

BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy

 ,
,  ,
, 
Abstract
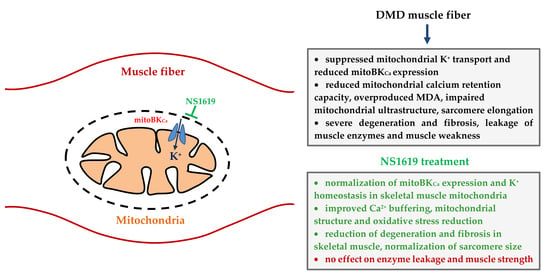
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Mitochondria Isolation and Determination of Respiration and Oxidative Phosphorylation
2.3. Determination of Mitochondrial K+ Transport and Total Content
2.4. Determination of Mitochondrial Ca2+ Retention Capacity, MPT Pore Opening Assay
2.5. Assessment of Lipid Peroxidation
2.6. RNA Isolation, Reverse Transcription and Quantitative Real-Time PCR
2.7. Quantification of Mitochondrial DNA
2.8. Electron Microscopy
2.9. Histological Examination of Skeletal Muscle Tissues
2.10. Wire-Hanging Test
2.11. Statistical Analysis
3. Results
3.1. NS1619 Suppresses Respiration and OXPHOS in Skeletal Muscle Mitochondria of Dystrophin-Deficient and WT Mice
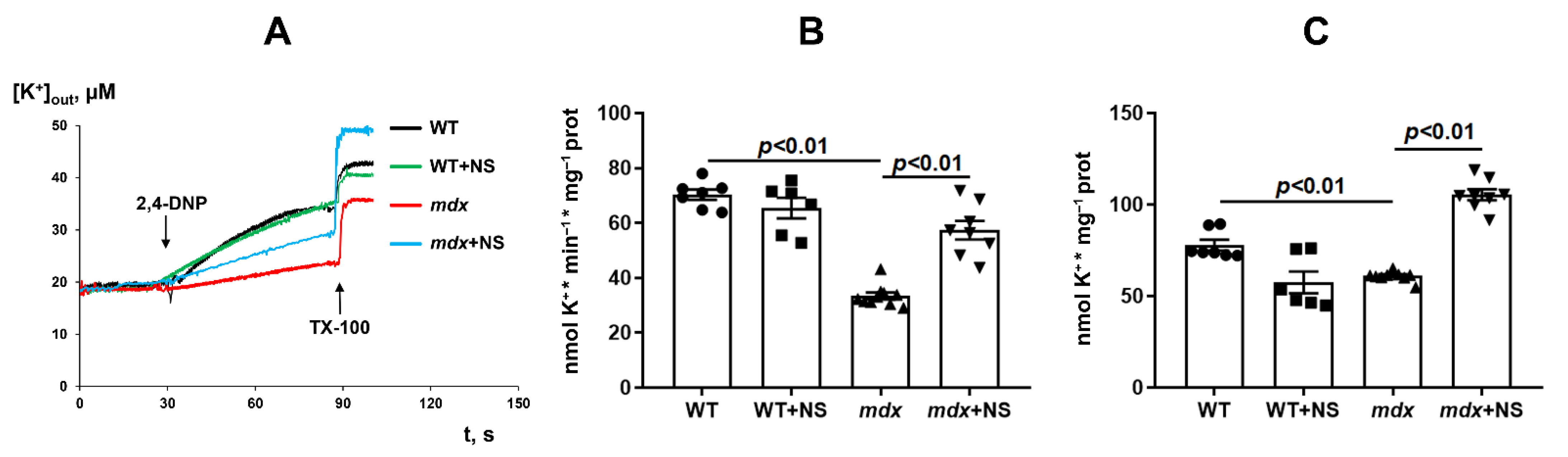
3.2. NS1619 Normalizes the Rate of Potassium Ion Transport and Total Ion Level in Skeletal Muscle Mitochondria of mdx Mice
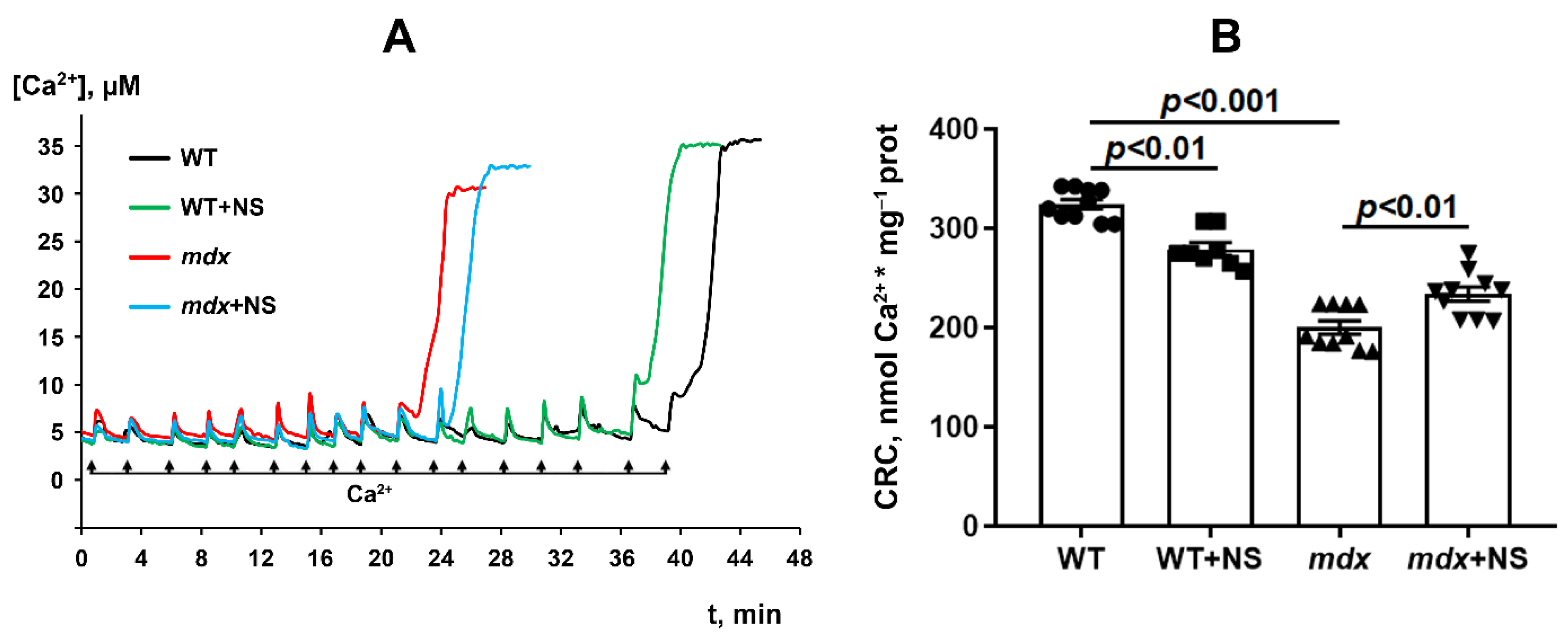
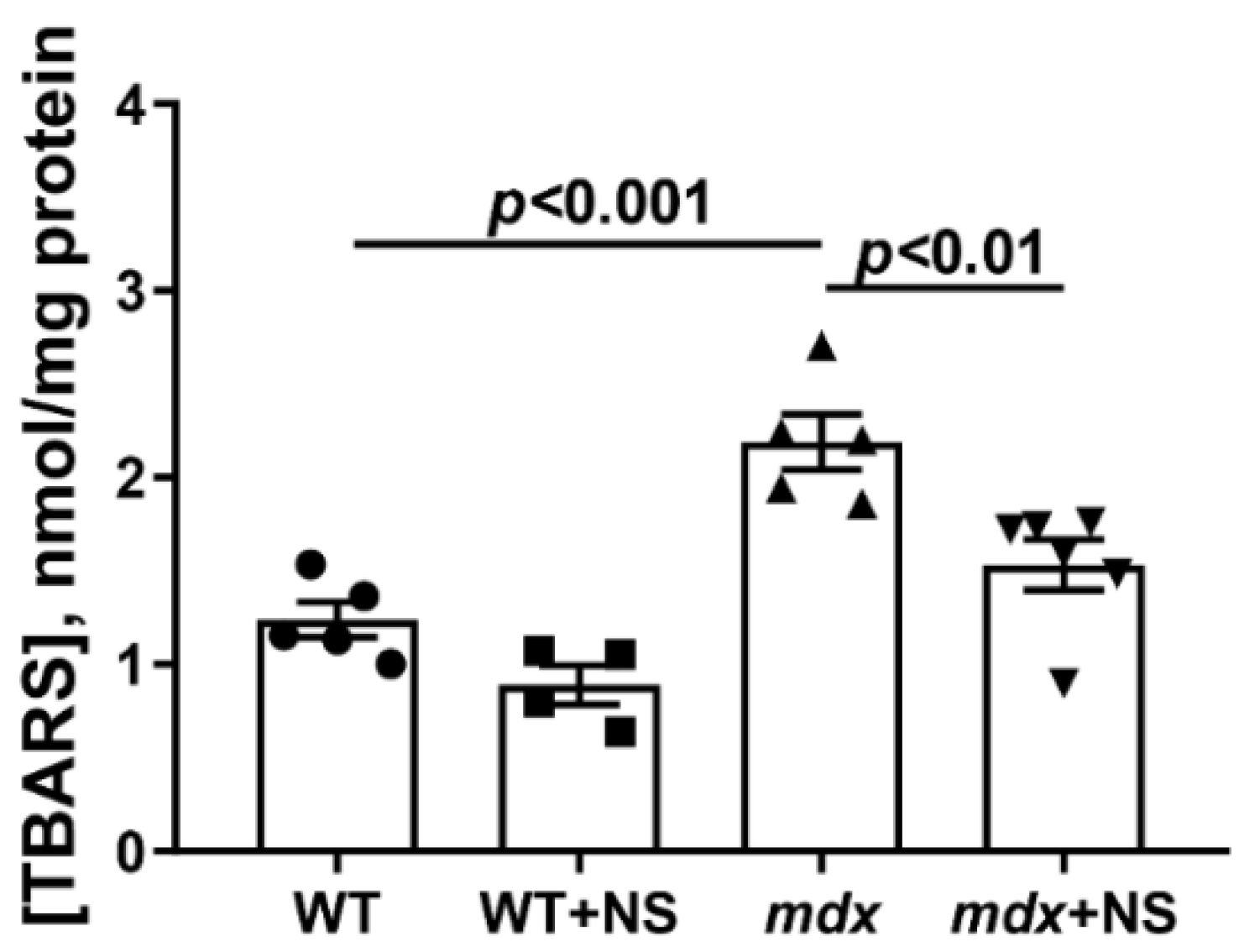
3.3. NS1619 Increases Mitochondrial Calcium Retention Capacity and Attenuates Oxidative Stress in Skeletal Muscle Mitochondria of mdx Mice
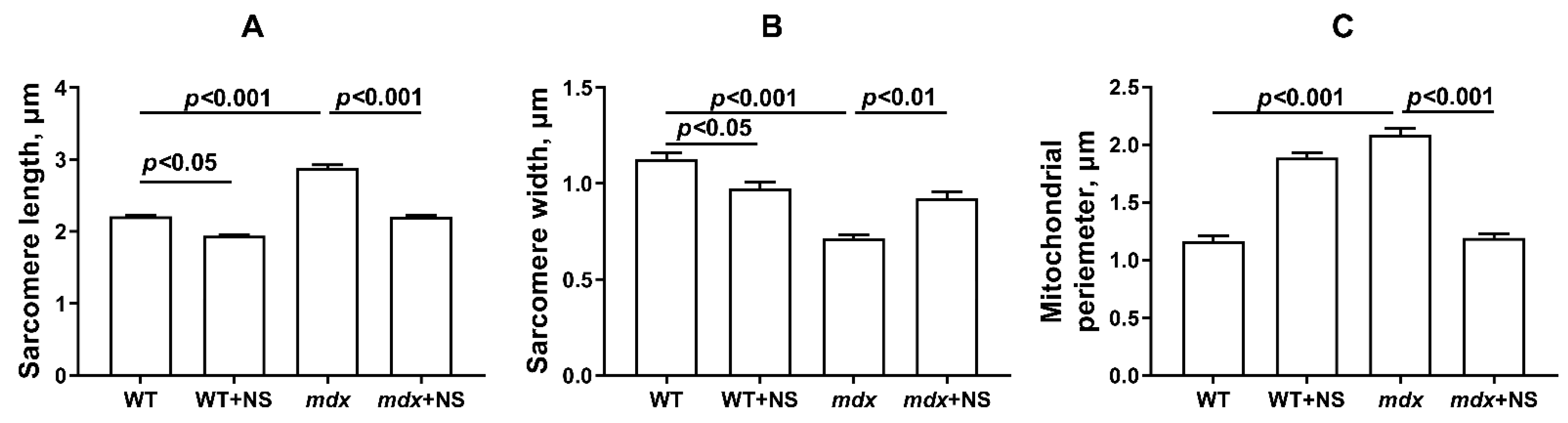
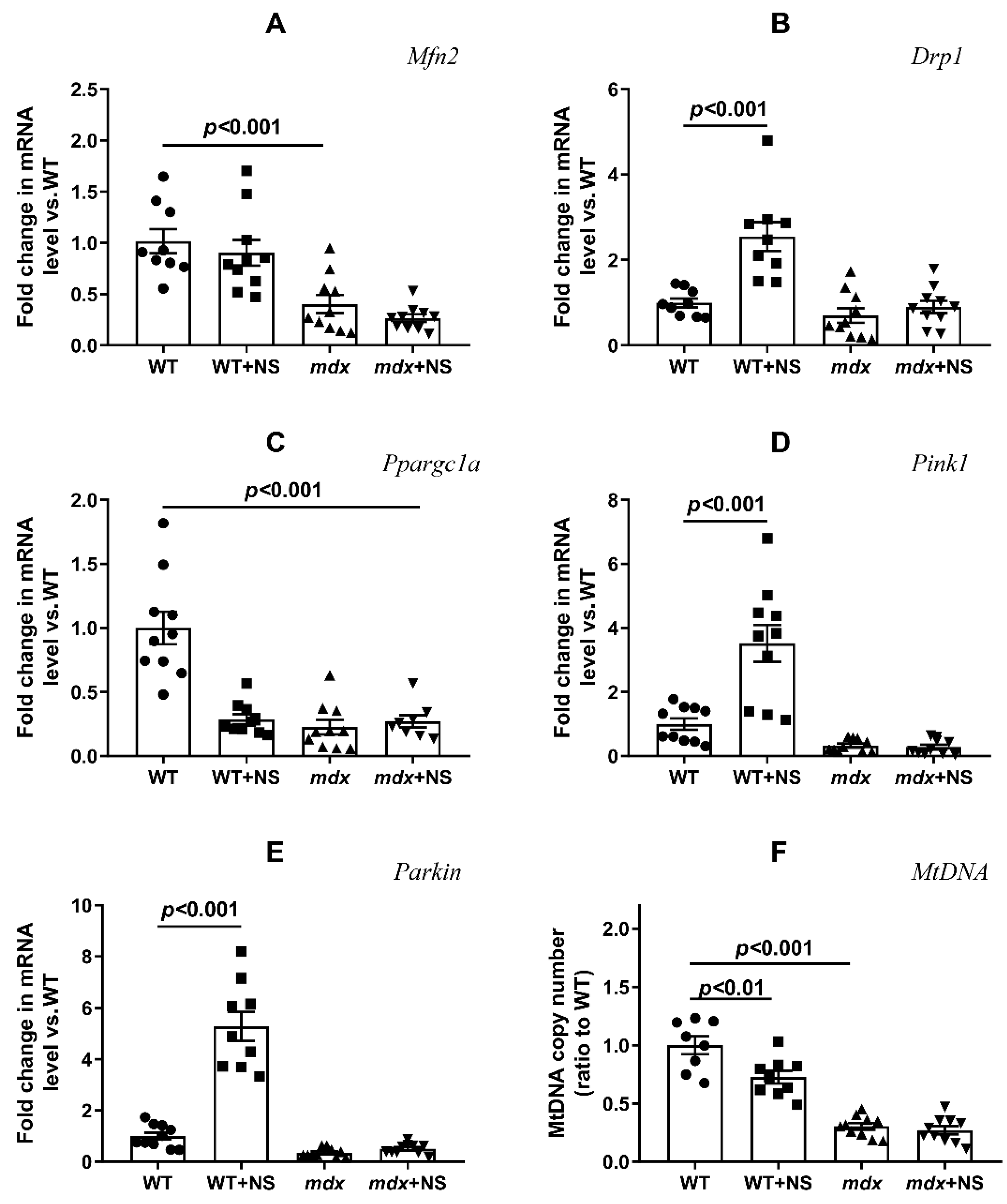
