3.2. Second-Generation Antipsychotics
First-generation antipsychotics induce a variety of adverse events, including some severe neurological side effects, in both acute and long-term exposure. Moreover, these drugs are almost ineffective in the treatment of negative and cognitive symptoms. This led to a search for new drugs with higher efficacy and/or safety. Therefore, the second-generation, or “atypical”, antipsychotics were developed, being a series of molecules devoid of neurological side effects, with a generally improved safety profile, and with overall superior efficacy and (some) effectiveness against negative and/or cognitive symptoms [
3,
4].
Risperidone is one of the oldest second-generation antipsychotic drugs, having been approved for the treatment of schizophrenia in 1993, and acting as an inhibitor of dopaminergic D2 and serotonergic 5-HT2A receptors [
19,
43,
44]. Nevertheless, its oral administration still leads to numerous side effects, such as dystonia, tremor, sedation, dizziness, and several gastrointestinal and upper respiratory tract symptoms, also displaying a slow onset of therapeutic action, which is especially relevant to counteract acute psychosis or emergency situations [
20,
21,
22,
44]. Moreover, risperidone has poor water solubility (predicted to be 0.171 mg/mL), and substantial first-pass metabolism and P-glycoprotein efflux [
19,
20,
43]. To tackle these issues, and specifically for emergency situations and when medicine swallowing is not an option for administration, Dordevic et al. [
19,
20] developed O/W nanoemulsions for parenteral delivery. The oil mixture was made of medium chain triglycerides and soybean oil (Lipoid Purified Soybean Oil 700), and the formulation also contained soybean lecithin (Lipoid S 75), benzyl alcohol, butylhydroxytoluene, glycerol, sodium oleate and water (pH 9). Additionally, the formulations also comprised a hydrophilic surfactant, that was either Tween
® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), Kolliphor
® P 188 (poloxamer 188) or Kolliphor
® HS 15 (macrogol 15 hydroxystearate) (detailed quantities in
Table 3). The nanoemulsions were prepared by high pressure homogenization, and drug strength was set at 1 mg/g, which is almost 6 times higher than risperidone’s aqueous solubility. Measured droplet size was 160 nm, PDI between 0.10–0.13, zeta potential –50 mV, pH between 8.2–8.4, and viscosity between 6–11 cP (being considered sufficiently low for parenteral administration purposes), for all formulations. All preparations were stable after a 4-month storage period. The in vivo pharmacokinetic evaluation (rats; 1 mg/kg) indicated that the Tween
® 80-based nanoemulsion showed the best brain targeting profile, since it led to the highest brain C
max and AUC values when compared to the other nanoemulsions and a drug solution. Since all nanoemulsions had similar characterization parameters (droplet size, PDI, zeta potential, viscosity) and composition aside from the hydrophilic surfactant type, Tween
® 80 was indicated as the most suitable hydrophilic emulsifier for brain targeting purposes (between the ones that were studied), since it originated the highest brain drug levels. Therefore, in vivo pharmacodynamics (rats, locomotor activity assessment) was carried out using this formulation, which indeed had a more sustained antipsychotic effect, with less sedation (side-effect), when compared to a drug solution, further confirming its potential for risperidone brain delivery.
Kumar et al. [
21,
22] also decided on preparing O/W risperidone nanoemulsions, but in this case for intranasal administration. Formulations were prepared by spontaneous emulsification, with their composition including Capmul
® MCM (medium chain mono- and diglycerides), Tween
® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), Transcutol
® P (diethylene glycol monoethyl ether), propylene glycol and water (specific quantities in
Table 3). The mucoadhesive nanoemulsion also had chitosan. Drug strength was 3.5 mg/mL, which is more than 20 times higher than risperidone’s water solubility. Formulation characterization parameters were 14–18 nm for droplet size, 0.12–0.17 for PDI, −9 to −12 mV for zeta potential, 225–250 cP for viscosity, and 4.5–5.3 for pH. Osmolarity was approximately 270 mOsmol/L, which is within the established limits for intranasal preparations. Both formulations were stable after a 3-month storage and under accelerated conditions. In ex vivo drug permeation studies (sheep nasal mucosa), a risperidone solution (made of ethanol, propylene glycol and water) had the highest permeation coefficient, probably due to low viscosity and having cosolvents in its composition, followed by the mucoadhesive nanoemulsion, which performed better than the non-mucoadhesive nanoemulsion, probably due to the permeation enhancing properties of chitosan. In vivo pharmacokinetic studies (rats; 0.09 mg/kg) showed that the intranasal mucoadhesive nanoemulsion had the best brain drug targeting, since it had the highest brain C
max and AUC values, and also the highest brain/blood ratios at all time points, when compared to the other intranasal and intravenous formulations. Hence, chitosan not only had a role in enhancing drug permeation, but also in decreasing mucociliary clearance, with a clear consequence on brain bioavailability. To further confirm the potential of this formulation, in vivo pharmacodynamics (mice; 0.325 mg/kg; locomotor activity) indicated that both intranasal nanoemulsions performed better than the intravenous group, and between them the mucoadhesive nanoemulsion was superior, demonstrating higher brain drug uptake. Moreover, the formulation’s components did no damage to the nasal mucosa, as indicated by the nasal ciliotoxicity studies (sheep nasal mucosa).
