
Bacteriomimetic Liposomes Improve Antibiotic Activity of a Novel Energy-Coupling Factor Transporter Inhibitor

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Liposome Preparation and Drug Encapsulation
2.2. Characterization of Liposomes
2.3. HPLC-MS
2.4. Calculation of Encapsulation Efficiency (EE)
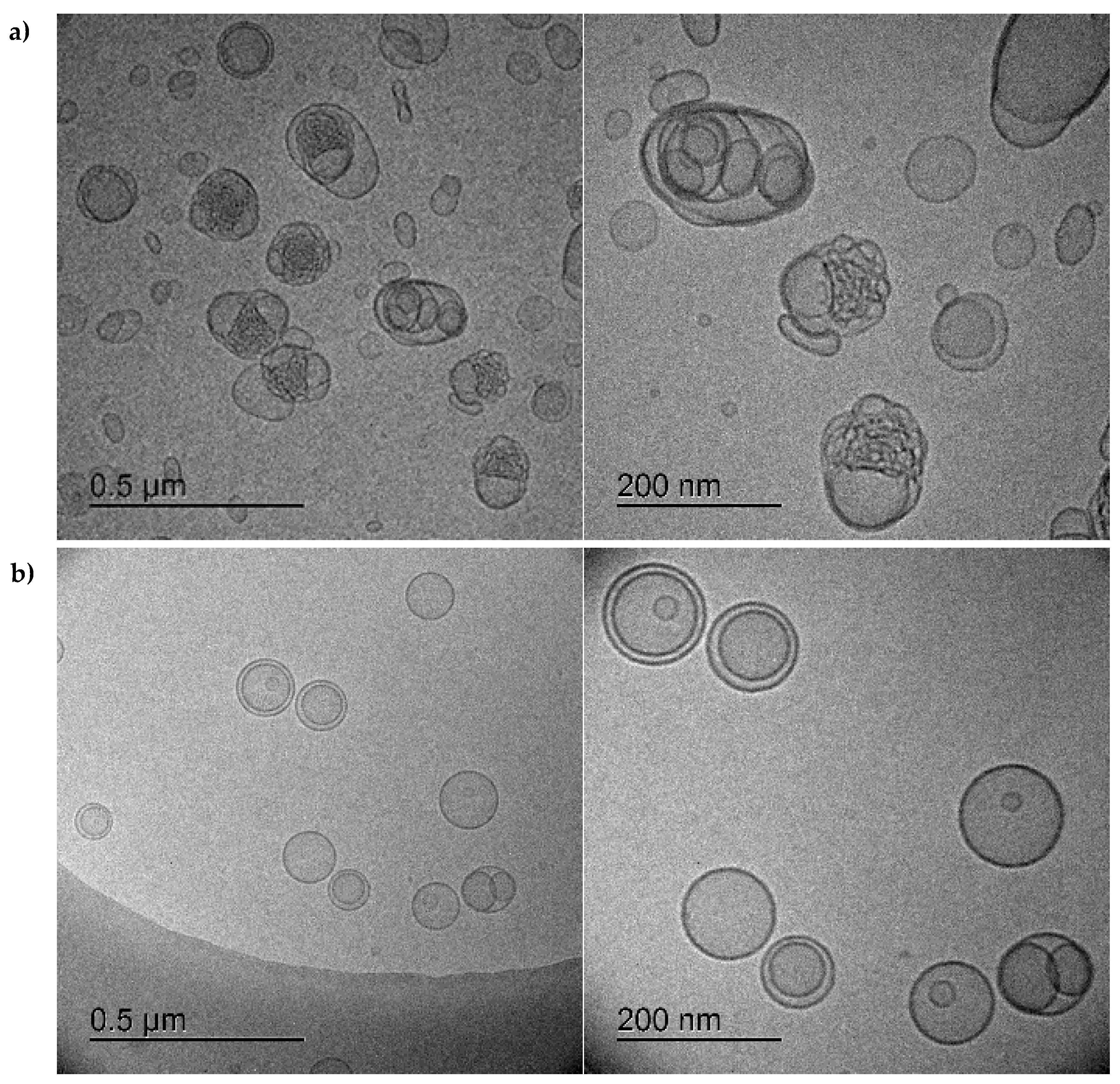
2.5. Cryo-TEM
2.6. ECF Transporter Inhibition Assay
2.7. Bacterial Culture, Bacterial Growth Assays and Viability Assay
2.8. B. subtilis 168 Biofilm for SEM & Fluorescence Imaging
2.9. Statistical Analysis
