
Polymorphism of Drug Transporters, Rather Than Metabolizing Enzymes, Conditions the Pharmacokinetics of Rasagiline

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Procedures
2.2. Pharmacokinetics and Safety
2.3. Genotyping and Phenotyping
2.4. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Linazasoro, G. Rasagiline in Parkinson’s disease. Neurologia 2008, 23, 238–245. [Google Scholar] [PubMed]
- Chau, K.Y.; Cooper, J.M.; Schapira, A.H.V. Rasagiline protects against alpha-synuclein induced sensitivity to oxidative stress in dopaminergic cells. Neurochem. Int. 2010, 57, 525–529. [Google Scholar] [CrossRef]
- Chen, J.J.; Swope, D.M. Clinical Pharmacology of Rasagiline: A Novel, Second-Generation Propargylamine for the Treatment of Parkinson Disease. J. Clin. Pharmacol. 2005, 45, 878–894. [Google Scholar] [CrossRef]
- Xtandi, E.M.A. Azilect: EPAR-Product Information 2022; European Medicines Agency (EMA): Amaterdam, The Netherlands, 2022. [Google Scholar]
- Food and Drug Administration (FDA). Highlights of Prescribing Information-Rasagiline Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021641s016s017lbl.pdf (accessed on 1 September 2022.).
- Bilal, R.; Ahmad, N.S.; Zaffar, S.; Mazhar, M.U.; Siddiqui, W.A.; Tariq, S. Rasagiline Pharmacokinetics in CYP1A2 Variant Healthy Smokers & Non-smokers in Different Doses. Pak. J. Med. Sci. 2022, 38, 589. [Google Scholar] [CrossRef]
- Masellis, M.; Collinson, S.; Freeman, N.; Tampakeras, M.; Levy, J.; Tchelet, A.; Eyal, E.; Berkovich, E.; Eliaz, R.E.; Abler, V.; et al. Dopamine D2 receptor gene variants and response to rasagiline in early Parkinson’s disease: A pharmacogenetic study. Brain 2016, 139, 2050–2062. [Google Scholar] [CrossRef]
- Zubiaur, P.; Mejía-Abril, G.; Navares-Gómez, M.; Villapalos-García, G.; Soria-Chacartegui, P.; Saiz-Rodríguez, M.; Ochoa, D.; Abad-Santos, F. PriME-PGx: La Princesa University Hospital Multidisciplinary Initiative for the Implementation of Pharmacogenetics. J. Clin. Med. 2021, 10, 3772. [Google Scholar] [CrossRef]
- Shamim, B. Good Clinical Practice (GCP): A Review. PharmaTutor 2014, 9, 20–29. [Google Scholar]
- European Medicines Agency. Guideline on Bioanalytical Method Validation (EMEA/CHMP/EWP/192217/2009); European Medicines Agency: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Aguirre, C.; García, M. Causality Assessment in Reports on Adverse Drug Reactions. Algorithm of Spanish Pharmacovigilance System. Med. Clin. 2016, 147, 461–464. [Google Scholar] [CrossRef]
- Cooper-DeHoff, R.M.; Niemi, M.; Ramsey, L.B.; Luzum, J.A.; Tarkiainen, E.K.; Straka, R.J.; Gong, L.; Tuteja, S.; Wilke, R.A.; Wadelius, M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and Statin-Associated Musculoskeletal Symptoms. Clin. Pharmacol. Ther. 2022, 111, 1007–1021. [Google Scholar] [CrossRef]