Olanzapine is an atypical antipsychotic drug which was approved for clinical use by the American Food and Drug Administration (FDA) in 1996, acting as an antagonist of dopamine (mainly D2) and serotonin (mainly 5-HT2) receptors [
23,
45]. Its oral administration in tablet form leads to an extensive first-pass metabolism, with a slow onset of action [
23,
24]. Moreover, both oral and intramuscular marketed forms have been linked to adverse events such as somnolence, mydriasis, blurred vision and respiratory depression [
24,
45]. Since olanzapine is also a low water solubility drug (predicted to be 0.0942 mg/mL) [
45], and in order to tackle some of its other issues, Kumar et al. [
23] decided to formulate it into O/W nanoemulsions, for intranasal administration, containing Capmul
® MCM (medium chain mono- and diglycerides), Tween
® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), ethanol, polyethylene glycol 400 and distilled water (detailed quantities in
Table 4). Additionally, the mucoadhesive nanoemulsion also had chitosan. Formulations were prepared by spontaneous emulsification, and drug strength was kept at 8.5 mg/mL, more than 90 times higher than olanzapine’s water solubility. Droplet size varied between 20–24 nm, PDI between 0.25–0.30, zeta potential between −4 and −11 mV, viscosity between 0.10–0.13 cP, and pH between 4.5–5.7. The results of the in vivo pharmacokinetic study (rats; 0.225 mg/kg) showed that all intranasally administered formulations led to a lower brain T
max when compared to the intravenous control (non-mucoadhesive nanoemulsion), which suggested preferential nose-to-brain transport. Moreover, both intranasal nanoemulsions led to significantly higher brain drug levels and brain/blood ratios at all time points, when compared to the intravenous group and the intranasal solution (made of ethanol and propylene glycol). Furthermore, the mucoadhesive nanoemulsion led to higher brain C
max and AUC than the non-mucoadhesive nanoemulsion, which in turn had a better performance than the intranasal solution. This indicated that both the nanoemulsion’s general composition, specifically the excipients with permeation enhancing capability, and the mucoadhesive agent led to an increase in brain drug uptake. Nevertheless, as before, the intravenous control should have been a drug solution (for example, such as the one that was administered intranasally), since the pharmacokinetic profile of a mucoadhesive nanoemulsion given intravenously could be quite altered. The in vivo pharmacodynamic assessment (locomotor activity in mice, 0.325 mg/kg; and paw test in rats, 0.225 mg/kg) further confirmed the potential of the developed formulations, with the intranasal route showing superiority when compared to intravenous administration, and with the mucoadhesive nanoemulsion also having a better performance within the intranasally administered formulations.
Patel et al. [
24] also chose to develop olanzapine O/W nanometric emulsions for intranasal delivery, containing oleic acid, Labrasol
® (polyethylene glycol-8 caprylic/capric glycerides), Kolliphor
® RH40 (polyoxyl 40 hydrogenated castor oil), Transcutol
® P (diethylene glycol monoethyl ether) and water (specific quantities in
Table 4). The mucoadhesive microemulsion also had polycarbophil AA-1 in its composition. The formulations were prepared by spontaneous emulsification, and drug strength was 8 mg/mL, which is 85 times higher than olanzapine’s water solubility. Measured droplet sizes were 23.87 and 31.66 nm, PDI 0.121 and 0.251, zeta potential −35.14 and −42.15 mV, viscosity 75 and 93 cP, and pH 5.87 and 5.95, for the non-mucoadhesive and mucoadhesive microemulsions, respectively. The developed preparations were deemed stable after storage for 6 months and under accelerated conditions. The in vivo pharmacokinetic evaluation (rats; 0.16–0.21 mg/kg) showed that all intranasally administered formulations (microemulsions and propylene glycol drug solution) had higher brain/blood ratios when compared to the intravenous group (non-mucoadhesive microemulsion), which suggests a more favorable efficacy vs. safety profile for this administration route. Moreover, brain drug concentrations obtained with the intranasal microemulsions were significantly higher at all time points when compared with the intravenous group, with the intranasal microemulsions also having significantly higher brain C
max and AUC values, and lower T
max, which indicates a faster and more effective brain drug delivery. Furthermore, the addition of the mucoadhesive polymer to the microemulsion led to higher brain drug uptake. Yet again, the intravenously administered formulation should have been a drug solution, and not a microemulsion, due to potentially altered pharmacokinetic profile and, in this specific case, a viscosity that is probably too high to be safely administered intravenously (without the risk of causing vascular adverse events). Nonetheless, the in vivo pharmacodynamics (mice, 3.20 mg/kg, apomorphine-induced compulsive behavior and spontaneous motor activity) confirmed an overall superiority of the intranasal route, and especially of the mucoadhesive microemulsion, in reversing induced schizophrenia-like symptoms, with the nanometric size, the presence of drug permeation enhancing excipients, and nasociliary clearance prevention by the mucoadhesive polymer being clearly important.