3.4. NS1619 Normalizes the Ultrastructure of Skeletal Muscle Mitochondria and Sarcomere Size in mdx Mice but Does Not Affect the Expression of Protein Genes Responsible for Mitochondrial Biogenesis, Dynamics, and Mitophagy
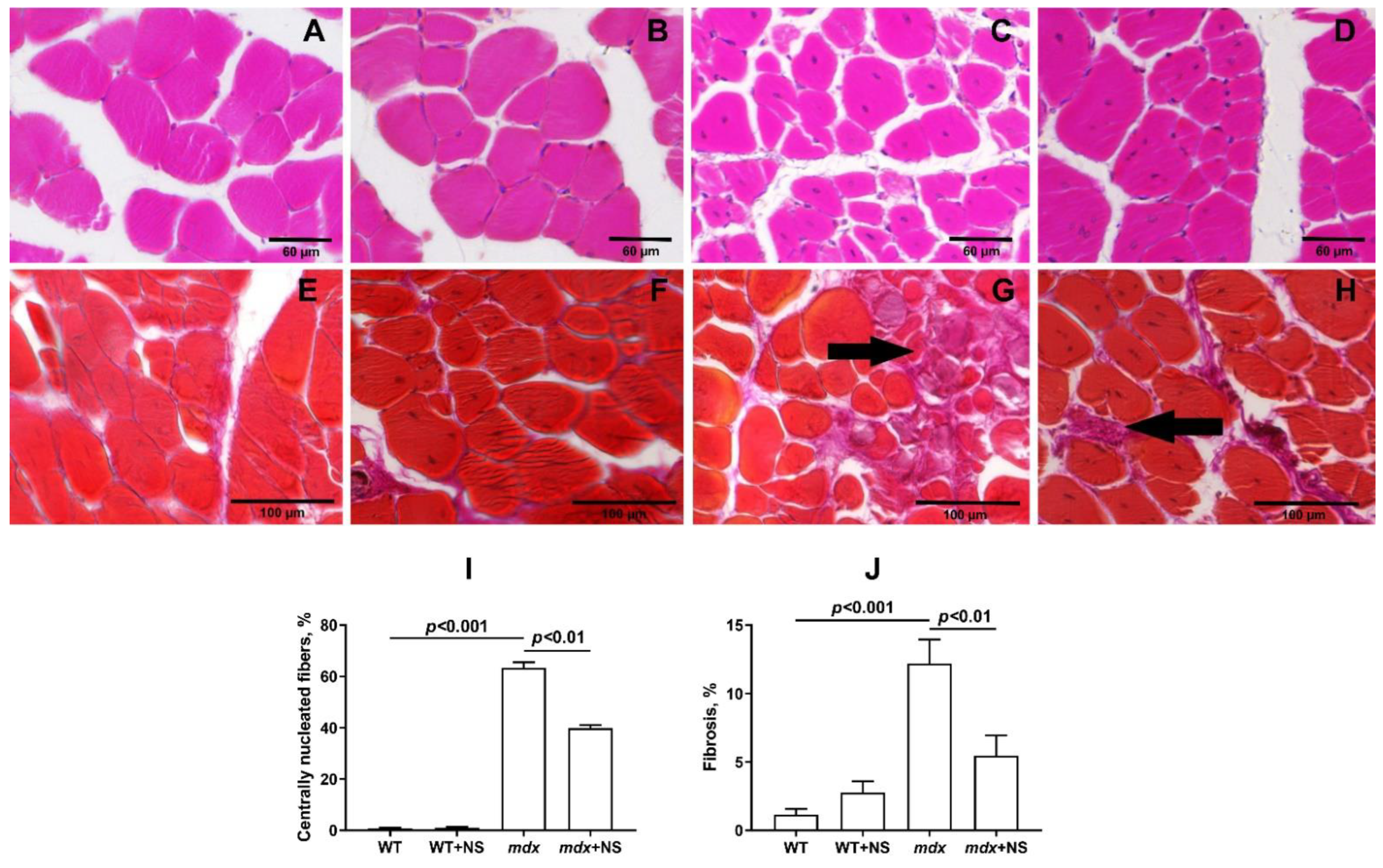
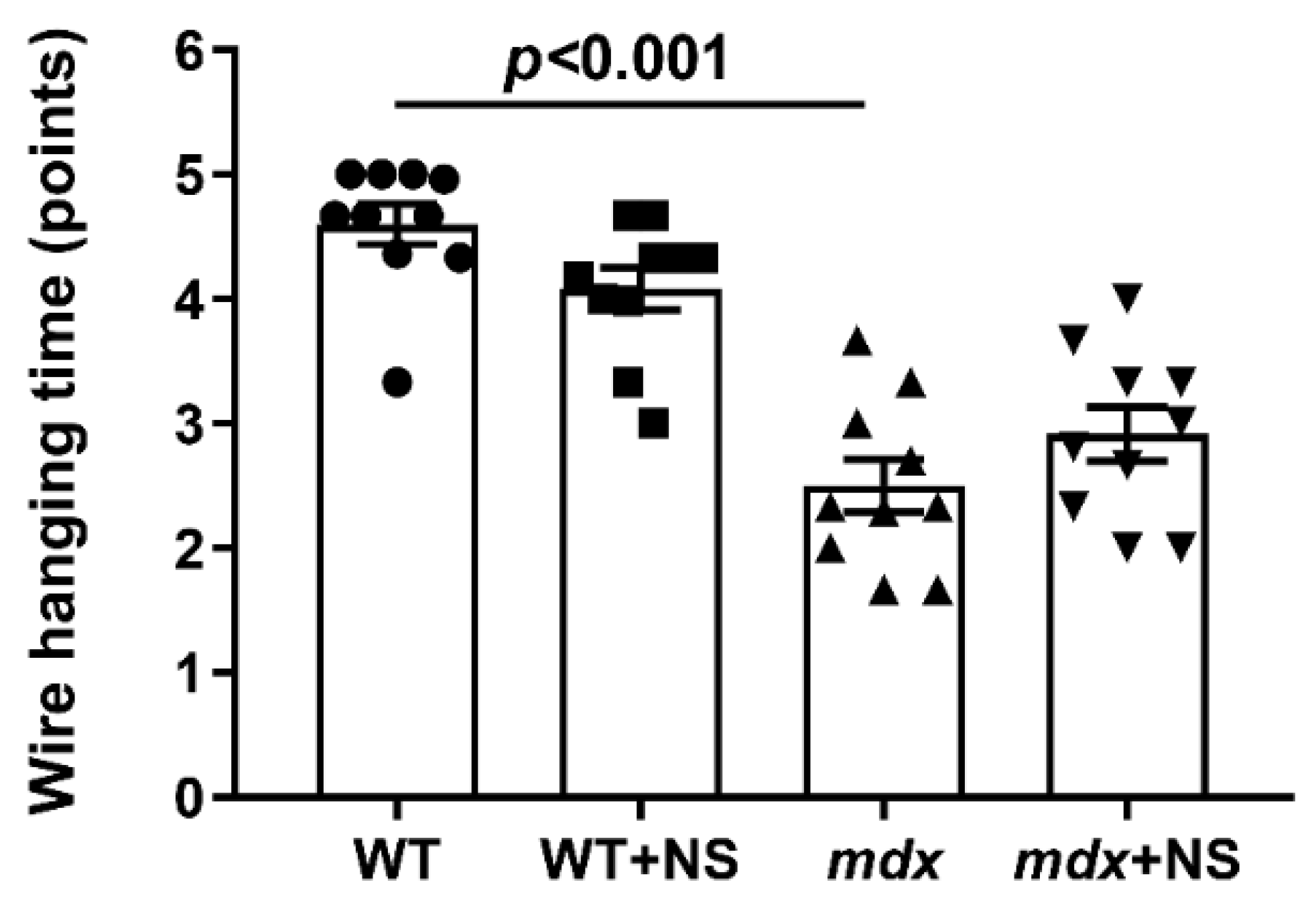
3.5. NS1619 Attenuates Skeletal Muscle Destruction in mdx Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, B.; Takeda, S.; Yokota, T. Nonmechanical roles of dystrophin and associated proteins in exercise, neuromuscular junctions, and brains. Brain Sci. 2015, 5, 275–298. [Google Scholar] [CrossRef] [Green Version]
- Leyva-Leyva, M.; Sandoval, A.; Felix, R.; González-Ramírez, R. Biochemical and functional interplay between ion channels and the components of the dystrophin-associated glycoprotein complex. J. Membr. Biol. 2018, 251, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, M. Duchenne muscular dystrophy. J. Am. Acad. Orthop. Surg. 2002, 10, 138–151. [Google Scholar] [CrossRef]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef]
- Willi, L.; Abramovich, I.; Fernandez-Garcia, J.; Agranovich, B.; Shulman, M.; Milman, H.; Baskin, P.; Eisen, B.; Michele, D.E.; Arad, M.; et al. Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2022, 23, 9808. [Google Scholar] [CrossRef]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; DE Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of systemic delivery of rAAVrh74.MHCK7 micro-dystrophin in children with Duchenne muscular dystrophy. JAMA Neurol. 2020, 77, 1122. [Google Scholar] [CrossRef]