3. Results & Discussion
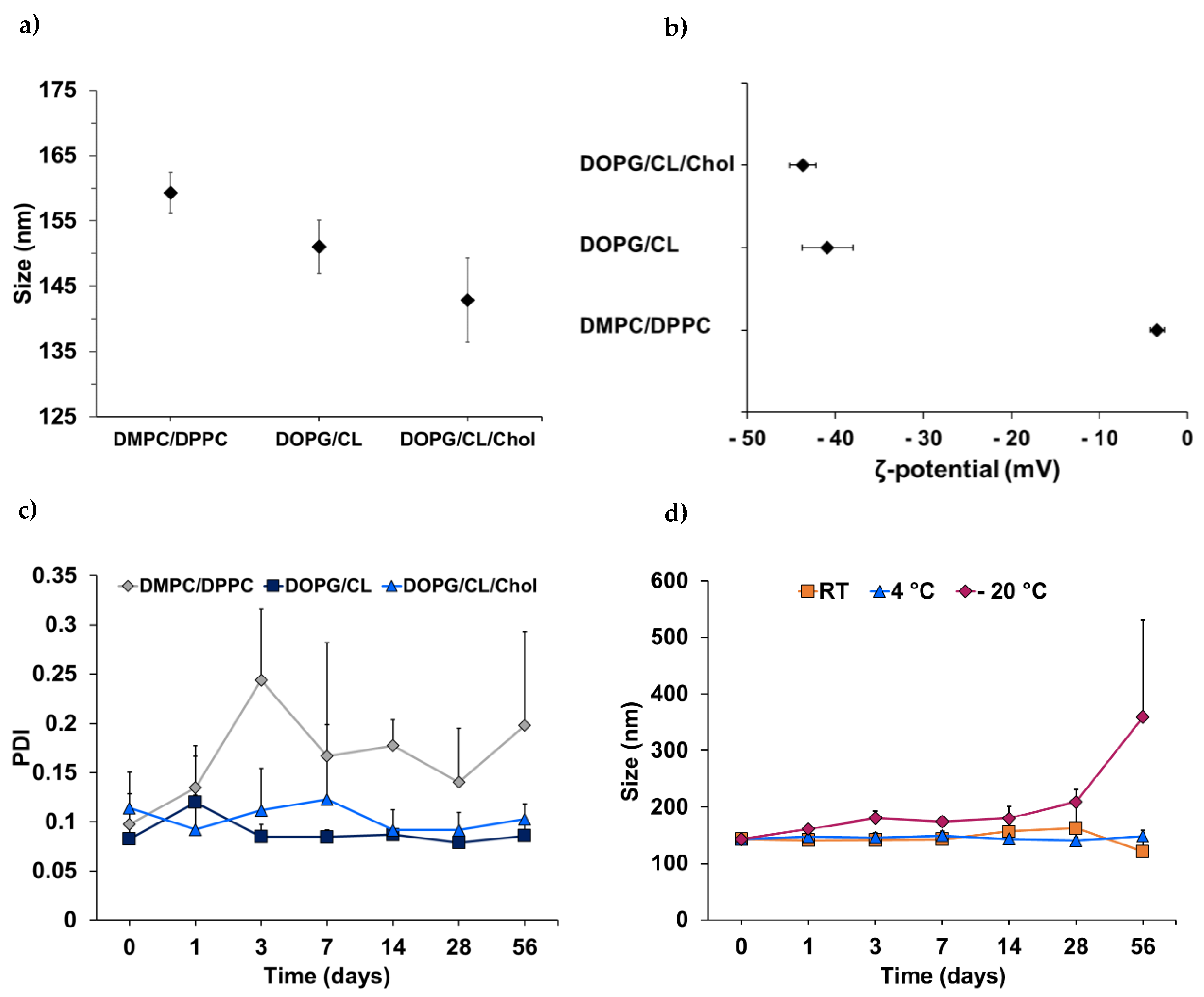
3.1. Characterization of Bacteriomimetic Liposomes
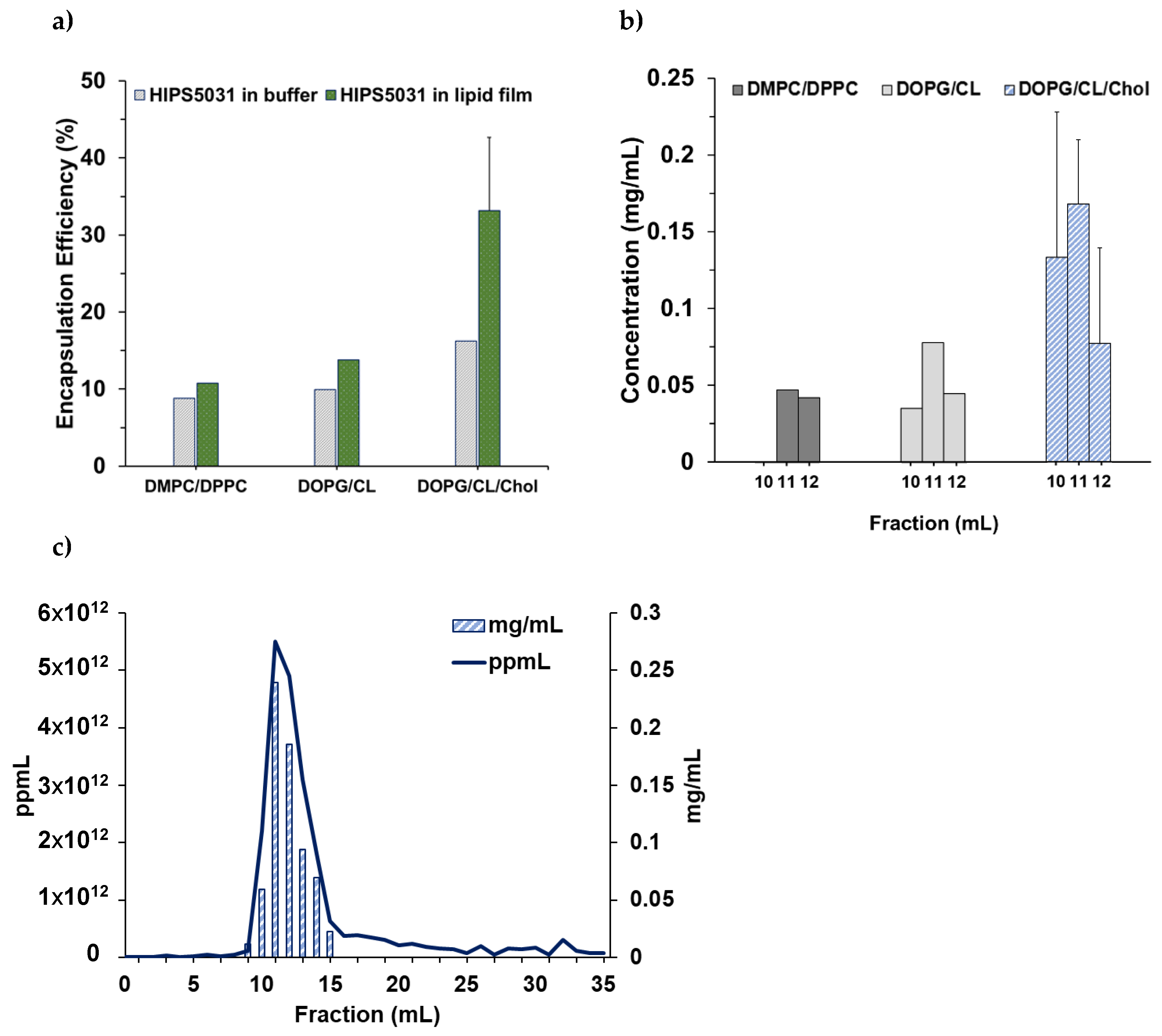
3.2. Loading of Liposomes with HIPS5031
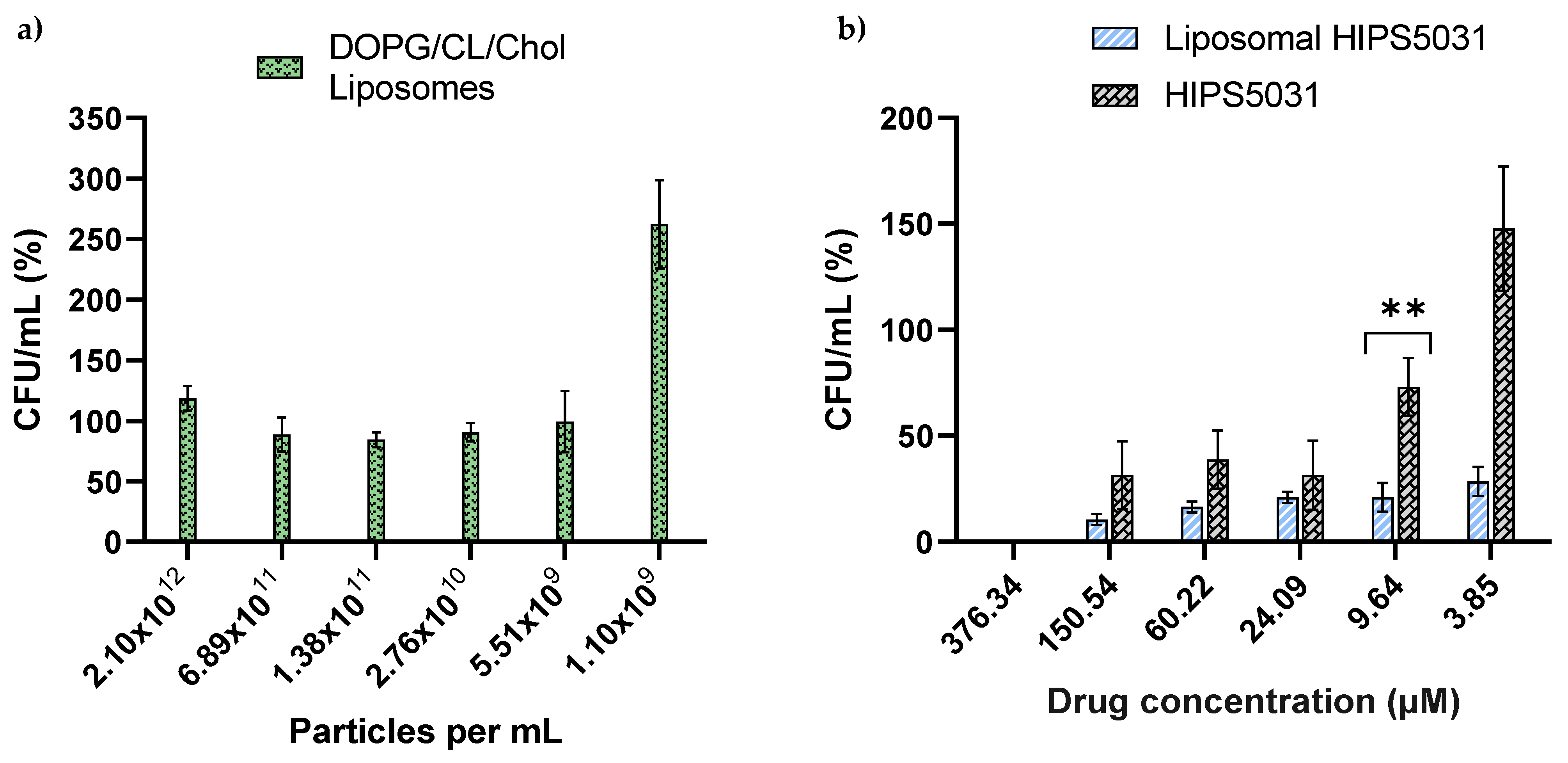
3.3. ECF Transporter Assay
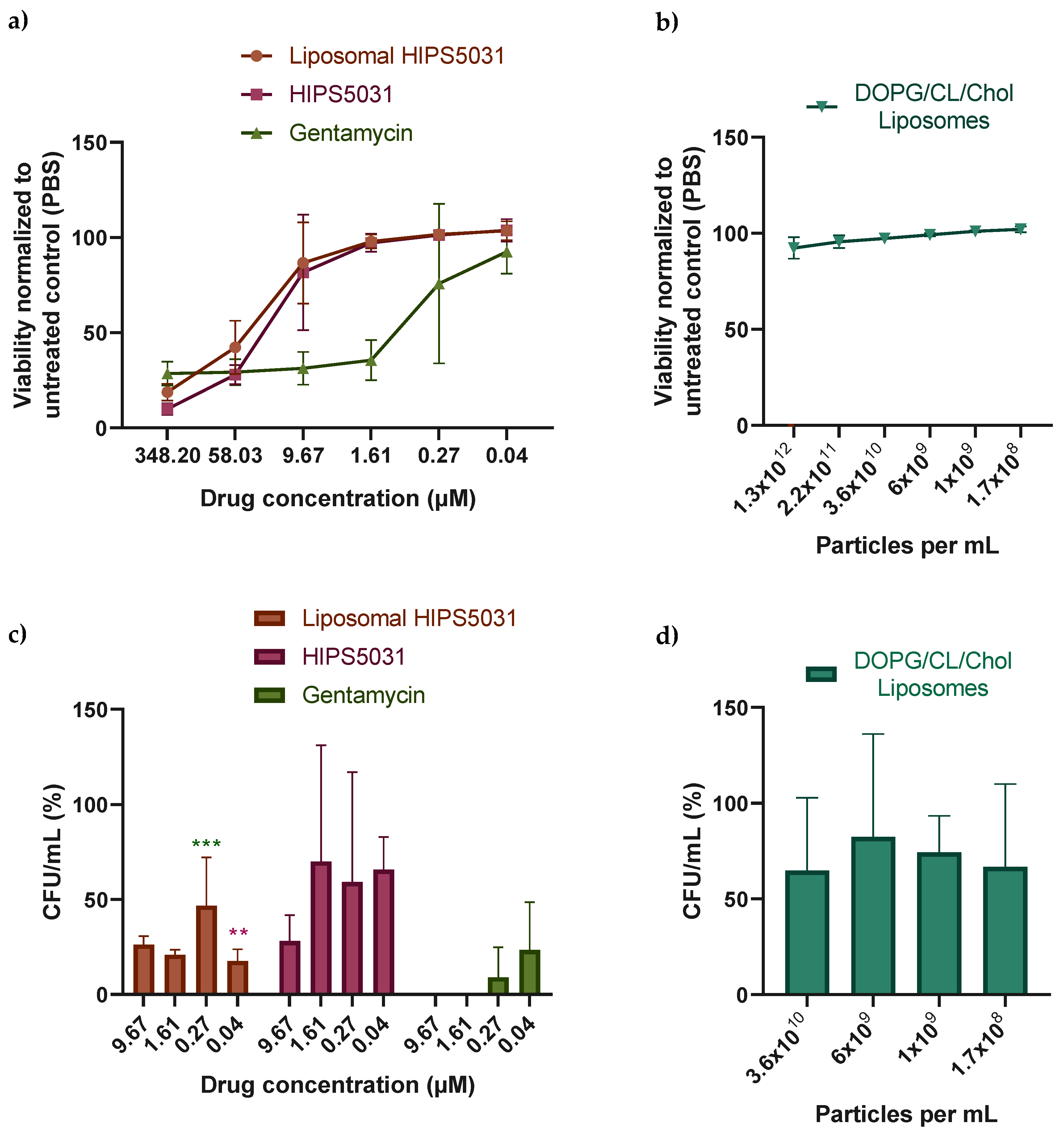