Quetiapine is another second-generation antipsychotic drug and was approved by the FDA in 1997 [
46]. Its action is thought to be mainly mediated by dopamine D2 and serotonin 5-HT2 receptors antagonism [
25,
46]. Despite being considered to have an acceptable safety profile, it has been reported to cause somnolence, orthostatic hypotension and anticholinergic effects, suicidal thinking or behavior in children under 10 years of age, and an increased death rate in elderly patients with dementia [
46]. Aside from side effects, this drug also has other issues, such as a short plasma half-life (hence requiring frequent administration), and low absorption and high metabolism after oral administration (leading to poor bioavailability) [
25,
26]. Moreover, it is a P-glycoprotein substrate, and is highly lipophilic (predicted water solubility 0.0403 mg/mL), also having a pH dependent solubility (higher solubility at lower pH) [
25,
26,
46]. In order to solve some of these issues, Boche et al. [
25] decided to develop an intranasal O/W nanoemulsion containing Capmul
® MCM (medium chain mono- and diglycerides), Tween
® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), Transcutol
® P (diethylene glycol monoethyl ether), propylene glycol and water, with quetiapine in fumarate salt form (higher water solubility) (specific quantities in
Table 5). Formulation preparation included a high sheer method (sonication), and measured droplet characteristics were 144 nm for droplet size, 0.193 for PDI, and −8.131 mV for zeta potential. The developed nanoemulsion was stable after a 6-month storage. The in vitro drug release assay (dialysis bag method) showed that the developed formulation had a more than 2-fold drug release increase when compared to a drug solution (water-ethanol). Moreover, the in vivo pharmacokinetic studies (rats; 1 mg/kg) indicated that the intranasal nanoemulsion had a better brain targeting, with higher brain C
max and AUC values, than the intranasal solution and both intravenous groups (solution and nanoemulsion). Moreover, brain T
max was lower for the intranasal groups, which means that intranasal delivery was faster in making the drug reach the brain. Additionally, brain/blood ratios were always equal to or higher than 1 for the intranasal nanoemulsion, which did not happen for any of the other groups, suggesting a higher efficacy vs. safety profile.
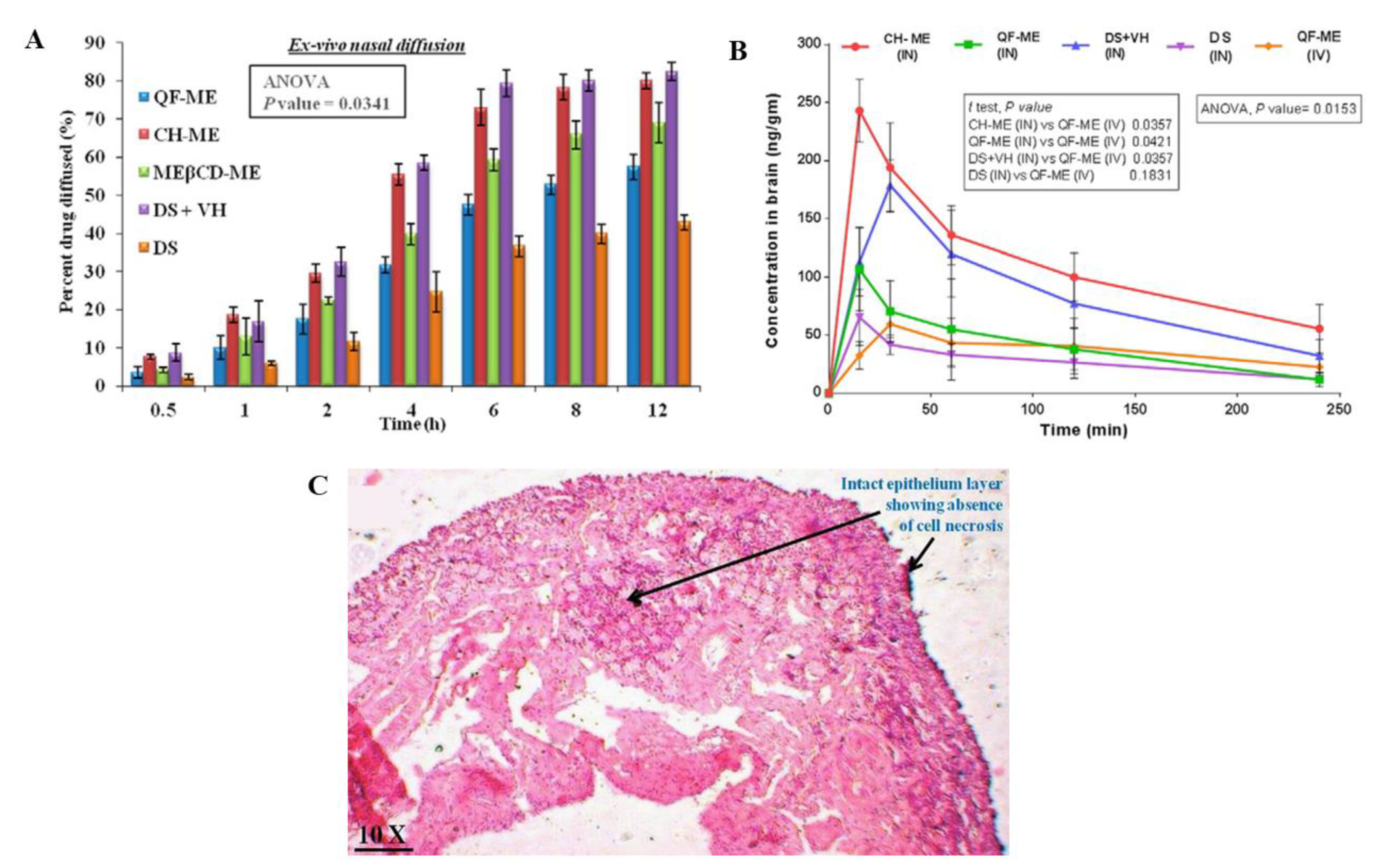
Shah et al. [
26] also developed quetiapine O/W nanometric emulsions, for intranasal administration, using Capmul
® MCM (medium chain mono- and diglycerides), Labrasol
® (polyethylene glycol-8 caprylic/capric glycerides), Tween
® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), Transcutol
® P (diethylene glycol monoethyl ether) and water (pH 5) (detailed amounts in
Table 5), but this time the preparation method was spontaneous emulsification. Drug strength was kept at 6 mg/mL, which is 149 times higher than quetiapine’s water solubility. Three formulas were developed, one being a non-mucoadhesive microemulsion, another being a mucoadhesive microemulsion (additionally having chitosan), and a third one having cyclodextrins (trimethyl-beta-cyclodextrins). Measured droplet sizes were between 29.75–46.55 nm, PDI between 0.221–0.249, zeta potential between 2.77–20.29 mV, viscosity between 17.5–38.5 cP, and pH between 5.5–6.5 (safe for the human nasal mucosa). All formulations were stable under accelerated conditions. The ex vivo mucoadhesive strength test (goat nasal mucosa) showed that both the mucoadhesive microemulsion and the microemulsion with cyclodextrins had an improved adhesion to the mucosal surface (although the mucoadhesive formulation had a more substantial improvement). This could be attributed to chitosan having an ionizable positively charged group, capable of interacting with the negatively charged groups of sialic acid on the mucosal surface, and cyclodextrins also interacting with the same residues, but by hydrogen bond formation. Ex vivo drug permeation (goat nasal mucosa,
Figure 3A) was highest for the mucoadhesive microemulsion, when compared to the other microemulsions, and all microemulsions had an increased drug permeation when compared to a drug solution. The superiority of mucoadhesive microemulsion performance could be attributed to the presence of chitosan and its reported ability to enhance paracellular transport by disruption of tight junctions. In vivo pharmacokinetics (rats, 2.3 mg/kg,
