
Novel Selectively Targeted Multifunctional Nanostructured Lipid Carriers for Prostate Cancer Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures
2.3. Methods
2.3.1. Preparation of NLCs
2.3.2. Synthesis of SA-PEG-TL and Preparation of Targeted NLCs
2.3.3. Preparation of Drug-Loaded NPs
2.3.4. Particle Size Distribution and Zeta-Potential Analyses
2.3.5. Analysis of Drug Loading Capacity and Encapsulation Efficiency
2.3.6. Cryogenic-Transmission Electron Microscopy Imaging (Cryo-TEM)
2.3.7. Time-Course of In Vitro Drug Release
2.3.8. Determination of NPs Concentration by Nanosight
2.3.9. Characterization of NPs Specificity by Confocal Laser Microscopy
2.3.10. Growth Inhibition Assays
3. Results and Discussion
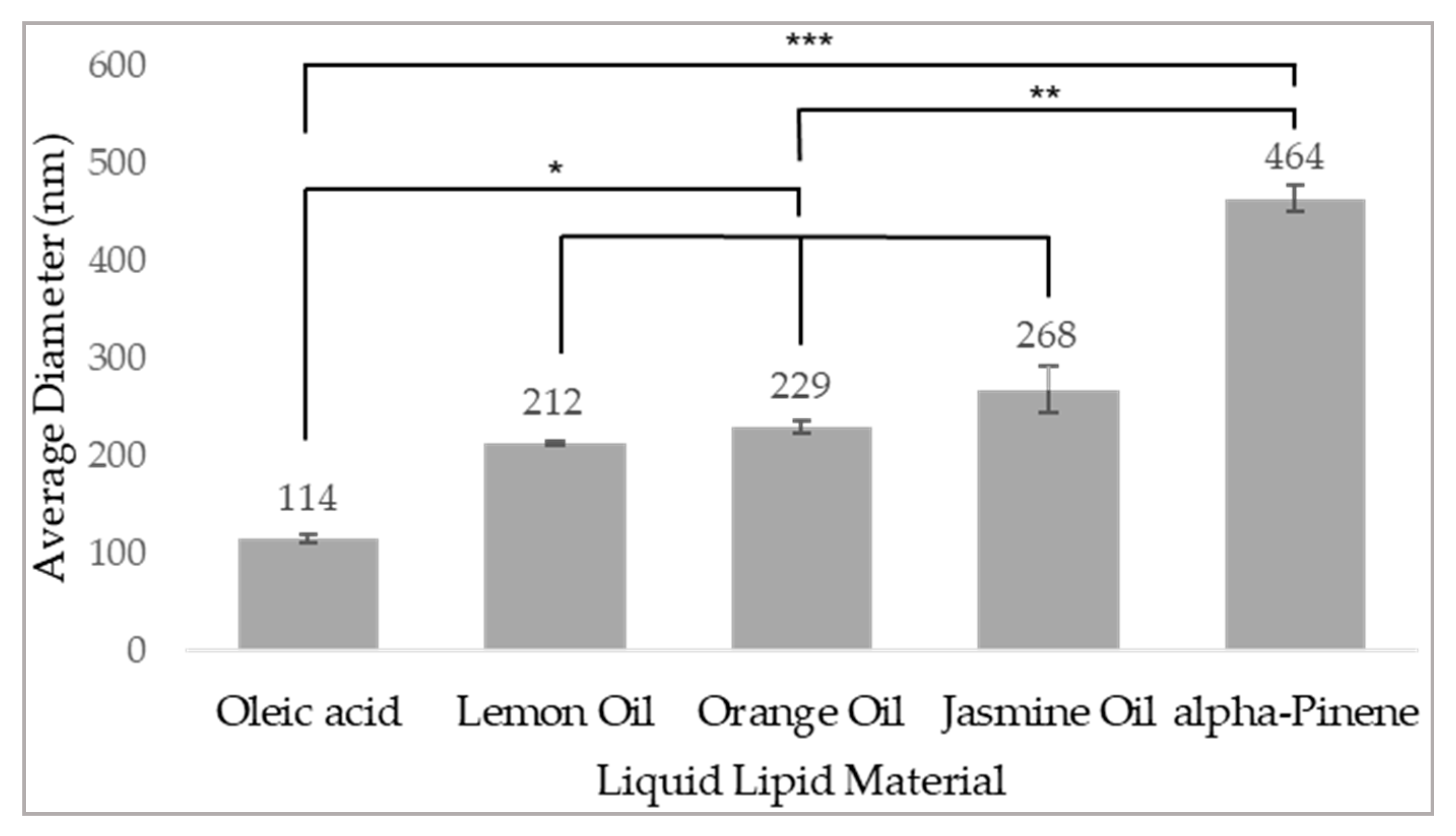
3.1. Self-Assembly of the Delivery System
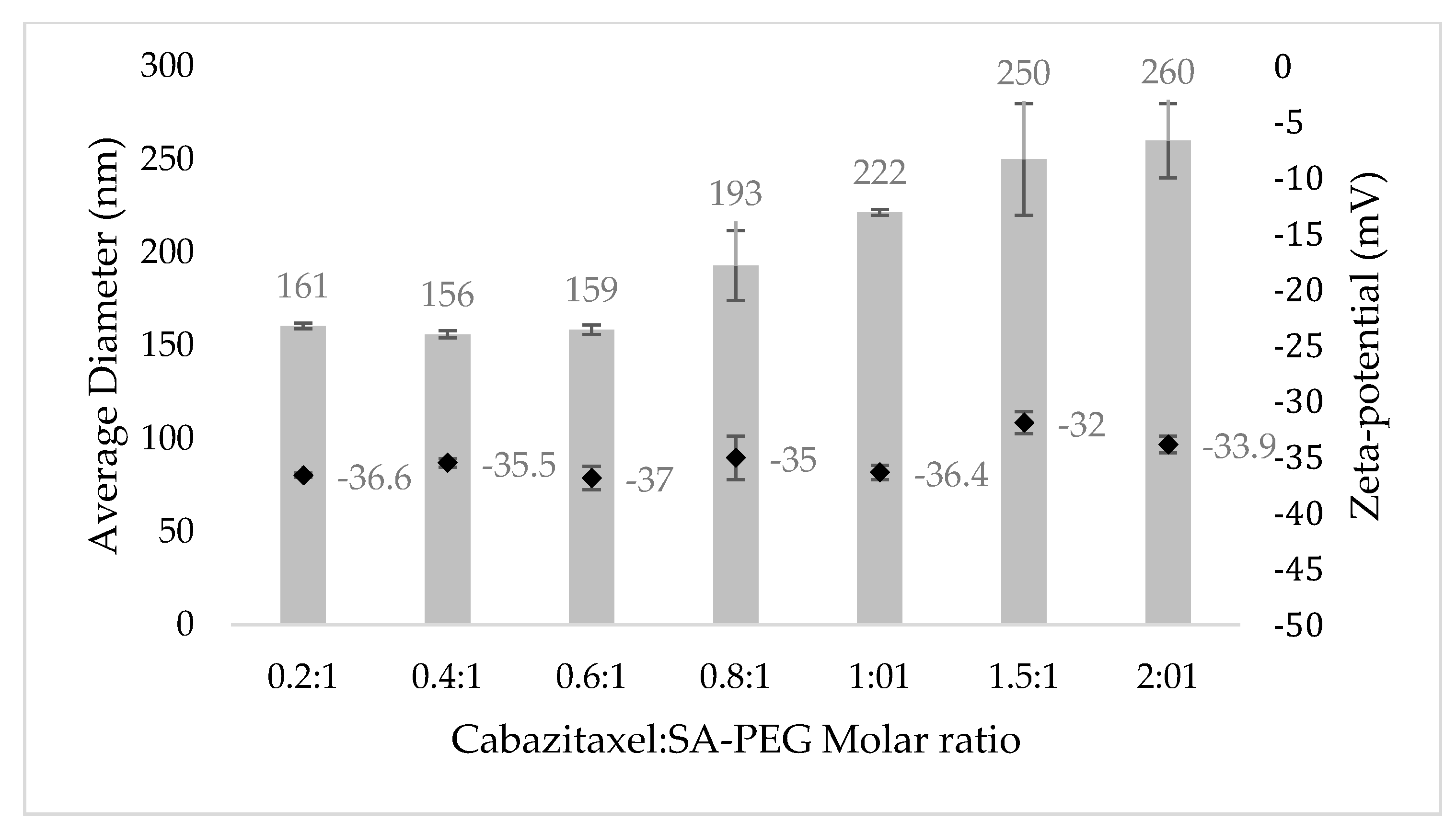
3.2. Physicochemical Characterization of NPs
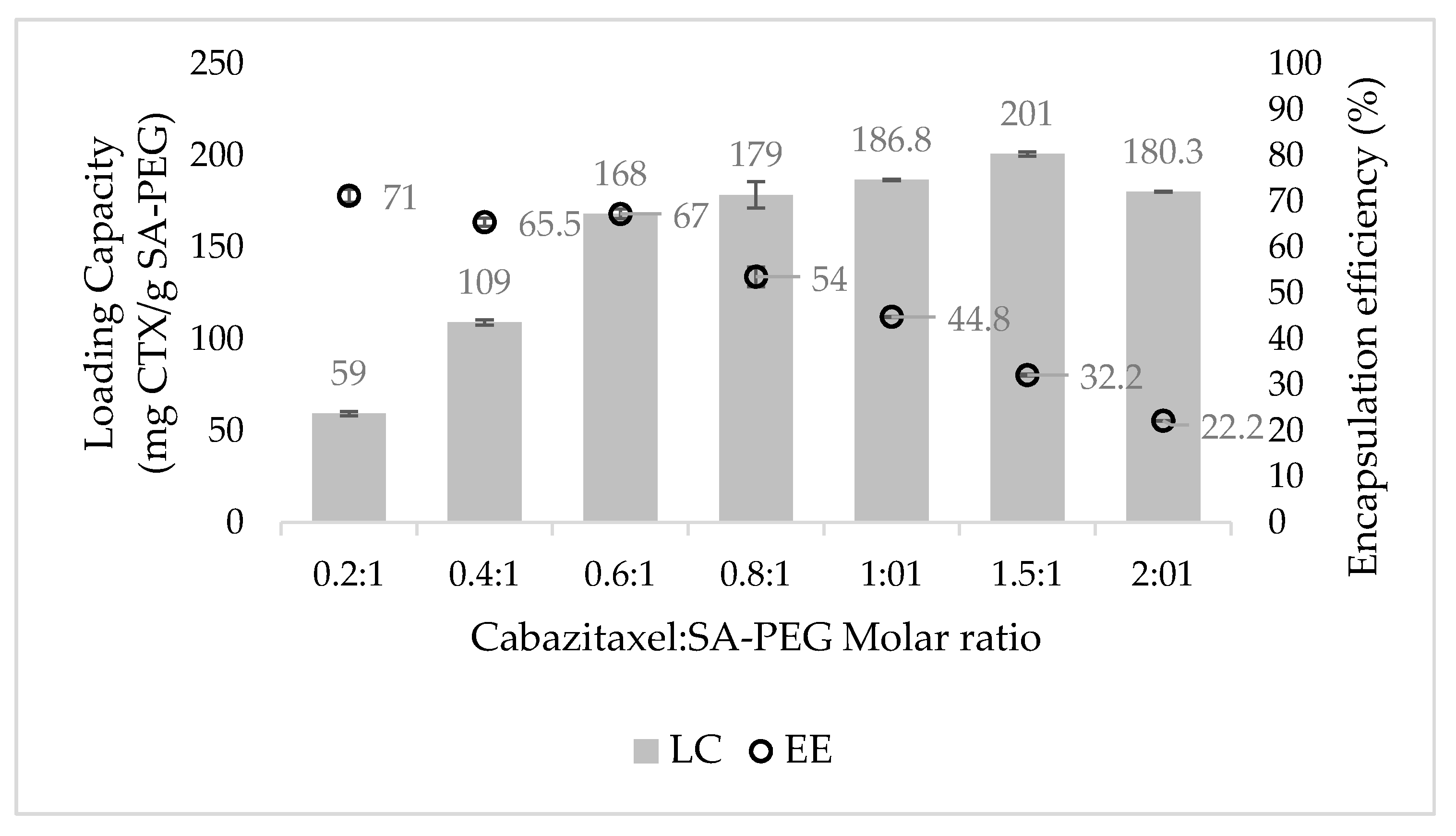
3.3. Drug Loading Capacity (LC) and Encapsulation Efficiency (EE)
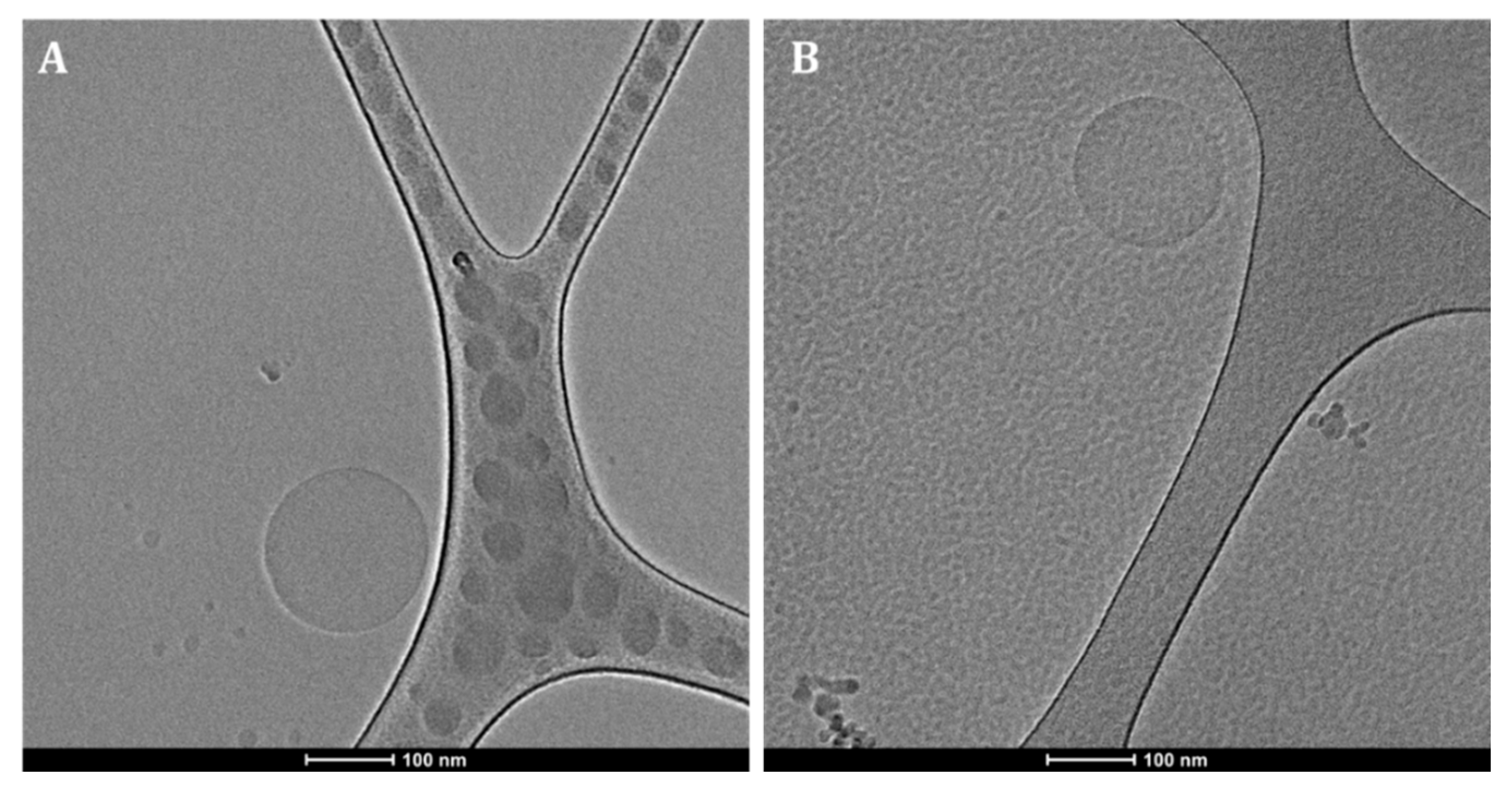
3.4. The Morphology of Drug-Loaded NPs
3.5. In Vitro Profile of CTX Release from the NPs
3.6. Selective Targeting and Internalization of NPs into LNCaP Target Cells
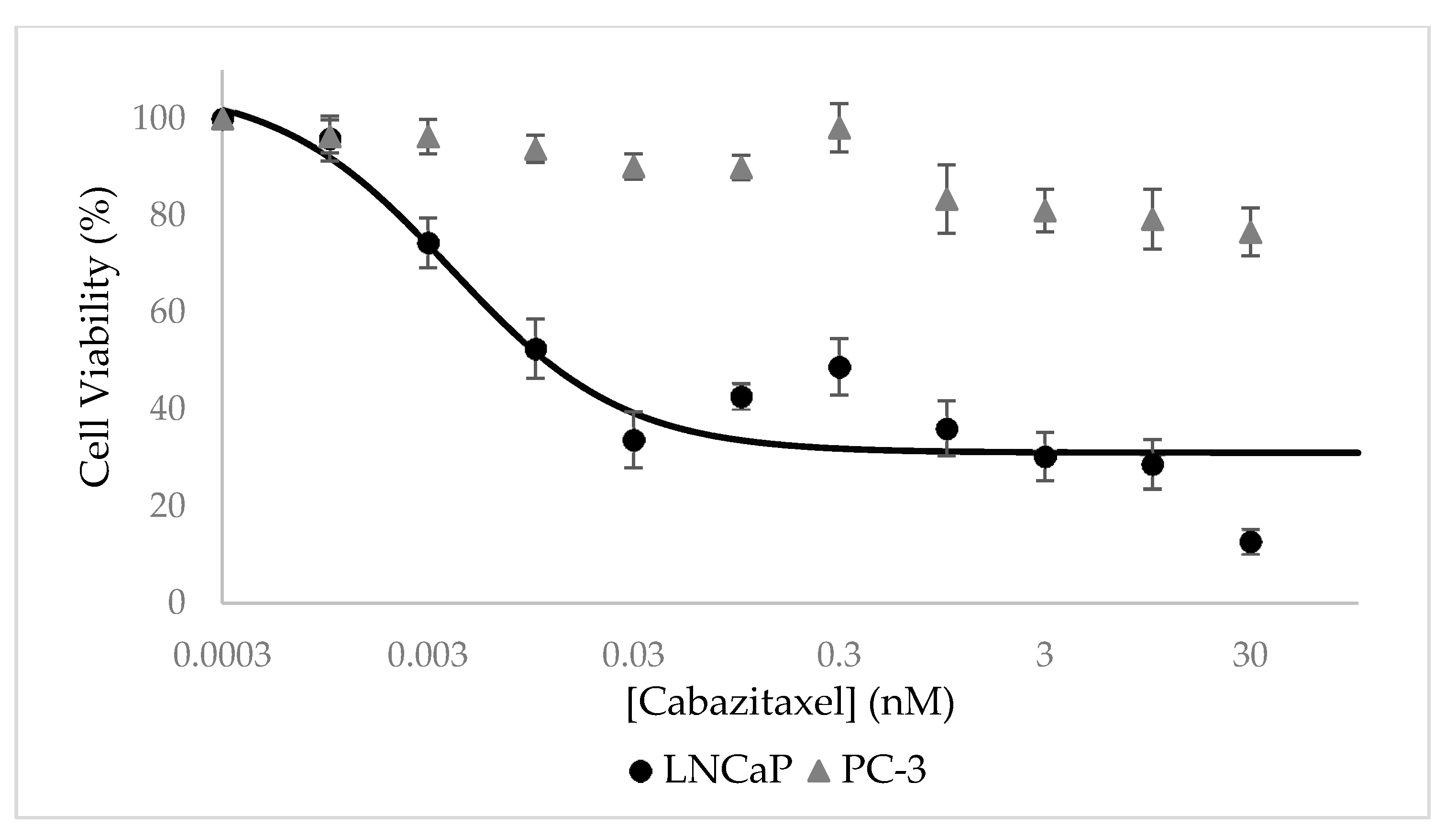
3.7. Selective Growth Inhibition of CTX-Loaded NPs In Vitro
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Qin, B.; Chen, Z.; Liu, H.; Barve, A.; Cheng, K. Discovery of PSMA-specific peptide ligands for targeted drug delivery. Int. J. Pharm. 2016, 513, 138–147. [Google Scholar] [CrossRef]
- Barve, A.; Jin, W.; Cheng, K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J. Control. Release 2014, 187, 118–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine modalities for prostate cancer treatment. Drug Resist. Updates 2021, 56, 100762. