
Functionalization of Morin-Loaded PLGA Nanoparticles with Phenylalanine Dipeptide Targeting the Brain
 , , , , , , , and
, , , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
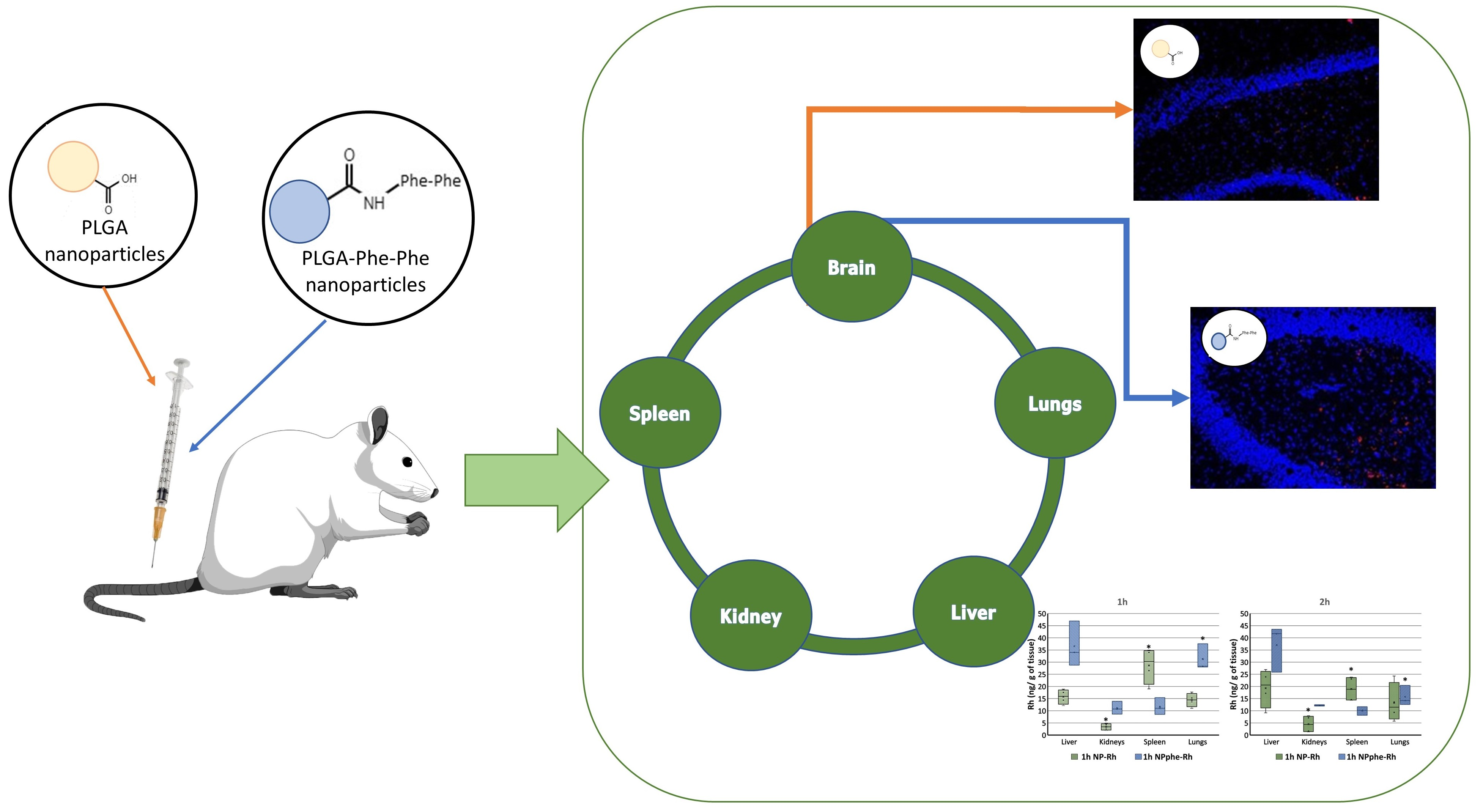
2.2. Synthesis of PLGA-phe-phe
2.2.1. Activation of PLGA
2.2.2. Conjugation of Phe-Phe Peptide to Activated PLGA