Figure 3B) further confirmed the superiority of the mucoadhesive microemulsion, since the intranasal administration of this formulation led to quetiapine brain concentrations that were significantly higher at all the time points when compared to all other groups (intranasal and intravenous), also having higher brain C
max and AUC values, and brain/blood ratios. Moreover, intranasal administration led to overall lower plasma drug levels when compared to the intravenous group which could indicate a higher safety profile (although, again the intravenously administered formulation should have been a drug solution instead of the non-mucoadhesive microemulsion). As for formulation safety (nasal ciliotoxicity in goat nasal mucosa,
Figure 3C), the mucoadhesive microemulsion showed no signs of cell necrosis or structural damage, indicating it to be apparently safe for intranasal administration.
Ziprasidone is a dopamine D2 and serotonin 5-HT2A receptor antagonist and was approved by the FDA for the treatment of schizophrenia in 2001 [
27,
47,
48]. It is commercialized as an oral capsule but has low bioavailability due extensive first-pass metabolism, and also leads to several systemic side effects (such as somnolence, respiratory tract infections, dizziness, akathisia, abnormal vision and asthenia). Moreover, its water solubility is quite low (predicted to be 0.00718 mg/mL), which poses a problem for the formulation process [
27,
48]. In order to solve some of these issues, Bahadur et al. [
27] decided to develop O/W nanoemulsions for intranasal delivery. The formulations contained Capmul
® MCM (medium chain mono- and diglycerides), Labrasol
® (polyethylene glycol-8 caprylic/capric glycerides), Transcutol
® (diethylene glycol monoethyl ether) and phosphate buffer (pH 8.0) (specific quantities in
Table 5). Additionally, the mucoadhesive nanoemulsions also had either chitosan or Carbopol
® 934 (carbomer 934) in their composition. Drug strength was set at 20 mg/mL, which is 2786 times higher than ziprasidone’s water solubility, and formulations were prepared by spontaneous emulsification. Mean droplet size, PDI, zeta potential, pH and viscosity of the non-mucoadhesive nanoemulsion were 145.24 nm, 0.186, −30.2 mV, 6.5 and 183 cP, respectively. This same formulation was considered to be stable under accelerated conditions. Nevertheless, these characterization and stability parameters were not measured for the mucoadhesive nanoemulsions, which leaves the question of whether these formulations were actually nanoemulsions, since the addition of a mucoadhesive polymer to a nanoemulsion can alter droplet size and PDI greatly, as well as the formulation’s viscosity (which the authors refer is an important parameter but do not measure for the mucoadhesive formulations). All formulations had an osmolarity of around 310 mOsmol/L, which is considered safe for the nasal mucosa. The ex vivo permeation assay (sheep nasal mucosa) indicated that the mucoadhesive nanoemulsions had higher diffusion coefficients (when compared to the non-mucoadhesive nanoemulsion), with the one containing chitosan having a better performance than the one containing Carbopol, which might be explained by chitosan’s capacity for permeation enhancing due to reversible opening of tight junctions (as previously mentioned) or modifying phospholipids bilayers. In vivo pharmacodynamic results (mice, locomotor activity) indicated that, in general, the intranasal administration of the developed nanoemulsions led to a more significant therapeutic effect than oral or intraperitoneal administrations, suggesting the superiority of intranasal delivery. Moreover, the mucoadhesive chitosan nanoemulsion had the best performance, which again could be due to chitosan not only increasing the residence time of the formulation in the nasal mucosa but also enhancing drug permeation. Nevertheless, again the intraperitoneally administered formulation should have been a drug solution, and it was the non-mucoadhesive nanoemulsion, which could have led to altered pharmacokinetics, also being a highly viscous preparation (which should not be given through this route). Yet, nasal ciliotoxicity studies (sheep nasal mucosa) showed that the developed nanoemulsions could be regarded as safe, since they did not cause any sort of acute damage to the nasal mucosa.