- Desta, Z.; Gammal, R.S.; Gong, L.; Whirl-Carrillo, M.; Gaur, A.H.; Sukasem, C.; Hockings, J.; Myers, A.; Swart, M.; Tyndale, R.F.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2B6 and Efavirenz-Containing Antiretroviral Therapy. Clin. Pharmacol. Ther. 2019, 106, 726–733. [Google Scholar] [CrossRef]
- Lee, C.R.; Luzum, J.A.; Sangkuhl, K.; Gammal, R.S.; Sabatine, M.S.; Stein, C.M.; Kisor, D.F.; Limdi, N.A.; Lee, Y.M.; Scott, S.A.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2C19 Genotype and Clopidogrel Therapy: 2022 Update. Clin. Pharmacol. Ther. 2022. [Google Scholar] [CrossRef]
- Crews, K.R.; Monte, A.A.; Huddart, R.; Caudle, K.E.; Kharasch, E.D.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Callaghan, J.T.; Samer, C.F.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2D6, OPRM1, and COMT Genotypes and Select Opioid Therapy. Clin. Pharmacol. Ther. 2021, 110, 888–896. [Google Scholar] [CrossRef]
- KNMP Pharmacogenomics Working Group. CYP2D6: Quetiapine (2393/2394/2395). KNMP: The Hague, The Netherlands, 2021. [Google Scholar]
- Birdwell, K.; Decker, B.; Barbarino, J.; Peterson, J.; Stein, C.; Sadee, W.; Wang, D.; Vinks, A.; He, Y.; Swen, J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef]
- Gammal, R.; Court, M.; Haidar, C.; Iwuchukwu, O.; Gaur, A.; Alvarellos, M.; Guillemette, C.; Lennox, J.L.; Whirl-Carrillo, M.; Brummel, S.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for UGT1A1 and Atazanavir Prescribing. Clin. Pharmacol. Ther. 2015, 99, 363–369. [Google Scholar] [CrossRef]
- KNMP Pharmacogenomics Working Group. UGT1A1: Irinotecan (1691 to 1694). KNMP: The Hague, The Netherlands, 2021. [Google Scholar]
- McDonagh, E.M.; Boukouvala, S.; Aklillu, E.; Hein, D.; Altman, R.; Klein, T.E. PharmGKB summary: Very Important Pharmacogene Information for N-Acetyltransferase 2. Pharmacogen. Genom. 2014, 24, 409–425. [Google Scholar] [CrossRef]
- Gaedigk, A.; Ingelman-Sundberg, M.; Miller, N.A.; Leeder, J.S.; Whirl-Carrillo, M.; Klein, T.E.; PharmVar Steering Committee. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 2018, 103, 399–401. [Google Scholar] [CrossRef]
- PharmVAR. The Pharmacogene Variation (PharmVar) Consortium: CYP1A2 Allele Nomenclature. Available online: https://www.pharmvar.org/gene/CYP1A2 (accessed on 1 September 2022).
- Soldin, O.P.; Mattison, D. Sex Differences in Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2009, 48, 143–157. [Google Scholar] [CrossRef]
- Graff, J.; Brinch, K.; Madsen, J.L. Gastrointestinal mean transit times in young and middle-aged healthy subjects. Clin. Physiol. 2001, 21, 253–259. [Google Scholar] [CrossRef]
- Sadik, R.; Abrahamsson, H.; Stotzer, P.-O. Gender Differences in Gut Transit Shown with a Newly Developed Radiological Procedure. Scand. J. Gastroenterol. 2003, 38, 36–42. [Google Scholar] [CrossRef]
- HMDB Metabocard Rasagiline (HMDB0015454) 2022. Available online: https://hmdb.ca/metabolites/HMDB0015454 (accessed on 1 September 2022).