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Health 2018, 37, 288–295. [Google Scholar] [CrossRef]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Clegg, N.J.; Scher, H.I. Anti-androgens and androgen-depleting therapies in prostate cancer: New agents for an established target. Lancet Oncol. 2009, 10, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Gelmon, K. The taxoids: Paclitaxel and docetaxel. Lancet 1994, 344, 1267–1272. [Google Scholar] [CrossRef]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Haldar, S.; Chintapalli, J.; Croce, C.M. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996, 56, 1253–1255. [Google Scholar]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomedicine 2018, 13, 3921–3935. [Google Scholar] [CrossRef] [Green Version]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.-D.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Passive and active drug targeting: Drug delivery to tumors as an example. Handb. Exp. Pharmacol. 2010, 197, 3–53. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist. Updates 2017, 31, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Minner, S.; Wittmer, C.; Graefen, M.; Salomon, G.; Steuber, T.; Haese, A.; Huland, H.; Bokemeyer, C.; Yekebas, E.; Dierlamm, J.; et al. High level PSMA expression is associated with early psa recurrence in surgically treated prostate cancer. Prostate 2011, 71, 281–288. [Google Scholar] [CrossRef]
- Mayor, N.; Sathianathen, N.J.; Buteau, J.; Koschel, S.; Juanilla, M.A.; Kapoor, J.; Azad, A.; Hofman, M.S.; Murphy, D.G. Prostate-specific membrane antigen theranostics in advanced prostate cancer: An evolving option. BJU Int. 2020, 126, 525–535. [Google Scholar] [CrossRef]
- Jones, W.; Griffiths, K.; Barata, P.C.; Paller, C.J. PSMA theranostics: Review of the current status of PSMA-targeted imaging and radioligand therapy. Cancers. 2020, 12, 1367. [Google Scholar] [CrossRef]