Paliperidone is another second-generation atypical antipsychotic and was approved by the FDA for the treatment of schizophrenia in 2006 [
49]. It is the primary active metabolite of risperidone (9-hydroxyrisperidone), and acts by blocking dopamine D2 and serotonin 5-HT2A receptors. Nevertheless, its oral bioavailability (extended-release tablet) is only 28%, and has low water solubility (predicted to be 0.297 mg/mL), making it hard to formulate at high strength [
28,
49]. Hence, Patel et al. [
28] decided to incorporate paliperidone into O/W microemulsions for intranasal administration. Both microemulsions had oleic acid, Labrasol
® (polyethylene glycol-8 caprylic/capric glycerides), Kolliphor
® RH40 (polyoxyl 40 hydrogenated castor oil), Transcutol
® P (diethylene glycol monoethyl ether) and water, and the mucoadhesive microemulsion additionally had polycarbophil AA-1 (detailed quantities in
Table 6). The chosen preparation method was spontaneous emulsification, and drug strength was 5 mg/mL, which is almost 17 times higher than paliperidone’s water solubility. Formulation characterization parameters were between 20.01–27.31 nm for droplet size, 0.049–0.117 for PDI, −36.59 and −38.65 mV for zeta potential, 77–96 cP for viscosity, and 5.85–5.98 for pH. The developed formulations were stable up to 6 months and under accelerated conditions. The in vivo pharmacokinetic study (rats; 0.18–0.20 mg/kg) indicated that, overall, the intranasal route was faster than the intravenous route at making the drug reach the brain (lower T
max for the non-mucoadhesive microemulsion). Moreover, both microemulsions were more effective in delivering the drug to the brain, having higher brain C
max, brain AUC and brain/blood ratio values than the intravenous group, with the mucoadhesive microemulsion having a better performance. Yet again, the intravenous control should not have been a microemulsion, especially since it is reasonably viscous. In vivo pharmacodynamics (mice; 0.20 mg/kg; apomorphine-induced compulsive behavior and spontaneous motor activity) confirmed the overall potential of the intranasal route, and especially of the developed microemulsions, with the mucoadhesive polymer clearly having an important role and resulting in higher therapeutic efficacy.
Pidaparthi et al. [
29] also formulated paliperidone into an O/W nanometric emulsion for intranasal delivery. The nanoemulsion’s composition included Labrafil
® M 1944 CS (oleoyl polyoxyl-6 glyceride), Tween
® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), Transcutol
® HP (diethylene glycol monoethyl ether), benzalkonium chloride and phosphate buffer (pH 6.4) (specific amounts in
Table 6). The preparation method was water titration followed by high-pressure homogenization, and measured characterization parameters were −0.009 mV for zeta potential, 6.5 for pH, 94.5 cP for viscosity and 38.25 nm for droplet size, with no PDI reported, which leaves a question about the formulation’s homogeneity. Drug strength was kept at 2.5 mg/mL, which is more than 8 times higher than paliperidone’s water solubility, and the developed nanoemulsion was stable under accelerated conditions. In vitro drug release (Franz diffusion apparatus) and ex vivo drug permeation (goat nasal mucosal) studies showed that the paliperidone nanoemulsion had a more than 4 times higher drug release and a more than twice higher drug permeation than a drug suspension. As for in vivo evaluation, pharmacodynamic studies (mice; 2 mg/kg; spontaneous motor activity) showed that the intranasal administration of the paliperidone nanoemulsion led to a more effective therapeutic action than the intranasal drug suspension. No other controls were carried out, but it would have been interesting to have an intravenous control group (drug solution), where the drug would have been readily systemically available. The developed formulation was also safe, given that in continuous intranasal administration to mice (every 3 h for 12 h), their nasal mucosa showed no signs of inflammation, hemorrhage, erosion or nasociliary damage.
Iloperidone is another atypical antipsychotic drug that acts as an antagonist for both dopamine D2 and serotonin 5-HT2 receptors, having been approved for schizophrenia treatment by the FDA in 2009. It is available as oral tablets, but extensive first-pass metabolism leads to low bioavailability, and it also has poor water solubility (predictably 0.0304 mg/mL) [
30,
50]. To help solve these issues, Narala et al. developed an O/W nanoemulsion, for oral administration, containing soybean oil (Lipoid), egg lecithin (egg phosphatidyl choline, EPC80), cholesterol, oleic acid, glycerol and water (detailed quantities in
Table 6). The formulation was prepared by hot homogenization and ultrasonication, and drug strength was 0.4 mg/mL, the equivalent to 13 times iloperidone’s water solubility. The measured droplet size was 222.3 nm, PDI 0.200, and zeta potential −28.9 mV, and stability studies deemed the developed nanoemulsion as fairly stable after storage for 3 months and under accelerated conditions. In vitro drug release (open tube dialysis method) appeared to be sustained, being below 50% (cumulative release) after 12 h, and only being close to full release after 48 h (approximately 90% cumulative release). Oral administration to rats (2.5 mg/kg) led to a higher systemic bioavailability than an oral drug suspension (higher plasma C
max and AUC), and also to a higher suppression of hyperlocomotor activity (pharmacokinetic and pharmacodynamic studies, respectively). Nevertheless, the comparison to an intravenous control (especially if it was a drug solution) would have been useful.