- Karastergiou, K.; Smith, S.R.; Greenberg, A.S.; Fried, S.K. Sex differences in human adipose tissues–the biology of pear shape. Biol. Sex Differ. 2012, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Dobrinas, M.; Cornuz, J.; Oneda, B.; Serra, M.K.; Puhl, M.; Eap, C. Impact of Smoking, Smoking Cessation, and Genetic Polymorphisms on CYP1A2 Activity and Inducibility. Clin. Pharmacol. Ther. 2011, 90, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved Brain Penetration and Antitumor Efficacy of Temozolomide by Inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Leschziner, G.D.; Andrew, T.; Pirmohamed, M.; Johnson, M.R. ABCB1 genotype and PGP expression, function and therapeutic drug response: A critical review and recommendations for future research. Pharmacogen. J. 2006, 7, 154–179. [Google Scholar] [CrossRef]
- Brambila-Tapia, A.J.-L. MDR1 (ABCB1) Polymorphisms: Functional Effects and Clinical Implications. Rev. Invest. Clin. 2013, 65, 445–454. [Google Scholar] [PubMed]
- Furuno, T.; Landi, M.-T.; Ceroni, M.; Caporaso, N.; Bernucci, I.; Nappi, G.; Martignoni, E.; Schaeffeler, E.; Eichelbaum, M.; Schwab, M.; et al. Expression polymorphism of the blood???brain barrier component P-glycoprotein (MDR1) in relation to Parkinson’s disease. Pharmacogenetics 2002, 12, 529–534. [Google Scholar] [CrossRef]
- Sychev, D.A.; Levanov, A.N.; Shelekhova, T.V.; Bochkov, P.O.; Denisenko, N.P.; Ryzhikova, K.A.; Mirzaev, K.B.; Grishina, E.A.; Gavrilov, M.A.; Ramenskaya, G.V.; et al. The impact of ABCB1 (rs1045642 and rs4148738) and CES1 (rs2244613) gene polymorphisms on dabigatran equilibrium peak concentration in patients after total knee arthroplasty. Pharmacogen. Pers. Med. 2018, 11, 127–137. [Google Scholar] [CrossRef]
- Pingili, R.; Vemulapalli, S.; Mullapudi, S.S.; Nuthakki, S.; Pendyala, S.; Kilaru, N. Pharmacokinetic interaction study between flavanones (hesperetin, naringenin) and rasagiline mesylate in wistar rats. Drug Dev. Ind. Pharm. 2015, 42, 1110–1117. [Google Scholar] [CrossRef]
- Laechelt, S.; Turrini, E.; Ruehmkorf, A.; Siegmund, W.; Cascorbi, I.; Haenisch, S. Impact of ABCC2 haplotypes on transcriptional and posttranscriptional gene regulation and function. Pharmacogen. J. 2010, 11, 25–34. [Google Scholar] [CrossRef]