2.3. Elaboration of Nanoparticles
2.3.1. Elaboration of Morin Hydrate/Rhodamine B-loaded PLGA Nanoparticles
2.3.2. Elaboration of Morin Hydrate/Rhodamine B-Loaded PLGA-Phe-Phe Nanoparticles
2.4. Characterization of Nanoparticles
2.4.1. Morphology
2.4.2. Zeta Potential
2.4.3. Encapsulation Efficiency
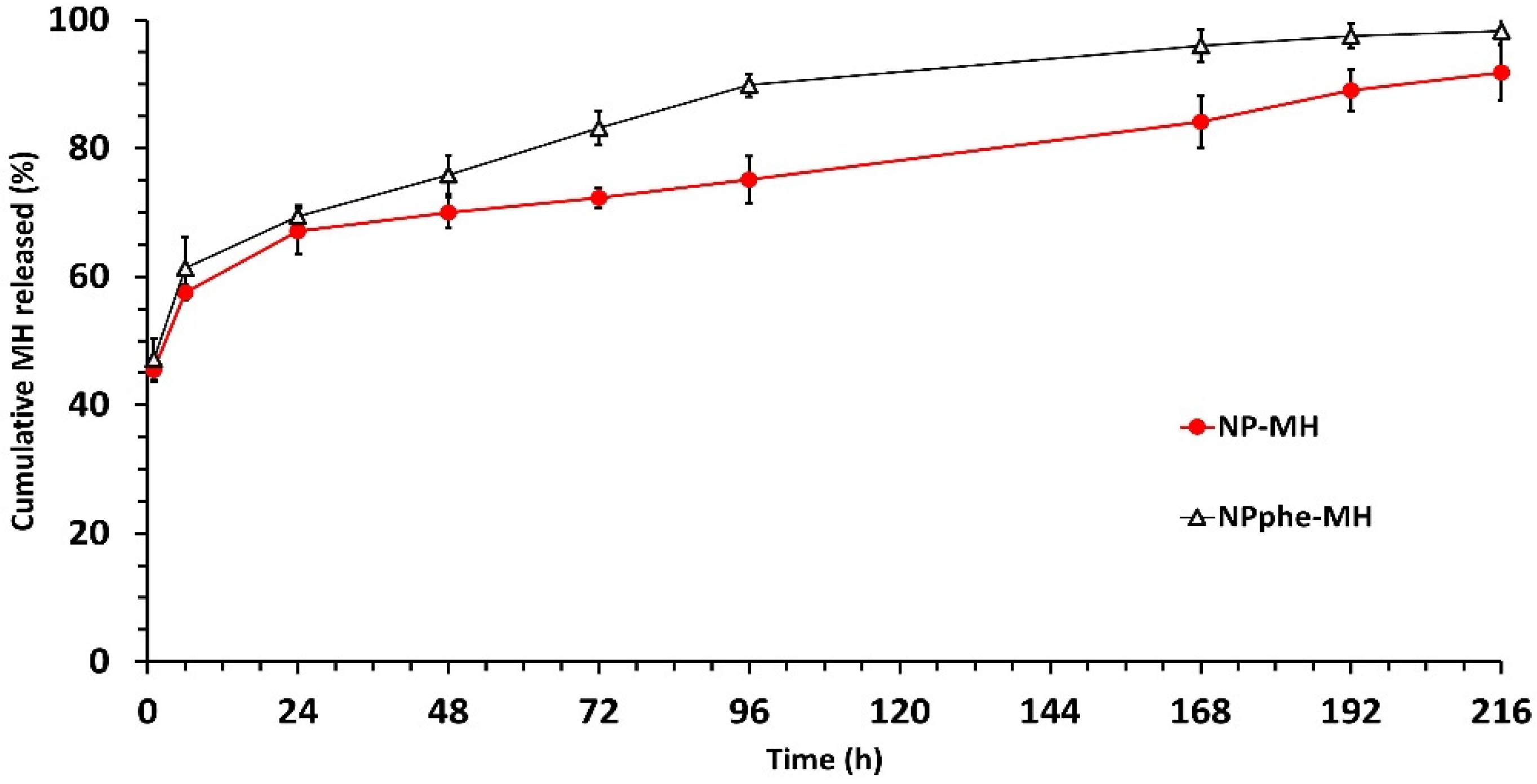
2.4.4. In Vitro Release Studies
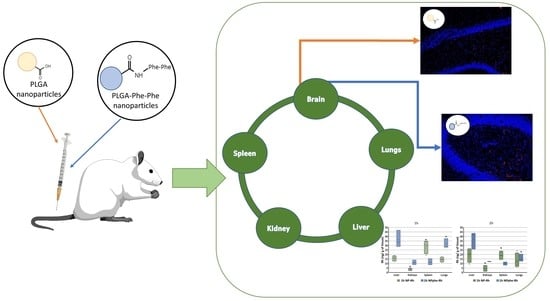
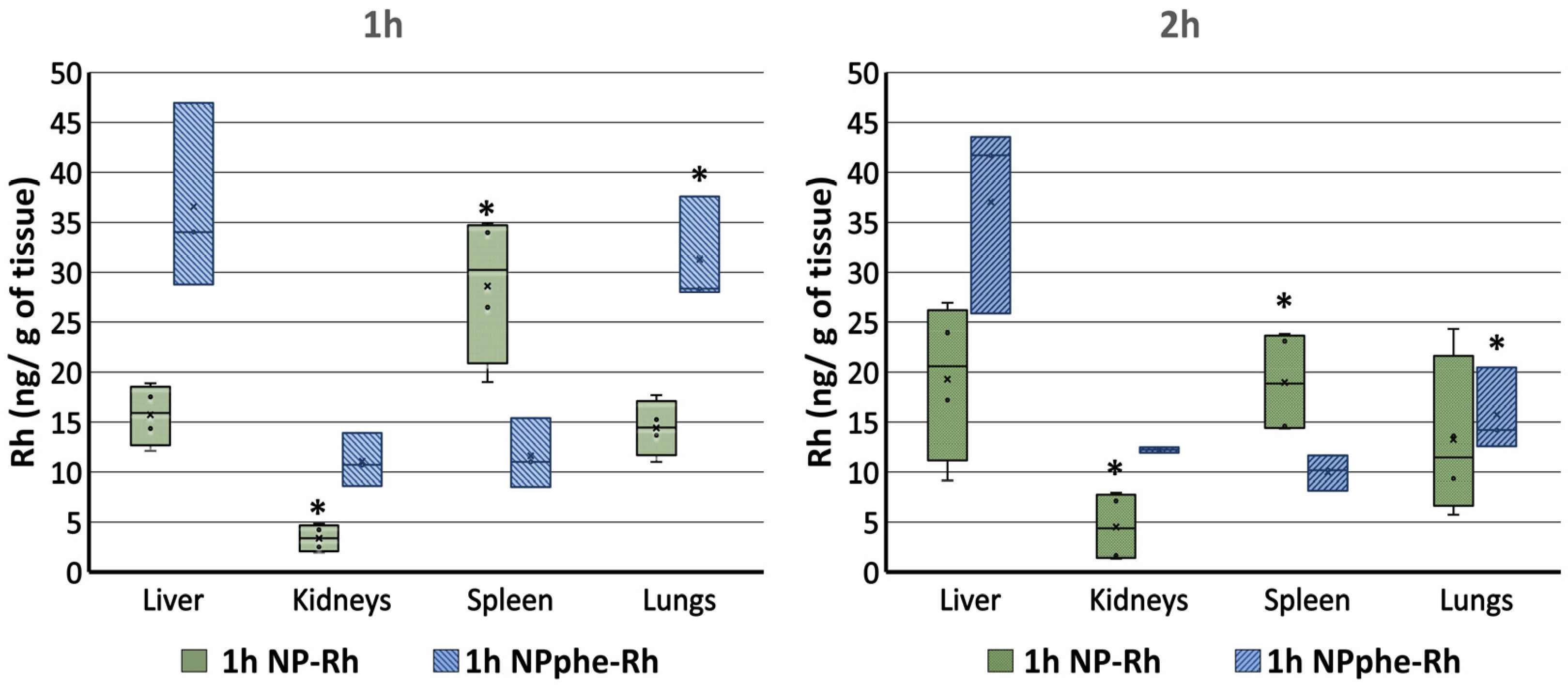
2.5. Biodistribution Studies
- -
- Group 1 (n = 4) received saline solution.
- -
- Group 2 (n = 8) received formulation NP-Rh.
- -
- Group 3 (n = 6) received formulation NPphe-Rh.