- HappiMbakam, C.; Lamothe, G.; Tremblay, G.; Tremblay, J.P. CRISPR-Cas9 gene therapy for Duchenne muscular dystrophy. Neurotherapeutics 2022, 19, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Mura, G.; Messina, G. Therapeutic approaches to preserve the musculature in Duchenne muscular dystrophy: The importance of the secondary therapies. Exp. Cell Res. 2022, 410, 112968. [Google Scholar] [CrossRef] [PubMed]
- Vandebrouck, A.; Ducret, T.; Basset, O.; Sebille, S.; Raymond, G.; Ruegg, U.; Gailly, P.; Cognard, C.; Constantin, B. Regulation of store operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. Faseb J. 2006, 20, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, R.; Mareedu, S.; Babu, G.J. Reducing sarcolipin expression improves muscle metabolism in mdx mice. Am. J. Physiol. Cell Physiol. 2022, 322, 260–274. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophindeficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef] [PubMed]
- De Mario, A.; Gherardi, G.; Rizzuto, R.; Mammucari, C. Skeletal muscle mitochondria in health and disease. Cell Calcium 2021, 94, 102357. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef]
- Servais, L.; Straathof, C.S.M.; Schara, U.; Klein, A.; Leinonen, M.; Hasham, S.; Meier, T.; De Waele, L.; Gordish-Dressman, H.; McDonald, C.M.; et al. Long-term data with idebenone on respiratoryfunction outcomes in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef] [PubMed]
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Farid, T.A.; Nair, K.; Massé, S.; Azam, M.A.; Maguy, A.; Lai, R.F.; Umapathy, K.; Dorian, P.; Chauhan, V.; Varró, A.; et al. Role of KATP channels in the maintenance of ventricular fibrillation in cardiomyopathic human hearts. Circ. Res. 2011, 109, 1309–1318. [Google Scholar] [CrossRef] [Green Version]
- Graciotti, L.; Becker, J.; Granata, A.L.; Procopio, A.D.; Tessarollo, L.; Fulgenzi, G. Dystrophin is required for the normal function of the cardio-protective K(ATP) channel in cardiomyocytes. PLoS ONE 2011, 6, e27034. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Penkina, D.K.; Vedernikov, A.A.; Belosludtsev, K.N. The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy. Int. J. Mol. Sci. 2022, 23, 10660. [Google Scholar] [CrossRef]
- Checchetto, V.; Leanza, L.; De Stefani, D.; Rizzuto, R.; Gulbins, E.; Szabo, I. Mitochondrial K+ channels and their implications for disease mechanisms. Pharmacol. Ther. 2021, 227, 107874. [Google Scholar] [CrossRef]