- Chen, J.; Su, Q.B.; Tao, Y.Q.; Qin, J.M.; Zhou, Y.; Zhou, S.; Li, H.L.; Chen, Z.J.; Zhou, Y.F.; Zhou, L.M.; et al. ABCC2 rs2273697 is associated with valproic acid concentrations in patients with epilepsy on valproic acid monotherapy. Pharmazie 2018, 73, 279–282. [Google Scholar] [CrossRef]
- Goswami, S.; Gong, L.; Giacomini, K.; Altman, R.; Klein, T.E. PharmGKB summary: Very Important Pharmacogene Information for SLC22A1. Pharmacogen. Genom. 2014, 24, 324–328. [Google Scholar] [CrossRef]
- Shu, Y.; Brown, C.; Castro, R.; Shi, R.; Lin, E.; Owen, R.; Sheardown, S.; Yue, L.; Burchard, E.; Brett, C.; et al. Effect of Genetic Variation in the Organic Cation Transporter 1, OCT1, on Metformin Pharmacokinetics. Clin. Pharmacol. Ther. 2007, 83, 273–280. [Google Scholar] [CrossRef]
- Zubiaur, P.; Saiz-Rodríguez, M.; Ochoa, D.; Navares-Gómez, M.; Mejía, G.; Román, M.; Koller, D.; Soria-Chacartegui, P.; Almenara, S.; Abad-Santos, F. Effect of Sex, Use of Pantoprazole and Polymorphisms in SLC22A1, ABCB1, CES1, CYP3A5 and CYP2D6 on the Pharmacokinetics and Safety of Dabigatran. Adv. Ther. 2020, 37, 3537–3550. [Google Scholar] [CrossRef]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.N.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H.C. OCT1 polymorphism is associated with response and survival time in anti-Parkinsonian drug users. Neurogenetics 2010, 12, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Hidestrand, M.; Oscarson, M.; Salonen, J.S.; Nyman, L.; Pelkonen, O.; Turpeinen, M.; Ingelman-Sundberg, M. CYP2B6 and CYP2C19 as the major enzymes responsible for the metabolism of selegiline, a drug used in the treatment of Parkinson’s disease, as revealed from experiments with recombinant enzymes. Drug Metab. Dispos. 2001, 29, 1480–1484. [Google Scholar]


{kind=link}
| Alelle | −3860G>A | −739T>G | −729C>T | −163C>A | 2499A>T | 5347T>C |
|---|---|---|---|---|---|---|
| rs2069514 | rs2069526 | rs12720461 | rs762551 | rs72547516 | rs2470890 | |
| *1A (normal activity) | G | T | C | C | A | T |
| *1B | C | |||||
| *1C (decreased activity) | A | |||||
| *1E | G | |||||
| *1F (high inducibility) | A | |||||
| *1G | G | C | ||||
| *1J | G | A | ||||
| *1K (decreased activity) | G | T | A | |||
| *1L | A | A | C | |||