Asenapine is a dopamine D2 and serotonin 5-HT2A receptors antagonist and was approved for the treatment of schizophrenia by the FDA in 2009. After oral administration it leads to a very low bioavailability, having extensive first-pass metabolism and low water solubility (predicted to be 0.0312 mg/mL) [
31,
51]. To address these issues, Kumbhar et al. [
31] developed O/W nanoemulsions for intranasal administration, containing Capmul
® PG-8 (propylene glycol monocaprylate), Kolliphor
® RH40 (polyoxyl 40 hydrogenated castor oil) and Transcutol
® HP (diethylene glycol monoethyl ether). Moreover, the mucoadhesive microemulsion also had Carbopol
® 971 (carbomer 971) in its composition (specific quantities in
Table 6). The non-mucoadhesive nanoemulsion had a droplet size of 21.2 nm, PDI of 0.355, zeta potential of −14.12 mV and viscosity of 91.67 cP. Aside from viscosity, the mucoadhesive microemulsion did not have these parameters determined, which again leaves the question of whether the formulation obtained after incorporation of the mucoadhesive polymer was still a microemulsion, and, if so, how homogeneous it was. The mucoadhesive microemulsion’s viscosity was 235.67 cP (higher, as expected from the addition of Carbopol). The formulations were prepared by aqueous titration and were stable under accelerated conditions. The in vitro drug release (Franz diffusion cells) from the developed nanoemulsions was higher than from an asenapine solution, and the mucoadhesive formulation, being more viscous, had a slightly lower cumulative drug release than the non-mucoadhesive nanoemulsion. Nevertheless, the ex vivo permeation assay (sheep nasal mucosa,
Figure 4A) showed that the mucoadhesive nanoemulsion had a higher overall drug permeation, which might, again, be due to transient opening of the tight junctions of the nasal mucosa due to the presence of the mucoadhesive Carbopol. Furthermore, the nanoemulsion’s mucoadhesive strength (ex vivo assay in sheep nasal mucosa) increased more than 3-fold with the addition of the mucoadhesive polymer. In vivo pharmacokinetic results (rats, 1 mg/kg,
Figure 4B) indicated the superiority of intranasal administration, since both intranasal nanoemulsions had significantly higher brain C
max and brain AUC and a lower brain T
max than the intravenous administration, suggesting that intranasal delivery was both faster and more effective in making the drug reach the brain. Moreover, the intranasal mucoadhesive nanoemulsion had a better performance than its non-mucoadhesive counterpart, probably due to enhanced viscosity and adhesion to the nasal mucosa, which resulted in prolonged retention time in the nasal cavity. Nevertheless, again, the intravenous control group should have probably been a drug solution, given the more predictable pharmacokinetics and lower viscosity. In vivo pharmacodynamics (rats; 1 mg/kg; catalepsy test, induced locomotor activity test and paw test–
Figure 4C) indicated that the developed nanoemulsions had therapeutic-like efficacy, showing better results that the drug solution, and with the mucoadhesive formulation performing better. As for the formulations’ safety (nasal ciliotoxicity, sheep nasal mucosa), they did not seem to cause morphological changes or other signs of toxicity in the nasal mucosa and were thus considered safe.
Amisulpride is another atypical antipsychotic drug which is not only highly selective (antagonist of dopamine D2 and D3 receptors), which decreases the likelihood of extrapyramidal symptoms, but also has the rare ability of reasonably improving both positive and negative symptoms of schizophrenia. Nevertheless, it has low intestinal permeability, which leads to low oral bioavailability, and is a P-glycoprotein substrate. Moreover, it has low aqueous solubility (predicted to be 0.293 mg/mL), which is also pH dependent, resulting in high solubility in the stomach (low pH) but poor solubility in the intestinal medium (higher pH), which could cause drug precipitation when reaching the intestine after oral administration [
32,
52]. Moreover, albeit selective, there have been reports of cardiovascular (prolongation of QT interval, bradycardia, hypotension), neuropsychiatric (sedation, seizures) and endocrine (increased prolactin levels) adverse events, as well as hematological toxicity with prolonged therapy (agranulocytosis or leukopenia) [
33,
52]. To solve some of these problems, Gamal et al. [
32] decided to develop an oral SNEDDS, made of Capryol™ 90 (propylene glycol monocaprylate (type II)), Cremophor
® RH40 (polyoxyl 40 hydrogenated castor oil) and Transcutol
® HP (diethylene glycol monoethyl ether), with varying proportions (detailed quantities in
Table 7). All 3 selected formulas had a drug strength of 50 mg/g, which is 170 times higher than amisulpride’s predicted water solubility. Formulations’ measured droplet size was between 16–22 nm, PDI between 0.092–0.193, and all formulas were stable under accelerated conditions. In vitro drug release (dialysis bag method) in a low pH medium (1.2) showed no statistically significant differences between the developed formulations and an aqueous drug suspension, but at neutral pH (7.4) there was a significantly higher amisulpride release from the developed SNEDDS, which suggests that these formulations could be useful in preventing drug precipitation at intestinal conditions. Drug permeation (ex vivo, rat intestine) was also significantly higher with the developed SNEDDS (compared to the drug solution), which shows the permeation enhancing effects of these formulation’s excipients and its favorable properties after dilution (nanometric droplet size). However, no in vivo evaluation was carried out, and hence the true potential of the developed SNEDDS was left undetermined.