- González-Sanabria, N.; Echeverría, F.; Segura, I.; Alvarado-Sánchez, R.; Latorre, R. BK in double-membrane organelles: A biophysical, pharmacological, and functional survey. Front. Physiol. 2021, 12, 761474. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Kosareva, E.A.; Talanov, E.Y.; Gudkov, S.V.; Dubinin, M.V. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie 2020, 176, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, P.C.; Kumar, M.A.; Resetar, A.; Harris, D.L. Mechanistic stoichiometry of mitochondrial oxidative phosphorylation. Biochemistry 1991, 30, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA ratio in mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, 75, e2609. [Google Scholar] [CrossRef]
- Baranova, O.V.; Skarga, Y.Y.; Negoda, A.E.; Mironova, G.D. Inhibition of 2,4-dinitrophenol-induced potassium efflux by adenine nucleotides in mitochondria. Biochemistry 2000, 65, 218–222. [Google Scholar]
- Singh, H.; Rong, L.; Bopassa, J.; Meredith, A.; Stefani, E.; Toro, L. MitoBK-Ca is encoded by the KCNMA1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. USA 2013, 110, 10836–10841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gałecka, S.; Kulawiak, B.; Bednarczyk, P.; Singh, H.; Szewczyk, A. Single channel properties of mitochondrial large conductance potassium channel formed by BK-VEDEC splice variant. Sci. Rep. 2021, 11, 10925. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [Green Version]