| *1N | A | C | ||||
| *4 (decreased expression) | T |
| Sex | n | Age (Years) | Height (m) | Weight (kg) | BMI (kg/m2) |
|---|---|---|---|---|---|
| Women | 65 | 25 (IQR = 9) | 1.62 ± 0.06 | 60.06 ± 7.89 | 22.87 ± 2.72 |
| Men | 53 | 24 (IQR = 10) | 1.76 ± 0.06 * | 75.18 ± 9.59 * | 24.30 ± 2.97 * |
| Ethnicity | |||||
| Caucasian | 83 | 24 (IQR = 7) | 1.70 ± 0.09 | 66.38 ± 10.89 | 22.91 ± 2.65 |
| Mixed | 35 | 30 (IQR = 15) * | 1.64 ± 0.09 * | 67.98 ± 12.89 | 24.96 ± 3.04 * |
| Total | 118 | 25 (IQR = 9) | 1.68 ± 0.09 | 66.85 ± 11.49 | 23.52 ± 2.91 |
| Sex | n | AUC0-∞/DW (h*ng*kg/mL*mg) | Cmax/DW (ng*kg/mL*mg) | tmax (h) | t1/2 (h) | Vd/F (L/kg) | Cl/F (L/kg*h) |
|---|---|---|---|---|---|---|---|
| Women | 65 | 254.19 ± 74.50 | 378.42 ± 156.70 | 0.48 ± 0.19 | 2.98 ± 0.80 | 17.98 ± 6.23 | 4.30 ± 1.39 |
| Men | 53 | 278.61 ± 73.51 | 447.91 ± 163.88 * | 0.52 ± 0.28 | 2.31 ± 0.85 *& | 12.18 ± 3.78 *& | 3.85 ± 1.08 |
| Ethnicity | |||||||
| Caucasian | 83 | 258.45 ± 71.81 | 418.18 ± 169.33 | 0.47 ± 0.19 | 2.56 ± 0.84 | 15.00 ± 5.72 | 4.18 ± 1.23 |
| Mixed | 35 | 281.05 ± 80.09 | 389.35 ± 147.25 | 0.55 ± 0.31 | 2.95 ± 0.94 * | 16.27 ± 6.62 | 3.92 ± 1.39 |
| Total | 118 | 265.16 ± 74.74 | 409.63 ± 163.00 | 0.49 ± 0.24 | 2.68 ± 0.89 | 15.38 ± 6.00 | 4.10 ± 1.28 |
| CYP1A2 Diplotype | n | AUC0-∞/DW (h*ng*kg/mL*mg) | Cmax/DW (ng*kg/mL*mg | tmax (h) | t1/2 (h) | Vd/F (L/kg) | Cl/F (L/kg*h) |
|---|---|---|---|---|---|---|---|
| *1F/*1F | 38 | 263.28 ± 60.08 | 403.23 ± 139.11 | 0.51 ± 0.21 | 2.69 ± 0.82 | 15.32 ± 5.34 | 3.98 ± 0.86 |
| *1B/*1F | 36 | 265.27 ± 80.68 | 426.5 ± 183.44 | 0.44 ± 0.19 | 2.66 ± 0.9 | 15.24 ± 5.73 | 4.15 ± 1.41 |
| *1L/*1F | 11 | 285.81 ± 77.84 | 389.59 ± 138.34 | 0.64 ± 0.48 | 2.55 ± 1.07 | 13.05 ± 4.16 | 3.82 ± 1.36 |
| *1B/*1B | 6 | 226.81 ± 52.00 | 411.84 ± 85.38 | 0.42 ± 0.18 | 1.87 ± 0.6 | 11.98 ± 3.33 | 4.62 ± 1.15 |
| *1L/*1L | 6 | 301.28 ± 89.3 | 518.16 ± 158.15 | 0.53 ± 0.17 | 3.06 ± 0.38 | 15.94 ± 5.55 | 3.61 ± 1.23 |
| *1B/*1N | 5 | 256.33 ± 116.11 | 327.66 ± 320.93 | 0.6 ± 0.28 | 3 ± 0.78 | 19.45 ± 9.55 | 4.63 ± 2.23 |
| *1J/*1N | 5 | 275.39 ± 91.37 | 435.74 ± 117.78 | 0.43 ± 0.09 | 2.15 ± 0.83 | 11.59 ± 3.81 | 3.9 ± 1.02 |
| *1B/*1L | 4 | 275.25 ± 108.77 | 392.87 ± 133.43 | 0.46 ± 0.16 | 3.32 ± 1.62 | 18.28 ± 7.57 | 4.11 ± 1.66 |