2.5.1. Organ Biodistribution
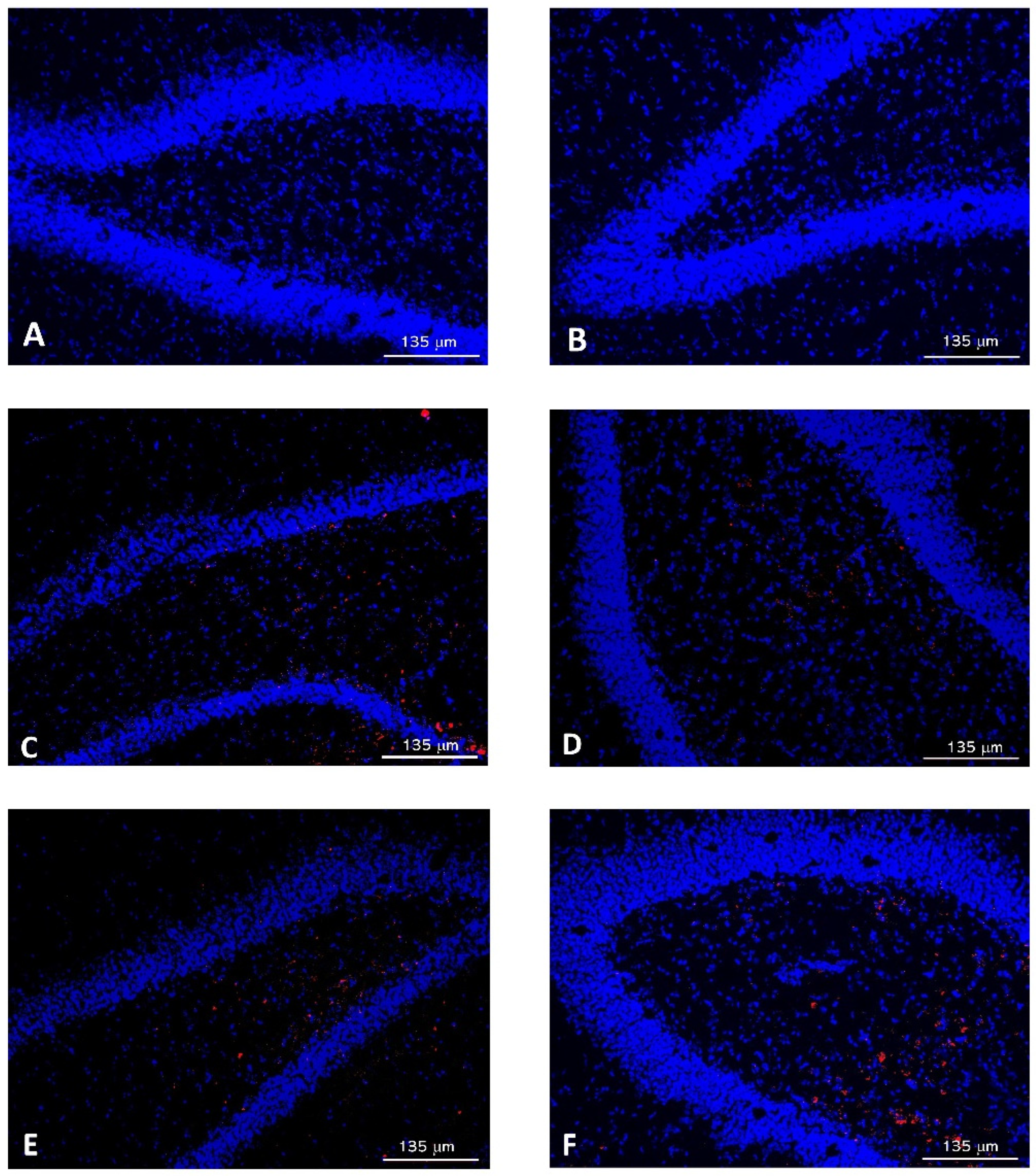
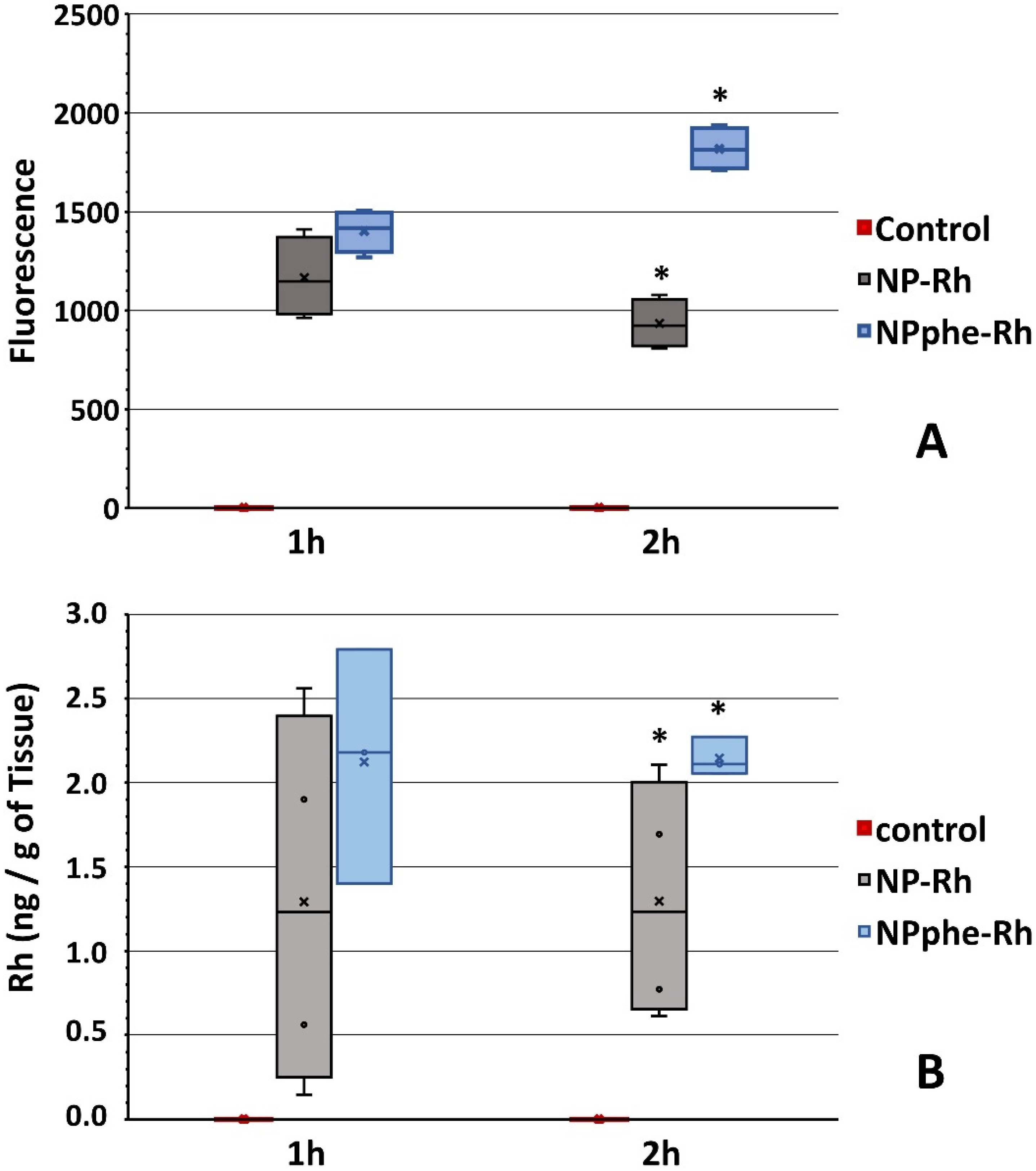
2.5.2. Passage of NPs through the BBB
2.5.3. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of PLGA phe-phe
3.2. Characterization of Nanoparticles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekhar, K.; Chakrabarti, M.; Govindaraju, T. Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 2015, 51, 13434–13450. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Waddad, A.Y.; Abbad, S.; Yu, F.; Munyendo, W.L.L.; Wang, J.; Lv, H.; Zhou, J. Formulation, characterization and pharmacokinetics of Morin hydrate niosomes prepared from various non-ionic surfactants. Int. J. Pharm. 2013, 456, 446–458. [Google Scholar] [CrossRef]
- Gong, E.J.; Park, H.R.; Kim, M.E.; Piao, S.; Lee, E.; Jo, D.G.; Chung, H.Y.; Ha, N.C.; Mattson, M.P.; Lee, J. Morin attenuates tau hyperphosphorylation by inhibiting GSK3β. Neurobiol. Dis. 2011, 44, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Qu, J.; Zhang, W.; Bai, M.; Zhou, Q.; Zhang, Z.; Li, Z.; Miao, J. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology 2016, 108, 1–13. [Google Scholar] [CrossRef]
- Mohammadi, N.; Asle-Rousta, M.; Rahnema, M.; Amini, R. Morin attenuates memory deficits in a rat model of Alzheimer’s disease by ameliorating oxidative stress and neuroinflammation. Eur. J. Pharmacol. 2021, 910, 174506. [Google Scholar] [CrossRef]
- Alberdi, E.; Gomez, M.V.S.; Ruiz, A.; Cavaliere, F.; Ortiz-Sanz, C.; Quintela, T.; Capetillo-Zarate, E.; Solé-Domènech, S.; Matute, C. Mangiferin and morin attenuate oxidative stress, mitochondrial dysfunction, and neurocytotoxicity, induced by amyloid beta oligomers. Oxid. Med. Cell. Longev. 2018, 2018, 2856063. [Google Scholar] [CrossRef]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.-I.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Selkoe, D. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Frandsen, J.; Choi, S.; Narayanasamy, P. Neural glyoxalase pathway enhancement by morin derivatives in an Alzheimer’s disease model. ACS Chem. Neurosci. 2020, 11, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Lu, L.; Ma, Q.; Zhang, J. Preparation, characterization and systemic application of self-assembled hydroxyethyl starch nanoparticles-loaded flavonoid Morin for hyperuricemia therapy. Int. J. Nanomed. 2018, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Thakur, V.; Deshmukh, R.; Sharma, A.; Rathore, M.S.; Kumar, A.; Mishra, N. Development and characterization of morin hydrate-loaded micellar nanocarriers for the effective management of Alzheimer’s disease. J. Microencapsul. 