- Niaz, M.O.; Sun, M.; Ramirez-Fort, M.; Niaz, M.J. Prostate-specific Membrane Antigen Based Antibody-Drug Conjugates for Metastatic Castration-resistance Prostate Cancer. Cureus 2020, 12, e7147. [Google Scholar] [CrossRef] [Green Version]
- Niaz, M.O.; Sun, M.; Ramirez-Fort, M.; Niaz, M.J. Review of Lutetium-177-labeled Anti-prostate-specific Membrane Antigen Monoclonal Antibody J591 for the Treatment of Metastatic Castration-resistant Prostate Cancer. Cureus 2020, 12, e7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iravani, A.; Violet, J.; Azad, A.; Hofman, M.S. Lutetium-177 prostate-specific membrane antigen (PSMA) theranostics: Practical nuances and intricacies. Prostate Cancer Prostatic Dis. 2020, 23, 38–52. [Google Scholar] [CrossRef]
- Chandran, S.S.; Ray, S.; Pomper, M.G.; Denmeade, S.R.; Mease, R.C. Prostate Specific Membrane Antigen (PSMA) Targeted Nanoparticles for Therapy of Prostate Cancer. U.S. Patent Application No. US9422234B2, 23 August 2016. [Google Scholar]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of Urea-Based Inhibitors as Active Site Probes of Glutamate Carboxypeptidase II: Efficacy as Analgesic Agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef]
- Harada, N.; Kimura, H.; Ono, M.; Saji, H. Preparation of asymmetric urea derivatives that target prostate-specific membrane antigen for SPECT imaging. J. Med. Chem. 2013, 56, 7890–7901. [Google Scholar] [CrossRef]
- Chandran, S.S.; Banerjee, S.R.; Mease, R.C.; Pomper, M.G.; Denmeade, S.R. Characterization of a targeted nanoparticle functionalized with a urea-based inhibitor of prostate-specific membrane antigen (PSMA). Cancer Biol. Ther. 2008, 7, 974–982. [Google Scholar] [CrossRef] [Green Version]
- Lepeltier, E.; Rijo, P.; Rizzolio, F.; Popovtzer, R.; Petrikaite, V.; Assaraf, Y.G.; Passirani, C. Nanomedicine to target multidrug resistant tumors. Drug Resist. Updates 2020, 52, 100704. [Google Scholar] [CrossRef]