3.4. Antibacterial Effect of Liposomal HIPS5031
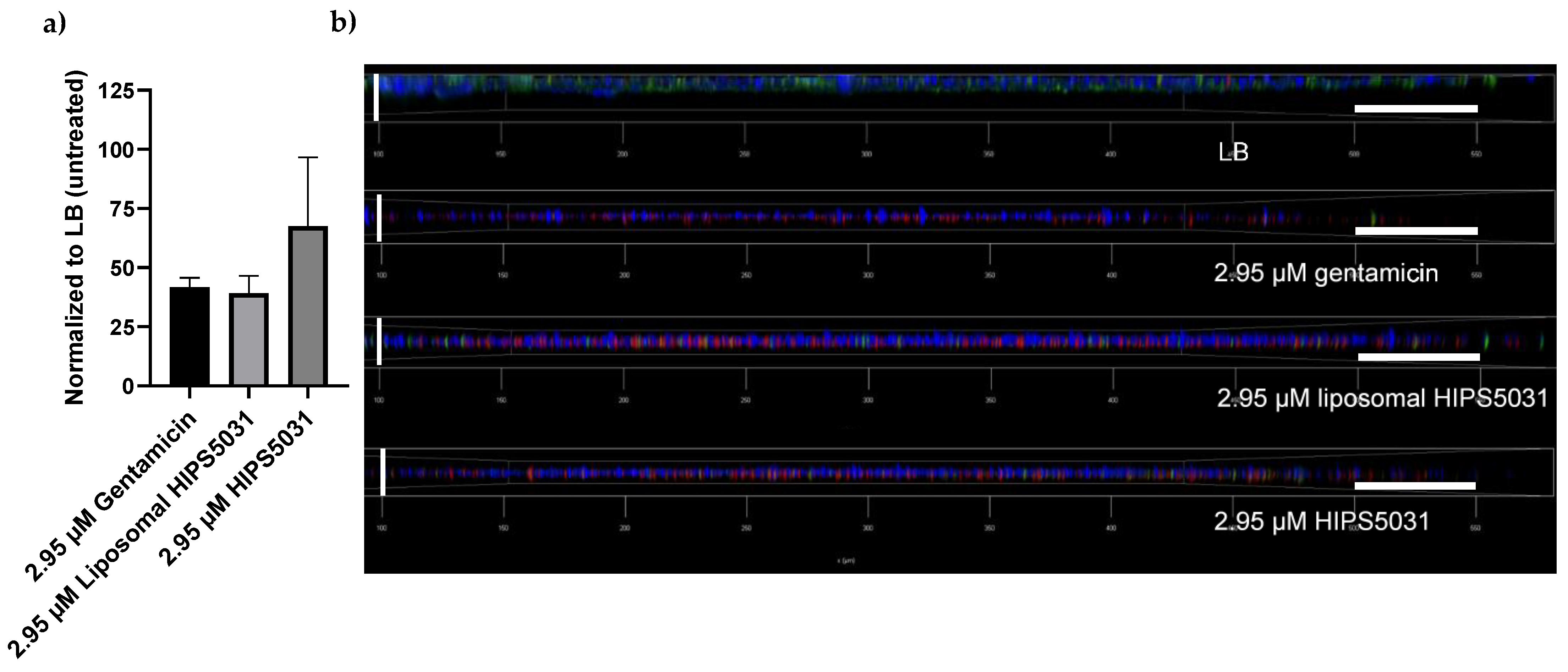
3.5. Anti-Biofilm Effects of Liposomal HIPS5031
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Cassini, A.; Hogberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Insfect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Antimicrobial Resistance in the EU/EEA (EARS-Net)-Annual Epidemiological Report for 2019. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/surveillance-antimicrobial-resistance-Europe-2019.pdf (accessed on 15 December 2021).