2018, 35, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Ning, E.; Peng, Y.; Zhang, J. Mechanisms of poor oral bioavailability of flavonoid Morin in rats: From physicochemical to biopharmaceutical evaluations. Eur. J. Pharm. Sci. 2019, 128, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Ohlow, M.J.; Sohre, S.; Granold, M.; Schreckenberger, M.; Moosmann, B. Why have clinical trials of antioxidants to prevent neurodegeneration failed?—A cellular investigation of novel phenothiazine-type antioxidants reveals competing objectives for pharmaceutical neuroprotection. Pharm. Res. 2017, 34, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, R.; Tosi, G.; Grabrucker, A. Emerging use of nanotechnology in the treatment of neurological disorders. Curr. Pharm. Des. 2015, 21, 3111–3130. [Google Scholar] [CrossRef]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.; Vilella, A.; Veratti, P.; Belletti, D.; Pederzoli, F.; Ruozi, B.; Vandelli, M.A.; Zoli, M.; Forni, M. Exploiting bacterial pathways for BBB crossing with PLGA nanoparticles modified with a mutated form of diphtheria toxin (CRM197): In vivo experiments. Mol. Pharm. 2015, 12, 3672–3684. [Google Scholar] [CrossRef]
- Bi, C.; Wang, A.; Chu, Y.; Liu, S.; Mu, H.; Liu, W.; Wu, Z.; Sun, K.; Li, Y. Intranasal delivery of rotigotine to the brain with lactoferrin-modified PEG-PLGA nanoparticles for Parkinson´s disease treatment. Int. J. Nanomed. 2016, 11, 6547–6559. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Jin, X.; Li, L.; Lv, F.; Liu, T. OX26 modified hyperbranched polyglycerol-conjugated poly(lactic-co-glycolic acid) nanoparticles: Synthesis, characterization and evaluation of its brain delivery ability. J. Mater. Sci. Mater. Med. 2012, 23, 1891–1901. [Google Scholar] [CrossRef]
- Ulbrich, K.; Michaelis, M.; Rothweiler, F.; Knobloch, T.; Sithisarn, P.; Cinatl, J.; Kreuter, J. Interaction of folate-conjugated human serum albumin (HSA) nanoparticles with tumour cells. Int. J. Pharm. 2011, 406, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Wagner, S.; Büchel, C.; von Briesen, H.; Kreuter, J. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J. Control. Release 2009, 137, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, A. Small molecular drug transfer across the blood-brain barrier via carrier-mediated transport systems. NeuroRx 2005, 2, 54–62. [Google Scholar] [CrossRef]