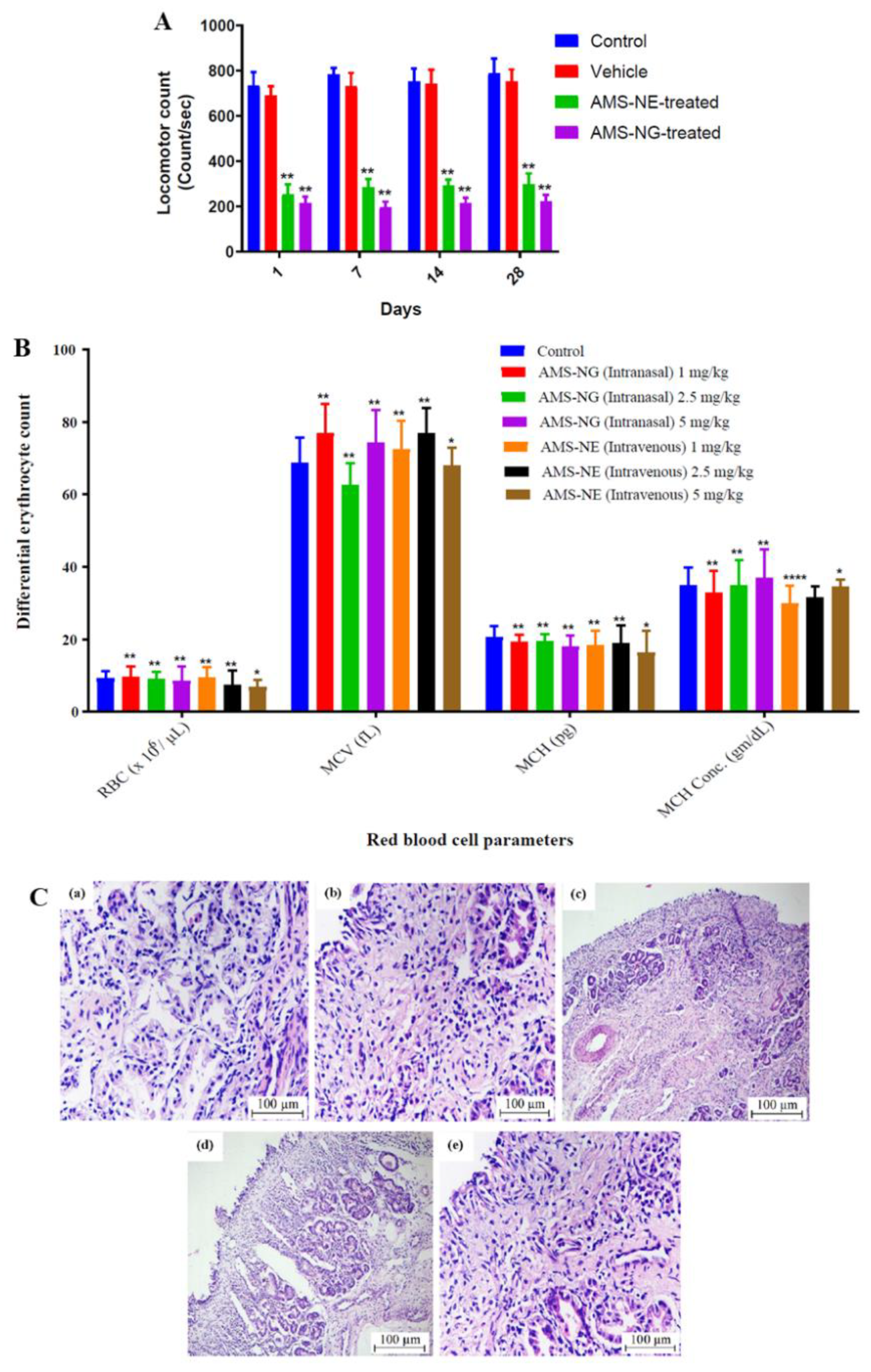
Gadhave et al. [
33] also developed amisulpride formulations, but for intranasal delivery. The developed nanoemulsions had Maisine
® CC (glyceryl monolinoleate), Labrasol
® (polyethylene glycol-8 caprylic/capric glycerides), Transcutol
® HP (diethylene glycol monoethyl ether) and water in their composition (concentration of each component in
Table 7). One of them also had poloxamer 407 and gellan gum (combination of thermosensitive and ion-sensitive polymers), making it a nanoemulgel. Nanoemulgels are nanoemulsions that have a gelling (and sometimes mucoadhesive) polymer in their composition, which gives them a higher viscosity that can increase their retention time in the nasal mucosa, consequently increasing the time available for drug absorption to occur. An aqueous-titration technique was the chosen preparation method, and drug strength was kept at 1 mg/mL, which is more than 3 times higher than amisulpride’s predicted water solubility. Measured formulation characteristics were 92.15 and 106.11 nm for droplet size, 0.46 and 0.51 for PDI, and 18.22 and –16.01 mV for zeta potential, for the non-mucoadhesive and mucoadhesive nanoemulsions, respectively. Although the other parameters could be considered to be within acceptable values, the PDI values were reasonably high, and hence the homogeneity of the developed nanometric emulsions could be questioned. Viscosity values were 8.69 cP (nanoemulsion) and 12.6 cP (nanoemulgel) at room temperature (25 °C), but at nasal temperature (34 °C) the viscosity of the nanoemulgel increased to 106.5 cP, due to having poloxamer 407 in its composition (thermosensitive polymer). The incorporation of the mucoadhesive polymer into formulation’s composition (gellan gum) also led to an increase in the mucoadhesive force (sheep nasal mucosa). Both formulas were stable under accelerated conditions. Compared to an amisulpride drug suspension (with 2% hydroxypropyl methylcellulose and 4% Tween
® 80), the developed formulations had a higher in vitro drug release (vertical Franz diffusion cell), and despite having a higher viscosity the nanoemulgel did not have a decrease in performance (when compared to the non-mucoadhesive nanoemulsion) but had a sustained-release effect (after 8 h). Additionally, ex vivo drug permeation (sheep nasal mucosa) led to a significantly higher cumulative permeation (also after 8 h) for the nanoemulgel, when compared to the nanoemulsion and the suspension, which indicates the advantage of not only having excipients with permeation enhancing capability (surfactants and cosolvents) and an increased surface area (nanodroplets), but also of the presence of the chosen polymers. In vivo pharmacokinetic studies (rats; 1 mg/kg) showed that the intranasal delivery of the developed formulations was better at making the drug reach the brain than the intravenous route, with the nanoemulgel having the highest brain drug delivery and least systemic drug distribution (leading to the most favorable brain/blood ratios and, consequently, a potentially higher efficacy and safety). In vivo pharmacodynamics also proved that the developed formulations were effective in reducing schizophrenia symptoms (catalepsy test, induced locomotor activity—
Figure 5A, paw test), with the nanoemulgel having a better performance. This could have been due to the mucoadhesive properties and in situ gelling of the nanoemulgel, which made it possible for the formulation to have a longer residence time in the nasal cavity, leading to a longer time for drug absorption to occur. Nevertheless, again the intravenous control should have been a drug solution, and not the developed nanoemulsion. As for safety evaluation (rats, chronic administration for 28 days), the intravenous administration led to nasal or respiratory toxicity, and behavioral changes, while the intranasal administration did not (
Figure 5C). Moreover, and the intranasal nanoemulgel led to a much lower hematological toxicity (