- Chambers, H.F.; Deleo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Bousis, S.; Setyawati, I.; Diamanti, E.; Slotboom, D.J.; Hirsch, A.K.H. Energy-Coupling Factor Transporters as Novel Antimicrobial Targets. Adv. Ther. 2019, 2, 1800066. [Google Scholar] [CrossRef] [Green Version]
- Slotboom, D.J. Structural and mechanistic insights into prokaryotic energy-coupling factor transporters. Nat. Rev. Microbiol. 2014, 12, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Qi, X.; Hong, S.; Xu, K.; He, F.; Zhang, M.; Chen, J.; Chao, D.; Zhao, W.; Li, D.; et al. Structure and mechanism of a group-I cobalt energy coupling factor transporter. Cell Res. 2017, 27, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the biopharmaceutics classification system. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Day, T.; Read, A.F. Does High-Dose Antimicrobial Chemotherapy Prevent the Evolution of Resistance? PLoS Comput. Biol. 2016, 12, e1004689. [Google Scholar] [CrossRef]
- Vasseur, M.V.; Laurentie, M.; Rolland, J.G.; Perrin-Guyomard, A.; Henri, J.; Ferran, A.A.; Toutain, P.L.; Bousquet-Melou, A. Low or high doses of cefquinome targeting low or high bacterial inocula cure Klebsiella pneumoniae lung infections but differentially impact the levels of antibiotic resistance in fecal flora. Antimicrob. Agents Chemother. 2014, 58, 1744–1748. [Google Scholar] [CrossRef] [Green Version]
- Diamanti, E.; Setyawati, I.; Bousis, S.; Souza, P.C.T.; Mojas, L.; Swier, L.; Haupenthal, J.; Gibson, P.; Volz, C.; Stanek, W.; et al. Targeting the energy-coupling factor (ECF) transporters: Identification of new tool compounds. ChemRXiv 2021. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez Gomez, A.; Hosseinidoust, Z. Liposomes for Antibiotic Encapsulation and Delivery. ACS Infect. Dis. 2020, 6, 896–908. [Google Scholar] [CrossRef]
- Ferreira, M.; Ogren, M.; Dias, J.N.R.; Silva, M.; Gil, S.; Tavares, L.; Aires-Da-Silva, F.; Gaspar, M.M.; Aguiar, S.I. Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance. Molecules 2021, 26, 2047. [Google Scholar] [CrossRef]
- Lee, Y.; Thompson, D.H. Stimuli-responsive liposomes for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1450. [Google Scholar] [CrossRef] [PubMed]
- Su, F.-Y.; Chen, J.; Son, H.-N.; Kelly, A.M.; Convertine, A.J.; West, T.E.; Skerrett, S.J.; Ratner, D.M.; Stayton, P.S. Polymer-augmented liposomes enhancing antibiotic delivery against intracellular infections. Biomater. Sci. 2018, 6, 1976–1985. [Google Scholar] [CrossRef]
- Goes, A.; Fuhrmann, G. Biogenic and Biomimetic Carriers as Versatile Transporters To Treat Infections. ACS Infect. Dis. 2018, 4, 881–892. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A. Liposomes as delivery systems for antibiotics. Int. J. Pharm. 2010, 387, 187–198. [Google Scholar] [CrossRef]
- Khameneh, B.; Diab, R.; Ghazvini, K.; Fazly Bazzaz, B.S. Breakthroughs in bacterial resistance mechanisms and the potential ways to combat them. Microb. Pathog. 2016, 95, 32–42. [Google Scholar] [CrossRef]
- Rukavina, Z.; Vanić, Ž. Current Trends in Development of Liposomes for Targeting Bacterial Biofilms. Pharmaceutics 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Alhariri, M.; Azghani, A.; Omri, A. Liposomal antibiotics for the treatment of infectious diseases. Expert Opin. Drug Deliv. 2013, 10, 1515–1532. [Google Scholar] [CrossRef] [PubMed]
- Diab, R.; Khameneh, B.; Joubert, O.; Duval, R. Insights in Nanoparticle-Bacterium Interactions: New Frontiers to Bypass Bacterial Resistance to Antibiotics. Curr. Pharm. Des. 2015, 21, 4095–4105. [Google Scholar] [CrossRef]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifat, N.; Fournier, J.B.; Angelova, M.I.; Puff, N. Lipid packing variations induced by pH in cardiolipin-containing bilayers: The driving force for the cristae-like shape instability. Biochim. Biophys. Acta 2011, 1808, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.N.; McElhaney, R.N. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta 2009, 1788, 2069–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomšiè, N.; Babnik, B.; Lombardo, D.; MavčIč, B.; Kandušer, M.; Iglič, A.; Kralj-Iglič, V. Shape and Size of Giant Unilamellar Phospholipid Vesicles Containing Cardiolipin. J. Chem. Inf. Modeling 2005, 45, 1676–1679. [Google Scholar] [CrossRef] [Green Version]