- Reggiani, C.; Marcucci, L. A controversial issue: Can mitochondria modulate cytosolic calcium and contraction of skeletal muscle fibers? J. Gen. Physiol. 2022, 154, e202213167. [Google Scholar] [CrossRef]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; Van Eyk, J.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef]
- Wang, X.; Yin, C.; Xi, L.; Kukreja, R.C. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2070H2077. [Google Scholar] [CrossRef]
- Dai, H.; Wang, M.; Patel, P.N.; Kalogeris, T.; Liu, Y.; Durante, W.; Korthuis, R.J. Preconditioning with the BKCa channel activator NS-1619 prevents ischemia-reperfusion-induced inflammation and mucosal barrier dysfunction: Roles for ROS and heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H988–H999. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Carvalho-De-Souza, J.; Wei, C.; Carrasquel-Ursulaez, W.; Lorenzo, Y.; Gonzalez, N.; Kubota, T.; Staisch, J.; Hain, T.; Petrossian, N.; et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc. Natl. Acad. Sci. USA 2020, 117, 6023–6034. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.; Domoki, F.; Lenti, L.; Katakam, P.; Snipes, J.; Bari, F.; Busija, D.W. Immediate neuronal preconditioning by NS1619. Brain Res. 2009, 1285, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Checchetto, V.; Biasutto, L.; Rossa, A.; Costa, R.; Bachmann, M.; Zoratti, M.; Szabo, I. Pharmacological modulation of mitochondrial ion channels. Br. J. Pharmacol. 