- Long, L.; Assaraf, Y.G.; Lei, Z.N.; Peng, H.; Yang, L.; Chen, Z.S.; Ren, S. Genetic biomarkers of drug resistance: A compass of prognosis and targeted therapy in acute myeloid leukemia. Drug Resist. Updates 2020, 52, 100703. [Google Scholar] [CrossRef] [PubMed]
- Nassir, A.M.; Ibrahim, I.A.A.; Md, S.; Waris, M.; Tanuja; Ain, M.R.; Ahmad, I.; Shahzad, N. Surface functionalized folate targeted oleuropein nano-liposomes for prostate tumor targeting: In vitro and in vivo activity. Life Sci. 2019, 220, 136–146. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updates 2020, 49, 100670. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Wever, B.; Mazzaschi, G.; Assaraf, Y.G.; Rolfo, C.; Quaini, F.; Tiseo, M.; Giovannetti, E. Molecular basis and rationale for combining immune checkpoint inhibitors with chemotherapy in non-small cell lung cancer. Drug Resist. Updates 2019, 46, 100644. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Jiang, W.; Xia, J.; Xie, S.; Zou, R.; Pan, S.; Wang, Z.W.; Assaraf, Y.G.; Zhu, X. Long non-coding RNAs as a determinant of cancer drug resistance: Towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist. Updates 2020, 50, 100683. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Livney, Y.D.; Assaraf, Y.G. Rationally designed nanovehicles to overcome cancer chemoresistance. Adv. Drug Deliv. Rev. 2013, 65, 1716–1730. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef]
- Shapira, A.; Livney, Y.D.; Broxterman, H.J.; Assaraf, Y.G. Nanomedicine for targeted cancer therapy: Towards the overcoming of drug resistance. Drug Resist. Updates 2011, 14, 150–163. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2020, 41, 525–555. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.Á.; Rodríguez-Gascón, A.; Almeida, A.J.; Préat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Selvamuthukumar, S.; Velmurugan, R. Nanostructured Lipid Carriers: A potential drug carrier for cancer chemotherapy. Lipids Health Dis. 2012, 11, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured lipid carriers for delivery of chemotherapeutics: A review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [Green Version]