- Clejan, S.; Krulwich, T.A.; Mondrus, K.R.; Seto-Young, D. Membrane lipid composition of obligately and facultatively alkalophilic strains of Bacillus spp. J. Bacteriol. 1986, 168, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Goes, A.; Lapuhs, P.; Kuhn, T.; Schulz, E.; Richter, R.; Panter, F.; Dahlem, C.; Koch, M.; Garcia, R.; Kiemer, A.K.; et al. Myxobacteria-Derived Outer Membrane Vesicles: Potential Applicability Against Intracellular Infections. Cells 2020, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Belin, B.J.; Busset, N.; Giraud, E.; Molinaro, A.; Silipo, A.; Newman, D.K. Hopanoid lipids: From membranes to plant–bacteria interactions. Nat. Rev. Microbiol. 2018, 16, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Gallegos-Monterrosa, R.; Mhatre, E.; Kovács, Á.T. Specific Bacillus subtilis 168 variants form biofilms on nutrient-rich medium. Microbiology 2016, 162, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Vlamakis, H.; Chai, Y.; Beauregard, P.; Losick, R.; Kolter, R. Sticking together: Building a biofilm the Bacillus subtilis way. Nat. Rev. Microbiol. 2013, 11, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Bousis, S.; Winkler, S.; Haupenthal, J.; Fulco, F.; Diamanti, E.; Hirsch, A.K.H. An efficient way to screen inhibitors of energy-coupling factor (ECF) transporters in bacteria uptake assay. ChemRXiv 2021. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Goes, A.; Vidakovic, L.; Drescher, K.; Fuhrmann, G. Interaction of myxobacteria-derived outer membrane vesicles with biofilms: Antiadhesive and antibacterial effects. Nanoscale 2021, 13, 14287–14296. [Google Scholar] [CrossRef]
- Branda, S.S.; Gonzalez-Pastor, J.E.; Ben-Yehuda, S.; Losick, R.; Kolter, R. Fruiting body formation by Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2001, 98, 11621–11626. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; van der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Lipid-Based Antimicrobial Delivery-Systems for the Treatment of Bacterial Infections. Front. Chem. 2019, 7, 872. [Google Scholar] [CrossRef]
- Berbel Manaia, E.; Paiva Abuçafy, M.; Chiari-Andréo, B.G.; Lallo Silva, B.; Oshiro-Júnior, J.A.; Chiavacci, L. Physicochemical characterization of drug nanocarriers. Int. J. Nanomed. 2017, 12, 4991–5011. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Omri, A. The Effect of Different Lipid Components on the In Vitro Stability and Release Kinetics of Liposome Formulations. Drug Deliv. 2004, 11, 33–39. [Google Scholar] [CrossRef]
- Duplessis, J.; Ramachandran, C.; Weiner, N.; Muller, D. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Int. J. Pharm. 1996, 127, 273–278. [Google Scholar] [CrossRef]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef] [Green Version]
- Briuglia, M.-L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Raffy, S.; Teissié, J. Control of lipid membrane stability by cholesterol content. Biophys. J. 1999, 76, 2072–2080. [Google Scholar] [CrossRef] [Green Version]
- Gregoriadis, G. Overview of liposomes. J. Antimicrob. Chemother. 1991, 28 (Suppl. B), 39–48. [Google Scholar] [CrossRef]
- Pietzyk, B.; Henschke, K. Degradation of phosphatidylcholine in liposomes containing carboplatin in dependence on composition and storage conditions. Int. J. Pharm. 2000, 196, 215–218. [Google Scholar] [CrossRef]
- Buffo, F.; Sierra, M.; Pedroni, V.; Morini, M. Lipidic nanoparticules: A model function to predict the transition temperature of DPPC-DMPC mixtures. Adv. Mater. Sci. 2017, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Furneri, P.M.; Fresta, M.; Puglisi, G.; Tempera, G. Ofloxacin-Loaded Liposomes: In Vitro Activity and Drug Accumulation in Bacteria. Antimicrob. Agents Chemother. 2000, 44, 2458–2464. [Google Scholar] [CrossRef] [Green Version]