| *1N/*1K, *1N/*1L, *1N/*1L and *1N/*1G | 4 | 220.53 ± 87.92 | 355.79 ± 242.1 | 0.5 ± 0.14 | 2.94 ± 0.69 | 22.89 ± 12.33 | 5.21 ± 2.41 |
| Total | 115 | 265.29 ± 75.63 | 411.77 ± 164.35 | 0.49 ± 0.24 | 2.66 ± 0.89 | 15.32 ± 6.06 | 4.11 ± 1.29 |
| Genotype or Phenotype | n | AUC0-∞/DW (h*ng*kg/mL*mg) | Cmax/DW (ng*kg/mL*mg) | tmax (h) | t1/2 (h) | Vd/F (L/kg) | Cl/F (L/kg*h) | |
|---|---|---|---|---|---|---|---|---|
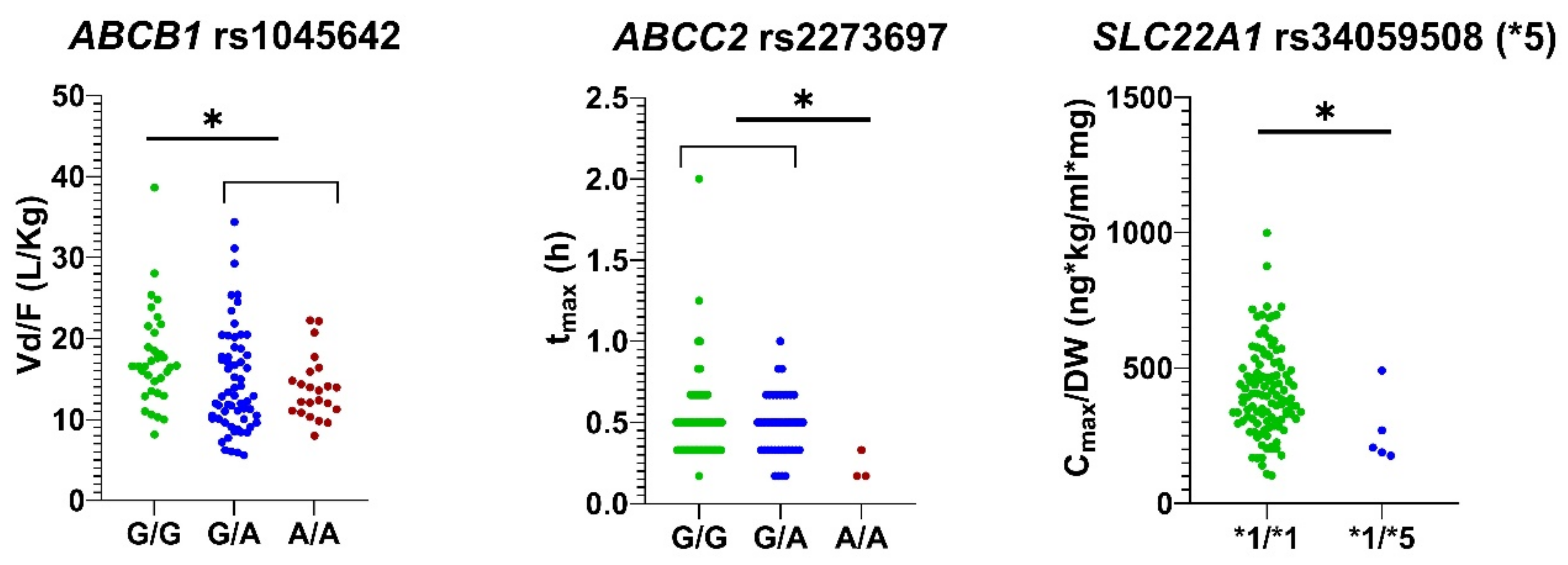
| ABCB1 rs1045642 | G/G | 34 | 244.10 ± 63.87 (pm = 0.012) | 359.70 ± 127.40 | 0.47 ± 0.19 | 2.83 ± 0.73 | 17.59 ± 5.97 (pu = 0.018, pm = 0.001) $ | 4.41 ± 1.31 (pm = 0.012) |
| G/A | 61 | 273.00 ± 83.60 | 426.48 ± 179.87 | 0.51 ± 0.27 | 2.63 ± 1.04 | 14.71 ± 6.41 | 4.05 ± 1.38 | |
| A/A | 23 | 275.48 ± 59.65 | 438.76 ± 152.32 | 0.50 ± 0.21 | 2.58 ± 0.63 | 13.89 ± 3.87 | 3.79 ± 0.79 | |
| ABCC2 rs2273697 | G/G | 73 | 262.04 ± 73.57 | 401.58 ± 167.92 | 0.51 ± 0.26 | 2.71 ± 0.89 | 15.73 ± 5.94 | 4.13 ± 1.23 |
| G/A | 42 | 264.50 ± 74.33 | 411.51 ± 152.39 | 0.48 ± 0.19 | 2.59 ± 0.90 | 14.95 ± 6.30 | 4.13 ± 1.36 | |
| A/A | 3 | 350.15 ± 86.34 | 579.28 ± 133.19 | 0.22 ± 0.09 (pu = 0.005, pm = 0.001) & | 3.01 ± 0.49 | 12.72 ± 2.28 | 2.99 ± 0.80 | |
| SLC22A1*5 (rs34059508) | *1/*1 | 113 | 267.52 ± 75.21 | 415.96 ± 161.85 | 0.49 ± 0.23 | 2.70 ± 0.89 | 15.38 ± 6.07 | 4.07 ± 1.29 |
| *1/*5 | 5 | 211.83 ± 36.15 | 266.58 ± 130.60 (pu = 0.022, pm = 0.003) | 0.67 ± 0.24 (pu = 0.018) | 2.24 ± 0.71 | 15.36 ± 4.85 | 4.83 ± 0.79 | |
| SLC22A1*3 (rs12208357) | *1/*1 | 104 | 265.80 ± 73.86 | 401.10 ± 159.35 | 0.51 ± 0.24 | 2.71 ± 0.89 | 15.52 ± 6.05 | 4.10 ± 1.32 |
| *1/*3 | 8 | 258.87 ± 34.73 | 473.14 ± 99.07 | 0.35 ± 0.11 (pu = 0.038 pm = 0.032) | 2.22 ± 0.81 | 12.35 ± 3.96 | 3.93 ± 0.58 | |
| CYP2C19 | UM | 3 | 205.53 ± 82.32 | 347.93 ± 233.78 | 0.61 ± 0.35 | 1.75 ± 0.67 | 15.39 ± 12.27 | 5.49 ± 2.41 |
| RM | 21 | 274.03 ± 86.22 | 426.69 ± 180.45 | 0.41 ± 0.11 | 2.66 ± 0.85 | 14.82 ± 6.62 | 4.09 ± 1.61 | |
| NM | 56 | 267.83 ± 70.38 | 402.65 ± 142.62 | 0.53 ± 0.27 | 2.67 ± 0.91 | 15.07 ± 5.84 | 4.02 ± 1.20 | |
| IM | 36 | 258.70 ± 75.90 | 415.50 ± 185.56 | 0.46 ± 0.22 | 2.85 ± 0.84 | 16.62 ± 5.25 | 4.16 ± 1.08 | |
| PM | 2 | 302.71 ± 14.98 | 412.92 ± 55.36 | 0.75 ± 0.11 | 1.55 ± 0.41 | 7.33 ± 1.58 | 3.31 ± 0.16 | |
