinolinedione. Molecules 2016, 21, 156. [Google Scholar] [CrossRef] [Green Version]
- Reda, A.; Refaat, A.; Abd-Rabou, A.A.; Mahmoud, A.M.; Adel, M.; Sabet, S.; Ali, S.S. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hormone-responsive breast cancer cells. Sci. Rep. 2019, 9, 13748–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhadi, P.; Yarani, R.; Valipour, E.; Kiani, S.; Hoseinkhani, Z.; Mansouri, K. Cell line-directed breast cancer research based on glucose metabolism status. Biomed. Pharmacother. 2022, 146, 112526–112533. [Google Scholar] [CrossRef] [PubMed]
- Denis-Pouxviel, C.; Gauthier, T.; Daviaud, D.; Murat, J.C. Phosphofructokinase 2 and glycolysis in HT29 human colon adenocarcinoma cell line. Regulation by insulin and phorbol esters. Biochem. J. 1990, 268, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Xie, T.; Tian, X.; Han, N.; Liu, X.; Chen, H.; Qi, J.; Gao, F.; Li, W.; Wu, Q.; et al. Betulinic Acid-Nitrogen Heterocyclic Derivatives: Design, Synthesis, and Antitumor Evaluation in Vitro. Molecules 2020, 25, 948. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.; Wang, J.; Zhang, W.; Xu, X. ADME Evaluation in Drug Discovery. 7. Prediction of oral absorption by correlation and classification. J. Chem. Inf. Model. 2007, 47, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’Driscoll, C.M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In silico prediction of blood–brain barrier permeability of compounds by machine learning and resampling methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef]
- Liu, M.C.; Cortés, J.; O’Shaughnessy, J. Challenges in the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer with brain metastases. Cancer Metastasis Rev. 2016, 35, 323–332. [Google Scholar] [CrossRef]
- Shah, N.; Mohammad, A.S.; Saralkar, P.; Sprowls, S.A.; Vickers, S.D.; John, D.; Tallman, R.M.; Lucke-Wold, B.; Jarrell, K.E.; Pinti, M.; et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol. Res. 2018, 132, 47–68. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.F.; Xue, C.C.; Yu, X.Q.; Li, C.; Wang, G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3a4 and the role of therapeutic drug monitoring. Ther. Drug Monit. 2007, 29, 687–710. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.; Sureshkumar, R.; Zehravi, M.; Akter, R.; Ali, F.; Ramproshad, S.; Mondal, B.; Tagde, P.; Ahmed, Z.; Khan, F.S.; et al. Multidrug resistance of cancer cells and the vital role of p-glycoprotein. Life 2022, 12, 897. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Leonessa, F.; Trock, B. Multidrug resistance/P-glycoprotein and breast cancer: Review and meta-analysis. Semin Oncol. 2005, 32 (Suppl. S7), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.R.; Kim, S.; Elghoul, Y.; Dou, Q.P. P-Glycoprotein Inhibition Sensitizes Human Breast Cancer Cells to Proteasome Inhibitors. J. Cell Biochem. 2017, 118, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Hagenbuch, B. Organic anion transporting polypeptides. Curr. Top. Membr. 2014, 73, 205–232. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Uddin, M.E.; DiGiacomo, D.; Lustberg, M.B.; Hu, S.; Alex Sparreboom, A. Role of SLC transporters in toxicity induced by anticancer drugs. Expert Opin. Drug Metab. Toxicol. 2020, 16, 493–506. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EB355A | EB365 | ||
|---|---|---|---|---|
| Parameter | Value | Probability | Value | Probability |
| Absorption | ||||
| Human intestinal absorption (HIA) | + | 0.9902 | + | 0.9933 |
| Caco-2 permeability | − | 0.8212 | − | 0.8326 |
| Human oral bioavailability | − | 0.6286 | − | 0.5714 |
| Distribution | ||||
| Subcellular localization | Mitochondria | 0.7321 | Mitochondria | 0.7744 |
| Blood–brain barrier (BBB) permeability organic anion-transporting | + | 0.9368 | + | 0.9609 |
| polypeptide (OATP) inhibitors: | ||||
| OATP 2B1 | ||||
| OATP 1B1 | − | 0.5672 | − | 0.5712 |
| OATP 1B3 | + | 0.8799 | + | 0.8451 |
| Multidrug and toxin extrusion Transporter 1 (MATE1) | + | 0.9157 | + | 0.8495 |
| Organic cation transport protein 2 (OCT2) inhibitor | ||||
| Bile salt export pump (BSEP) inhibitor | − | 0.94 | − | 0.8 |
| P-glycoprotein inhibitor | ||||
| P-glycoprotein substrate | − | 0.725 | − | 0.6364 |
| + | 0.9928 | + | 0.9874 | |
| + | 0.7226 | + | 0.7883 | |
| + | 0.552 | + | 0.5777 | |
| Metabolism | ||||
| Cytochrome P450: | ||||
| CYP450 3A4 substrate | + | 0.7486 | + | 0.7497 |
| CYP450 2C9 substrate | − | 1 | − | 0.6091 |
| CYP450 2D6 substrate | − | 0.8088 | − | 0.8292 |
| CYP450 3A4 inhibition | + | 0.7587 | + | 0.7781 |
| CYP450 2C9 inhibition | − | 0.6231 | − | 0.545 |
| CYP450 2C19 inhibition | − | 0.5276 | − | 0.644 |
| CYP450 2D6 inhibition | − | 0.8537 | − | 0.8629 |
| CYP450 1A2 inhibition | + | 0.5924 | + | 0.6778 |
| CYP inhibitory promiscuity | + | 0.7921 | + | 0.8891 |
| Toxicity | ||||
| Carcinogenicity | − | 0.9143 | − | 0.8857 |
| Eye corrosion | − | 0.9927 | − | 0.9913 |
| Eye irritation | − | 0.9265 | − | 0.9152 |
| Ames mutagenesis | − | 0.68 | − | 0.68 |
| Hepatotoxicity | + | 0.525 | + | 0.525 |
| Nephrotoxicity | − | 0.9145 | − | 0.8732 |
| Acute oral toxicity | III | 0.6406 | III | 0.7173 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzepka, Z.; Bębenek, E.; Chrobak, E.; Wrześniok, D. Synthesis and Anticancer Activity of Indole-Functionalized Derivatives of Betulin. Pharmaceutics 2022, 14, 2372. https://doi.org/10.3390/pharmaceutics14112372
Rzepka Z, Bębenek E, Chrobak E, Wrześniok D. Synthesis and Anticancer Activity of Indole-Functionalized Derivatives of Betulin. Pharmaceutics. 2022; 14(11):2372. https://doi.org/10.3390/pharmaceutics14112372
Chicago/Turabian StyleRzepka, Zuzanna, Ewa Bębenek, Elwira Chrobak, and Dorota Wrześniok. 2022. "Synthesis and Anticancer Activity of Indole-Functionalized Derivatives of Betulin" Pharmaceutics 14, no. 11: 2372. https://doi.org/10.3390/pharmaceutics14112372