- Montizaan, D.; Yang, K.; Reker-Smit, C.; Salvati, A. Comparison of the uptake mechanisms of zwitterionic and negatively charged liposomes by HeLa cells. Nanomedicine 2020, 30, 102300. [Google Scholar] [CrossRef]
- Bharatiya, B.; Wang, G.; Rogers, S.E.; Pedersen, J.S.; Mann, S.; Briscoe, W.H. Mixed liposomes containing gram-positive bacteria lipids: Lipoteichoic acid (LTA) induced structural changes. Colloids Surf. B Biointerfaces 2021, 199, 111551. [Google Scholar] [CrossRef]
- Desvaux, M.L.; Dumas, E.; Chafsey, I.; Hebraud, M. Protein cell surface display in Gram-positive bacteria: From single protein to macromolecular protein structure. FEMS Microbiol. Lett. 2006, 256, 1–15. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Gao, W.; Thamphiwatana, S.; Angsantikul, P.; Zhang, L. Nanoparticle approaches against bacterial infections. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 532–547. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Verma, M.; Karandikar, P.; Furin, J.; Langer, R.; Traverso, G. Nanotechnology approaches for global infectious diseases. Nat. Nanotechnol. 2021, 16, 369–384. [Google Scholar] [CrossRef]
- Kent, B.; Garvey, C.J.; Cookson, D.; Bryant, G. The inverse hexagonal-inverse ribbon-lamellar gel phase transition sequence in low hydration DOPC:DOPE phospholipid mixtures. Chem. Phys. Lipids 2009, 157, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; McNeil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Gubernator, J.; Dorotkiewicz-Jach, A.; Doroszkiewicz, W.; Kozubek, A. A comparison of the in vitro antimicrobial activity of liposomes containing meropenem and gentamicin. Cell. Mol. Biol. Lett. 2006, 11, 360–375. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Aiello, S.; Pagano, L.; Ceccacci, F.; Simonis, B.; Sennato, S.; Bugli, F.; Mancini, G. Mannosyl, glucosyl or galactosyl liposomes to improve resveratrol efficacy against Methicillin Resistant Staphylococcus aureus biofilm. Colloids Surf. A: Physicochem. Eng. Asp. 2021, 617, 126321. [Google Scholar] [CrossRef]
- Sande, L.; Sanchez, M.; Montes, J.; Wolf, A.J.; Morgan, M.A.; Omri, A.; Liu, G.Y. Liposomal encapsulation of vancomycin improves killing of methicillin-resistant Staphylococcus aureus in a murine infection model. J. Antimicrob. Chemother. 2012, 67, 2191–2194. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, L.N.D.M.; De Paula, E.; Rossi, D.A.; Monteiro, G.P.; Júnior, E.C.V.; Silva, R.R.; Franco, R.R.; Espíndola, F.S.; Goulart, L.R.; Fonseca, B.B. Hybrid Pectin-Liposome Formulation against Multi-Resistant Bacterial Strains. Pharmaceutics 2020, 12, 769. [Google Scholar] [CrossRef]
- Makabenta, J.M.V.; Nabawy, A.; Li, C.-H.; Schmidt-Malan, S.; Patel, R.; Rotello, V.M. Nanomaterial-based therapeutics for antibi-otic-resistant bacterial infections. Nat. Rev. Microbiol. 2021, 19, 23–36. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drost, M.; Diamanti, E.; Fuhrmann, K.; Goes, A.; Shams, A.; Haupenthal, J.; Koch, M.; Hirsch, A.K.H.; Fuhrmann, G. Bacteriomimetic Liposomes Improve Antibiotic Activity of a Novel Energy-Coupling Factor Transporter Inhibitor. Pharmaceutics 2022, 14, 4. https://doi.org/10.3390/pharmaceutics14010004
Drost M, Diamanti E, Fuhrmann K, Goes A, Shams A, Haupenthal J, Koch M, Hirsch AKH, Fuhrmann G. Bacteriomimetic Liposomes Improve Antibiotic Activity of a Novel Energy-Coupling Factor Transporter Inhibitor. Pharmaceutics. 2022; 14(1):4. https://doi.org/10.3390/pharmaceutics14010004
Chicago/Turabian StyleDrost, Menka, Eleonora Diamanti, Kathrin Fuhrmann, Adriely Goes, Atanaz Shams, Jörg Haupenthal, Marcus Koch, Anna K. H. Hirsch, and Gregor Fuhrmann. 2022. "Bacteriomimetic Liposomes Improve Antibiotic Activity of a Novel Energy-Coupling Factor Transporter Inhibitor" Pharmaceutics 14, no. 1: 4. https://doi.org/10.3390/pharmaceutics14010004