- Emami, J.; Rezazadeh, M.; Varshosaz, J.; Tabbakhian, M.; Aslani, A. Formulation of LDL Targeted Nanostructured Lipid Carriers Loaded with Paclitaxel: A Detailed Study of Preparation, Freeze Drying Condition, and In Vitro Cytotoxicity. J. Nanomater. 2012, 2012, 358782. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Pan, L.; Jiang, M.; Li, D.; Jin, L. Nanostructured lipid carriers enhance the bioavailability and brain cancer inhibitory efficacy of curcumin both in vitro and in vivo. Drug Deliv. 2016, 23, 1383–1392. [Google Scholar] [CrossRef]
- Jiang, H.; Geng, D.; Liu, H.; Li, Z.; Cao, J. Co-delivery of etoposide and curcumin by lipid nanoparticulate drug delivery system for the treatment of gastric tumors. Drug Deliv. 2016, 23, 3665–3673. [Google Scholar] [CrossRef] [Green Version]
- Bin, Z.; Yueying, Z.; Yu, D. Lung cancer gene therapy: Transferrin and hyaluronic acid dual ligand-decorated novel lipid carriers for targeted gene delivery. Oncol. Rep. 2017, 37, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.J.; Juang, L.W.; Lin, C.C. Stability and release performance of a series of pegylated copolymeric micelles. Pharm. Res. 2003, 20, 668–673. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, Z.; Yang, D.; Fan, W.; Wang, J.; Li, X.; Chen, X.; Wang, Q.; Song, X. Preparation and affinity identification of glutamic acid-urea small molecule analogs in prostate cancer. Oncol. Lett. 2016, 12, 1001–1006. [Google Scholar] [CrossRef] [Green Version]