| NAT2 | RA | 9 | 230.53 ± 67.28 | 315.46 ± 178.72 | 0.50 ± 0.24 | 2.72 ± 0.68 | 17.96 ± 5.87 | 4.69 ± 1.46 |
| SA | 103 | 266.53 ± 75.05 | 420.47 ± 161.94 (pu = 0.018, pm = 0.018) | 0.49 ± 0.24 | 2.62 ± 0.83 | 15.07 ± 6.05 | 4.08 ± 1.27 | |
| UGT1A6 rs7592281 | G/G | 111 | 264.10 ± 73.64 | 409.07 ± 159.30 | 0.48 ± 0.22 | 2.66 ± 0.89 | 15.25 ± 5.93 | 4.10 ± 1.23 |
| G/T | 7 | 281.88 ± 95.76 | 418.57 ± 229.87 | 0.70 ± 0.32 (pu = 0.021, pm = 0.029) | 3.03 ± 0.72 | 17.43 ± 7.29 | 4.08 ± 1.96 | |
| COMT rs4680 | G/G | 36 | 253.96 ± 59.13 | 385.75 ± 140.56 | 0.52 ± 0.32 | 2.58 ± 0.68 | 15.39 ± 5.13 | 4.14 ± 0.93 |
| G/A | 56 | 285.72 ± 81.43 | 450.03 ± 166.19 | 0.47 ± 0.18 | 2.71 ± 0.95 | 14.40 ± 6.06 | 3.84 ± 1.33 | |
| A/A | 26 | 236.37 ± 68.34 (pu = 0.018, pm = 0.008) | 355.69 ± 168.61 (pu = 0.012, pm = 0.035) | 0.50 ± 0.22 | 2.74 ± 1.00 | 17.45 ± 6.66 (pm=0.027) | 4.61 ± 1.45 (pm=0.008) | |
| TOTAL | 118 | 265.16 ± 74.74 | 409.63 ± 163.01 | 0.49 ± 0.24 | 2.68 ± 0.89 | 15.38 ± 6.00 | 4.10 ± 1.28 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubiaur, P.; Matas, M.; Martín-Vílchez, S.; Soria-Chacartegui, P.; Villapalos-García, G.; Figueiredo-Tor, L.; Calleja, S.; Navares-Gómez, M.; de Miguel, A.; Novalbos, J.; et al. Polymorphism of Drug Transporters, Rather Than Metabolizing Enzymes, Conditions the Pharmacokinetics of Rasagiline. Pharmaceutics 2022, 14, 2001. https://doi.org/10.3390/pharmaceutics14102001
Zubiaur P, Matas M, Martín-Vílchez S, Soria-Chacartegui P, Villapalos-García G, Figueiredo-Tor L, Calleja S, Navares-Gómez M, de Miguel A, Novalbos J, et al. Polymorphism of Drug Transporters, Rather Than Metabolizing Enzymes, Conditions the Pharmacokinetics of Rasagiline. Pharmaceutics. 2022; 14(10):2001. https://doi.org/10.3390/pharmaceutics14102001
Chicago/Turabian StyleZubiaur, Pablo, Miriam Matas, Samuel Martín-Vílchez, Paula Soria-Chacartegui, Gonzalo Villapalos-García, Laura Figueiredo-Tor, Sofía Calleja, Marcos Navares-Gómez, Alejandro de Miguel, Jesús Novalbos, and et al. 2022. "Polymorphism of Drug Transporters, Rather Than Metabolizing Enzymes, Conditions the Pharmacokinetics of Rasagiline" Pharmaceutics 14, no. 10: 2001. https://doi.org/10.3390/pharmaceutics14102001