Figure 5B) than the nanoemulsion, which further emphasizes the potential of the intranasal delivery of the developed nanoemulgel.
Lurasidone is another second-generation antipsychotic, having been approved by the FDA for the treatment of schizophrenia in 2010 [
53]. It acts as an antagonist of dopamine D2 and serotonin 5-HT2A receptors, being a potent drug and having only a weak affinity towards other receptors, hence leading to less extrapyramidal side effects and a reasonably good tolerability profile. Yet, there have still been reports of systemic side effects (elevated levels of serum prolactin, orthostatic hypotension, muscle tremor), and due to having low water solubility (predictably 0.00789 mg/mL) and being highly metabolized, lurasidone also has low oral bioavailability (although improved when taken with food) [
34,
53]. To improve at least some of these features, Patel et al. [
34] decided to formulate lurasidone into O/W nanoemulsions for intranasal administration. Their composition included Capmul
® MCM EP (medium chain mono- and diglycerides), Capmul
® MCM C8 EP (glyceryl monocaprylate), Cremophor
® EL (polyoxyl 35 castor oil) and water (detailed quantities in
Table 7). The mucoadhesive nanoemulsion also had polycarbophil AA-1, and the chosen preparation method was high pressure homogenization. Drug strength was kept at 10 mg/mL, which is 1267 times higher than lurasidone’s predicted water solubility. The non-mucoadhesive nanoemulsion had a droplet size of 48.07 nm and PDI of 0.31, but these parameters were not mentioned for the mucoadhesive formulation, which leaves it unclear as to whether it was in fact nanometric or homogeneous. For both formulations, zeta potential varied between −0.19 and −0.41 mV and pH between 5–6. The incorporation of the mucoadhesive polymer into the nanoemulsion made the viscosity increase 8-fold (from 119.46 to 892.33 cP), and the retention time (mucoadhesion assay) 4-fold (5 min to 21 min). The formulations were stable under accelerated conditions and after storage for 6 months. Ex vivo permeation studies (sheep nasal mucosa) showed that the developed nanoemulsions led to a more effective and faster drug permeation than a drug solution (water:Transcutol
® P mixture 1:1
v/
v), with the mucoadhesive nanoemulsion having a more sustained release due to being more viscous. Nevertheless, they did not perform better than a microemulsion (containing Capmul
® MCM EP, Labrasol
®, Cremophor
® RH 40, Transcutol
® P and water), that was also evaluated for comparison purposes, probably due to this microemulsion having a higher amount of permeation enhancing excipients (surfactants and cosolvents), but which also make it potentially less safe. In vivo pharmacodynamic evaluation (mice; 4.06 mg/kg; apomorphine induced compulsive behavior and spontaneous locomotor activity) showed that intranasal administration was, overall, more therapeutically effective than the intravenous one (although a nanoemulsion was, again, the intravenous control, and not a drug solution), and that the intranasal nano and microemulsions performed better than the intranasal drug solution, despite the solution having a cosolvent in its composition. This could be due to the high surface area ratio that the nanometric emulsions provide, and also the presence of surfactants. Moreover, the mucoadhesive nanoemulsion performed better than the non-mucoadhesive nanoemulsion, which suggests that the mucoadhesive polymer did in fact increase the formulation’s retention time in the nasal mucosa, decreasing and/or slowing down the nasociliary clearance. Nevertheless, the microemulsion still had the highest therapeutic effect, again probably due to the higher amount of cosolvents, which enhanced drug absorption and, consequently, brain bioavailability. As for safety evaluation (nasal ciliotoxicity, sheep nasal mucosa), the developed formulations did not appear to damage the nasal mucosa, hence being potentially safe for intranasal administration.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}