- Boado, R.J.; Li, J.Y.; Nagaya, M.; Zhang, C.; Pardridge, W.M. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 12079–12084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Welty, D.F. The simultaneous estimation of the influx and efflux blood-brain barrier permeabilities of gabapentin using a microdialysis-pharmacokinetic approach. Pharm. Res. 1996, 13, 398–403. [Google Scholar] [CrossRef]
- Kageyama, T.; Nakamura, M.; Matsuo, A.; Yamasaki, Y.; Takakura, Y.; Hashida, M.; Kanai, Y.; Naito, M.; Tsuruo, T.; Minato, N.; et al. The 4F2hc/LAT1 complex transports L-DOPA across the blood-brain barrier. Brain Res. 2000, 879, 115–121. [Google Scholar] [CrossRef]
- Gonzalez-Carter, D.A.; Ong, Z.Y.; McGilvery, C.M.; Dunlop, I.E.; Dexter, D.T.; Porter, A.E. L-DOPA functionalized, multi-branched gold nanoparticles as brain-targeted nano-vehicles. Nanomed. Nanotechnol. Biol. Med. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Wang, C.; Chu, C.; Ji, X.; Luo, G.; Xu, C.; He, H.; Yao, J.; Wu, J.; Hu, J.; Jin, Y. Biology of peptide transporter 2 in mammals: New insights into its function, structure and regulation. Cells 2022, 11, 2874. [Google Scholar] [CrossRef]
- Gardiner, R.M. Transport of amino acids across the blood-brain barrier: Implications for treatment of maternal phenylketonuria. J. Inherit. Metab. Dis. 1990, 13, 627–633. [Google Scholar] [CrossRef]
- Vyas, A.; Jain, A.; Hurkat, P.; Jain, A.; Jain, S.K. Targeting of AIDS related encephalopathy using phenylalanine anchored lipidic nanocarrier. Colloids Surf. B Biointerfaces 2015, 131, 155–161. [Google Scholar] [CrossRef]
- Alonso, M.; Barcia, E.; Córdoba-Díaz, M.; Negro, S.; Córdoba-Díaz, D.; Fernández-Carballido, A. Development and validation of an HPLC method for the quantification of morin flavonoid encapsulated within PLGA nanoparticles. Curr. Pharm. Anal. 2021, 17, 1178–1187. [Google Scholar] [CrossRef]
- Marcianes, P.; Negro, S.; Garcia-Garcia, L.; Montejo, C.; Barcia, E.; Fernandez-Carballido, A. Surface-modified gatifloxacin nanoparticles with potential for treating central nervous system tuberculosis. Int. J. Nanomed. 2017, 12, 1959–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Pilch, D.S.; Srinivasan, A.R.; Olson, W.K.; Geacintov, N.E.; Breslauer, K.J. Modulation of nucleic acid structure by ligand binding: Induction of a DNA· RNA· DNA hybrid triplex by DAPI intercalation. Bioorg. Med. Chem 1997, 5, 1137–1147. [Google Scholar] [CrossRef]
- Vera, M.; Barcia, E.; Negro, S.; Marcianes, P.; García-García, L.; Slowing, K.; Fernández-Carballido, A. New celecoxib multiparticulate systems to improve glioblastoma treatment. Int. J. Pharm. 2014, 473, 518–527. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ. National Institutes of Health, Bethesda, MD, USA. 1997–2018. Available online: https://imagej.nih.gov/ij/ (accessed on 27 September 2022).