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Keith, D.; Barone, C.; Joyal, J.L.; Zimmerman, C.N.; Kozikowski, A.P.; Barrett, J.A.; Eckelman, W.C.; et al. A Series of Halogenated Heterodimeric Inhibitors of Prostate Specific Membrane Antigen (PSMA) as Radiolabeled Probes for Targeting Prostate Cancer. J. Med. Chem. 2009, 52, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.G.T.; Hudoklin, S.S.; Kreft, M.E.; Kostevsek, N.; Stuart, M.C.A.; Al-Jamal, W.T. Intracellular Activation of a Prostate Specific Antigen-Cleavable Doxorubicin Prodrug: A Key Feature toward Prodrug-Nanomedicine Design. Mol. Pharm. 2019, 16, 1573–1585. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Qu, M.; Huang, S.; Fu, Y.; Yang, L.; He, S.; Li, L.; Zhang, Z.; Lin, Q.; Zhang, L. A novel α-enolase-targeted drug delivery system for high efficacy prostate cancer therapy. Nanoscale 2018, 10, 13673–13683. [Google Scholar] [CrossRef]
- Ikemoto, K.; Shimizu, K.; Ohashi, K.; Takeuchi, Y.; Shimizu, M.; Oku, N. Bauhinia purprea agglutinin-modified liposomes for human prostate cancer treatment. Cancer Sci. 2016, 107, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhou, Y.; Zhuang, Q.; Cui, L.; Xu, X.; Xu, R.; He, X. Anti-tumor effect of RGD modified PTX loaded liposome on prostatic cancer. Int. J. Clin. Exp. Med. 2015, 8, 12182–12191. [Google Scholar]
- Zhang, L.; Shan, X.; Meng, X.; Gu, T.; Lu, Q.; Zhang, J.; Chen, J.; Jiang, Q.; Ning, X. The first integrins β3-mediated cellular and nuclear targeting therapeutics for prostate cancer. Biomaterials 2019, 223, 119471. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.; Shmeeda, H.; Amitay, Y.; Ohana, P.; Kumar, S.; Gabizon, A. Targeting of folate-conjugated liposomes with co-entrapped drugs to prostate cancer cells via prostate-specific membrane antigen (PSMA). Nanomedicine 2018, 14, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- EP3799888A1—Liposomes Comprising Anti-Lox Antibody. Available online: https://www.patentguru.com/EP3799888A1 (accessed on 26 October 2021).