2019, 176, 4258–4283. [Google Scholar] [CrossRef] [PubMed]
- Debska, G.; Kicinska, A.; Dobrucki, J.; Dworakowska, B.; Nurowska, E.; Skalska, J.; Dolowy, K.; Szewczyk, A. Large-conductance K+ channel openers NS1619 and NS004 as inhibitors of mitochondrial function in glioma cells. Biochem. Pharmacol. 2003, 65, 1827–1834. [Google Scholar] [CrossRef]
- Kicinska, A.; Szewczyk, A. Large-conductance potassium cation channel opener NS1619 inhibits cardiac mitochondrial respiratory chain. Toxicol. Mech. Methods 2004, 14, 59–61. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. In Cochrane Database of Systematic Reviews; John Wiley & Sons: Hoboken, NJ, USA, 2016; CD003725. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| BKCa-DEC | GGTTTACAGATGAGCCGGATA | CATCTTCAACTTCTCTGATTGG |
| Drp1 | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Pink1 | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Parkin | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
| Nd4 | ATTATTATTACCCGATGAGGGAACC | ATTAAGATGAGGGCAATTAGCAGT |
| Gapdh | GTGAGGGAGATGCYCAGTGT | CTGGCATTGCTCTCAATGAC |
| Group | V Respiration, nmol O2 * min−1 * mg−1 Protein | RCR | ADP/O | |||