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Egleton, R.D.; Davis, T.P. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx 2005, 2, 44–53. [Google Scholar] [CrossRef]
- Abascal, N.C.; Regan, L. The past, present and future of protein-based materials. Open Biol. 2018, 8, 180113. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Banskota, S.; Roberts, S.; Kirmani, N.; Chilkoti, A. Engineering the architecture of elastin-like polypeptides: From Unimers to hierarchical self-assembly. Adv. Ther. 2020, 3, 1900164. [Google Scholar] [CrossRef]
- Harper, J.D.; Lansbury, P.T. Models of amyloid seeding in Alzheimers’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C.C.F. From de globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q Rev. Biophys. 1998, 31, 137–148. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valetti, S.; Mura, S.; Noiray, M.; Arpicco, S.; Dosio, F.; Vergnaud, J.; Desmaële, D.; Stella, B.; Couvreur, P. Peptide conjugation: Before or after nanoparticle formation? Bioconjug. Chem. 2014, 25, 1971–1983. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current strategies for brain drug delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Kasinathan, N.; Jagani, H.V.; Alex, A.T.; Volety, S.M.; Venkata Rao, J. Strategies for drug delivery to the central nervous system by systemic route. Drug Deliv. 2015, 22, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.; Stolnik, S.; Illum, L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009, 379, 146–157. [Google Scholar] [CrossRef]
- Sadeghi, R.; Etemad, S.G.; Keshavarzi, E.; Haghshenasfard, M. Investigation of alumina nanofluid stability by UV-vis spectrum. Microfluid. Nanofluid. 2015, 18, 1023–1030. [Google Scholar] [CrossRef]
- Joseph, E.; Singhvi, G. Multifunctional nanocrystals for cancer therapy: A potential nanocarrier. In Nanomaterials for Drug Delivery and Therapy, 1st ed.; Grumezescu, A.M., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 91–116. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Torres-Suárez, A.I.; Cohen, M.; Delie, F.; Bastida-Ruiz, D.; Yart, L.; Martin-Sabroso, C.; Fernández-Carballido, A. PLGA nanoparticles for the intraperitoneal administration of CBD in the treatment of ovarian cancer: In vitro and in ovo assessment. Pharmaceutics 2020, 12, 439. [Google Scholar] [CrossRef]