- Saroj, S.; Rajput, S.J. Etoposide encased folic acid adorned mesoporous silica nanoparticles as potent nanovehicles for enhanced prostate cancer therapy: Synthesis, characterization, cellular uptake and biodistribution. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1115–1130. [Google Scholar] [CrossRef] [Green Version]
- Tambe, P.; Kumar, P.; Paknikar, K.M.; Gajbhiye, V. Decapeptide functionalized targeted mesoporous silica nanoparticles with doxorubicin exhibit enhanced apoptotic effect in breast and prostate cancer cells. Int. J. Nanomed. 2018, 13, 7669–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero-Buceta, E.; Vidaurre-Agut, C.; Vera-Donoso, C.D.; Benlloch, J.M.; Moreno-Manzano, V.; Botella, P. PSMA-Targeted Mesoporous Silica Nanoparticles for Selective Intracellular Delivery of Docetaxel in Prostate Cancer Cells. ACS Omega 2019, 4, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Huo, S.; Zhang, X.; Liu, J.; Tan, A.; Li, S.; Jin, S.; Xue, X.; Zhao, Y.; Ji, T.; et al. Neuropilin-1-targeted gold nanoparticles enhance therapeutic efficacy of platinum(IV) drug for prostate cancer treatment. ACS Nano 2014, 8, 4205–4220. [Google Scholar] [CrossRef] [PubMed]
- Hrkach, J.; Von Hoff, D.; Ali, M.M.; Andrianova, E.; Auer, J.; Campbell, T.; De Witt, D.; Figa, M.; Figueiredo, M.; Horhota, A.; et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci. Transl. Med. 2012, 4, 128ra39. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Mita, M.M.; Ramanathan, R.K.; Weiss, G.J.; Mita, A.C.; Lorusso, P.M.; Burris, H.A.; Hart, L.L.; Low, S.C.; Parsons, D.M.; et al. Phase I study of PSMA-targeted docetaxel-containing nanoparticle BIND-014 in patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 3157–3163. [Google Scholar] [CrossRef] [Green Version]
- Autio, K.A.; Dreicer, R.; Anderson, J.; Garcia, J.A.; Alva, A.; Hart, L.L.; Milowsky, M.I.; Posadas, E.M.; Ryan, C.J.; Graf, R.P.; et al. Safety and Efficacy of BIND-014, a Docetaxel Nanoparticle Targeting Prostate-Specific Membrane Antigen for Patients with Metastatic Castration-Resistant Prostate Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Autio, K.A.; Garcia, J.A.; Alva, A.S.; Hart, L.L.; Milowsky, M.I.; Posadas, E.M.; Ryan, C.J.; Summa, J.M.; Youssoufian, H.; Scher, H.I.; et al. A phase 2 study of BIND-014 (PSMA-targeted docetaxel nanoparticle) administered to patients with chemotherapy-naïve metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2016, 34, 233. [Google Scholar] [CrossRef]
- Bharali, D.J.; Sudha, T.; Cui, H.; Mian, B.M.; Mousa, S.A. Anti-CD24 nano-targeted delivery of docetaxel for the treatment of prostate cancer. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tai, Z.; Gu, F.; Hu, C.; Zhu, Q.; Gao, S. Aptamer-mediated delivery of docetaxel to prostate cancer through polymeric nanoparticles for enhancement of antitumor efficacy. Eur. J. Pharm. Biopharm. 2016, 107, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Karandish, F.; Haldar, M.K.; You, S.; Brooks, A.E.; Brooks, B.D.; Guo, B.; Choi, Y.; Mallik, S. Prostate-Specific Membrane Antigen Targeted Polymersomes for Delivering Mocetinostat and Docetaxel to Prostate Cancer Cell Spheroids. ACS Omega 2016, 1, 952–962. [Google Scholar] [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288. [Google Scholar] [CrossRef]
- Yin, X.; Luo, L.; Li, W.; Yang, J.; Zhu, C.; Jiang, M.; Qin, B.; Yuan, X.; Yin, H.; Lu, Y.; et al. A cabazitaxel liposome for increased solubility, enhanced antitumor effect and reduced systemic toxicity. Asian J. Pharm. Sci. 2019, 14, 658–667. [Google Scholar] [CrossRef]
- Sun, B.; Straubinger, R.M.; Lovell, J.F. Current taxane formulations and emerging cabazitaxel delivery systems. Nano Res. 2018, 11, 5193–5218. [Google Scholar] [CrossRef]
- Engelberg, S.; Netzer, E.; Assaraf, Y.G.; Livney, Y.D. Selective eradication of human non-small cell lung cancer cells using aptamer-decorated nanoparticles harboring a cytotoxic drug cargo. Cell Death Dis. 2019, 10, 702. [Google Scholar] [CrossRef]
- Zhao, X.; Tang, D.; Yang, T.; Wang, C. Facile preparation of biocompatible nanostructured lipid carrier with ultra-small size as a tumor-penetration delivery system. Colloids Surf. B Biointerfaces 2018, 170, 355–363. [Google Scholar] [CrossRef]
- Ding, X.; Xu, X.; Zhao, Y.; Zhang, L.; Yu, Y.; Huang, F.; Yin, D.; Huang, H. Tumor targeted nanostructured lipid carrier co-delivering paclitaxel and indocyanine green for laser triggered synergetic therapy of cancer. RSC Adv. 2017, 7, 35086–35095. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; Pinho, S.C.; Souto, E.B.; Santana, M.H.A. Polymorphism, crystallinity and hydrophilic–lipophilic balance of stearic acid and stearic acid–capric/caprylic triglyceride matrices for production of stable nanoparticles. Colloids Surf. B Biointerfaces 2011, 86, 125–130. [Google Scholar] [CrossRef]