|---|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | |||
| WT | 23.3 ± 1.7 | 199.9 ± 7.0 | 44.2 ± 3.3 | 217.7 ± 8.4 | 4.7 ± 0.2 | 1.2 ± 0.1 |
| WT + NS | 18.4 ± 1.2 * | 116.4 ± 8.6 * | 34.1 ± 0.9 | 141.7 ± 8.0 * | 3.4 ± 0.3 * | 1.2 ± 0.1 |
| mdx | 20.0 ± 1.3 | 168.2 ± 7.6 * | 55.5 ± 7.9 | 189.4 ± 6.8 * | 3.8 ± 0.5 | 1.2 ± 0.1 |
| mdx + NS | 21.4 ± 0.8 | 147.0 ± 5.4 * | 34.1 ± 1.9 # | 171.6 ± 5.0 * | 4.5 ± 0.3 | 1.4 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Igoshkina, A.D.; Vafina, A.B.; Vedernikov, A.A.; Belosludtsev, K.N. BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy. Pharmaceutics 2022, 14, 2336. https://doi.org/10.3390/pharmaceutics14112336
Dubinin MV, Starinets VS, Belosludtseva NV, Mikheeva IB, Chelyadnikova YA, Igoshkina AD, Vafina AB, Vedernikov AA, Belosludtsev KN. BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy. Pharmaceutics. 2022; 14(11):2336. https://doi.org/10.3390/pharmaceutics14112336
Chicago/Turabian StyleDubinin, Mikhail V., Vlada S. Starinets, Natalia V. Belosludtseva, Irina B. Mikheeva, Yuliya A. Chelyadnikova, Anastasia D. Igoshkina, Aliya B. Vafina, Alexander A. Vedernikov, and Konstantin N. Belosludtsev. 2022. "BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy" Pharmaceutics 14, no. 11: 2336. https://doi.org/10.3390/pharmaceutics14112336