- Hoyos-Ceballos, G.P.; Ruozi, B.; Ottonelli, I.; da Ros, F.; Vandelli, M.A.; Forni, F.; Daini, E.; Vlella, A.; Zoli, M.; Tosi, G.; et al. PLGA-PEG-ANG-2 Nanoparticles for blood–brain barrier crossing: Proof-of-concept study. Pharmaceutics 2020, 12, 72. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Patwari, J.; Dasgupta, S. Complexation with human serum albumin facilitates sustained release of morin from polylactic-co-glycolic acid nanoparticles. J. Phys. Chem. B 2017, 121, 1758–1770. [Google Scholar] [CrossRef]
- Mohammad, A.K.; Reineke, J.J. Quantitative detection of PLGA nanoparticle degradation in tissues following intravenous administration. Mol. Pharm. 2013, 10, 2183–2189. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Tsai, T.H. Pharmacokinetics and biodistribution of the illegal food colorant rhodamine B in rats. Agric. Food Chem. 2017, 65, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | PLGA (mg) | PLGA-Phe-Phe (mg) | MH (mg) | Rh-B (mg) |
|---|---|---|---|---|
| NP-0 | 50 | - | - | - |
| NP-MH | 50 | - | 5 | - |
| NP-Rh | 50 | - | - | 2.5 |
| NPphe-0 | 30 | 20 | - | - |
| NPphe-MH | 30 | 20 | 5 | - |
| NPphe-Rh | 30 | 20 | - | 2.5 |
| Formulation | Particle Size ± SD (nm) | PDI | Zeta Potential ± SD (mV) | DL (µg/100 mg NPs) ± SD | EE% ± SD |
|---|---|---|---|---|---|
| NP-0 | 176.4 ± 13.4 | 0.2 | −25.4 ± 1.4 | - | |
| NP-MH | 188.6 ± 16.7 | 0.3 | −20.8 ± 0.5 | 7503.6 ± 88.1 | 82.5 ± 1.4 |
| NPphe-0 | 192.8 ± 1.4 | 0.2 | −18.3 ± 2.1 | - | |
| NPphe-MH | 189.1 ± 8.2 | 0.3 | −23.3 ± 0.7 | 9023.8 ± 203.4 | 96.5 ± 3.2 |
| NP-Rh | 187.7 ± 6.9 | 0.2 | −22.6 ± 0.4 | 362.8 ± 17.3 | 7.6 ± 0.4 |
| NPphe-Rh | 191.0 ± 6.5 | 0.25 | −7.88 ± 0.2 | 715.3 ± 8.6 | 15 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, M.; Barcia, E.; González, J.-F.; Montejo, C.; García-García, L.; Villa-Hermosilla, M.-C.; Negro, S.; Fraguas-Sánchez, A.-I.; Fernández-Carballido, A. Functionalization of Morin-Loaded PLGA Nanoparticles with Phenylalanine Dipeptide Targeting the Brain. Pharmaceutics 2022, 14, 2348. https://doi.org/10.3390/pharmaceutics14112348
Alonso M, Barcia E, González J-F, Montejo C, García-García L, Villa-Hermosilla M-C, Negro S, Fraguas-Sánchez A-I, Fernández-Carballido A. Functionalization of Morin-Loaded PLGA Nanoparticles with Phenylalanine Dipeptide Targeting the Brain. Pharmaceutics. 2022; 14(11):2348. https://doi.org/10.3390/pharmaceutics14112348
Chicago/Turabian StyleAlonso, Mario, Emilia Barcia, Juan-Francisco González, Consuelo Montejo, Luis García-García, Mónica-Carolina Villa-Hermosilla, Sofía Negro, Ana-Isabel Fraguas-Sánchez, and Ana Fernández-Carballido. 2022. "Functionalization of Morin-Loaded PLGA Nanoparticles with Phenylalanine Dipeptide Targeting the Brain" Pharmaceutics 14, no. 11: 2348. https://doi.org/10.3390/pharmaceutics14112348