- Malhotra, M.; Tomaro-Duchesneau, C.; Prakash, S. Synthesis of TAT peptide-tagged PEGylated chitosan nanoparticles for siRNA delivery targeting neurodegenerative diseases. Biomaterials 2013, 34, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Delgado, A.V.; González-Caballero, F.; Hunter, R.J.; Koopal, L.K.; Lyklema, J. Measurement and Interpretation of Electrokinetic Phenomena (IUPAC Technical Report). Pure Appl. Chem. 2005, 77, 1753–1805. [Google Scholar] [CrossRef] [Green Version]
- Edelman, R.; Assaraf, Y.G.; Levitzky, I.; Shahar, T.; Livney, Y.D. Hyaluronic acid-serum albumin conjugate-based nanoparticles for targeted cancer therapy. Oncotarget 2017, 8, 24337–24353. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Chen, L.; Gu, J.; Ren, X.; Wei, Z.; Luo, J.; Chen, Y.; Jiang, X.; Sha, X.; Fang, X. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ε-caprolactone) nanoparticles: In vitro and in vivo evaluation. Int. J. Pharm. 2010, 402, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Lee, R.J.; Sun, Y.; Cai, G.; Wang, J.; Wang, M.; Lu, J.; Meng, Q.; Teng, L.; Wang, D.; et al. Cabazitaxel-loaded human serum albumin nanoparticles as a therapeutic agent against prostate cancer. Int. J. Nanomedicine 2016, 11, 3451–3459. [Google Scholar] [CrossRef] [Green Version]
- Aravind, A.; Varghese, S.H.; Veeranarayanan, S.; Mathew, A.; Nagaoka, Y.; Iwai, S.; Fukuda, T.; Hasumura, T.; Yoshida, Y.; Maekawa, T.; et al. Aptamer-labeled PLGA nanoparticles for targeting cancer cells. Cancer Nanotechnol. 2012, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Engelberg, S.; Lin, Y.; Assaraf, Y.G.; Livney, Y.D. Targeted nanoparticles harboring jasmine-oil-entrapped paclitaxel for elimination of lung cancer cells. Int. J. Mol. Sci. 2021, 22, 1019. [Google Scholar] [CrossRef]
- Qian, W.; Murakami, M.; Ichikawa, Y.; Che, Y. Highly efficient and controllable PEGylation of gold nanoparticles prepared by femtosecond laser ablation in water. J. Phys. Chem. C 2011, 115, 23293–23298. [Google Scholar] [CrossRef]
- Standard, A. ASTM D4187-82: Zeta Potential of Colloids in Water and Waste Water; American Society for Testing and Materials: West Conshohocken, PA, USA, 1985. [Google Scholar]
- Champion, J.A.; Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaari, Z.; Da Silva, D.; Zinger, A.; Goldman, E.; Kajal, A.; Tshuva, R.; Barak, E.; Dahan, N.; Hershkovitz, D.; Goldfeder, M.; et al. Theranostic barcoded nanoparticles for personalized cancer medicine. Nat. Commun. 2016, 7, 13325. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yung, L.Y.L. Selectivity of folate conjugated polymer micelles against different tumor cells. Int. J. Pharm. 2008, 349, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res. 2010, 62, 90–99. [Google Scholar] [CrossRef]
- Harush-Frenkel, O.; Debotton, N.; Benita, S.; Altschuler, Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem. Biophys. Res. Commun. 2007, 353, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Dreifuss, T.; Ben-Gal, T.-S.; Shamalov, K.; Weiss, A.; Jacob, A.; Sadan, T.; Motiei, M.; Popovtzer, R. Uptake mechanism of metabolic-targeted gold nanoparticles. Nanomedicine 2018, 13, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Han, X.; Kawaguchi, T.; Babasaki, T.; Goto, K.; Inoue, S.; Hayashi, T.; Teishima, J.; Shiota, M.; Yasui, W.; et al. TUBB3 Reverses Resistance to Docetaxel and Cabazitaxel in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Nakouzi, N.; Le Moulec, S.; Albigès, L.; Wang, C.; Beuzeboc, P.; Gross-Goupil, M.; De La Motte Rouge, T.; Guillot, A.; Gajda, D.; Massard, C.; et al. Cabazitaxel Remains Active in Patients Progressing After Docetaxel Followed by Novel Androgen Receptor Pathway Targeted Therapies. Eur. Urol. 2015, 68, 228–235. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, L.; Assaraf, Y.G.; Livney, Y.D. Novel Selectively Targeted Multifunctional Nanostructured Lipid Carriers for Prostate Cancer Treatment. Pharmaceutics 2022, 14, 88. https://doi.org/10.3390/pharmaceutics14010088
Cohen L, Assaraf YG, Livney YD. Novel Selectively Targeted Multifunctional Nanostructured Lipid Carriers for Prostate Cancer Treatment. Pharmaceutics. 2022; 14(1):88. https://doi.org/10.3390/pharmaceutics14010088
Chicago/Turabian StyleCohen, Lital, Yehuda G. Assaraf, and Yoav D. Livney. 2022. "Novel Selectively Targeted Multifunctional Nanostructured Lipid Carriers for Prostate Cancer Treatment" Pharmaceutics 14, no. 1: 88. https://doi.org/10.3390/pharmaceutics14010088