
Microencapsulated Chitosan-Based Nanocapsules: A New Platform for Pulmonary Gene Delivery

 ,
,
Abstract
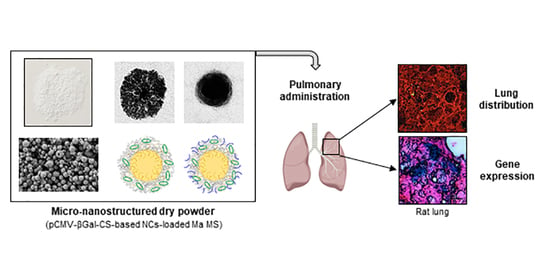
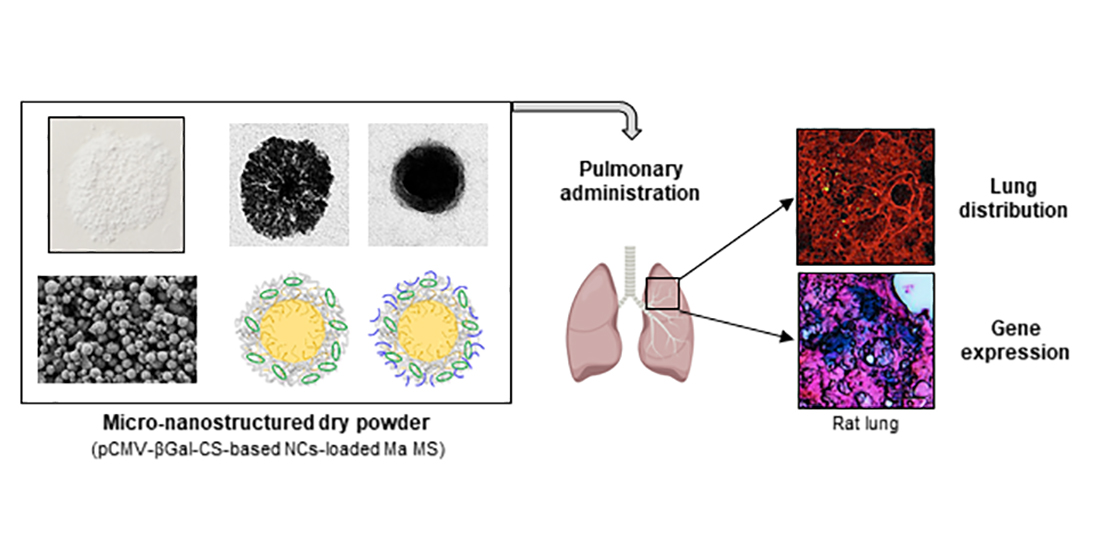
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Production of pCMV-βGal
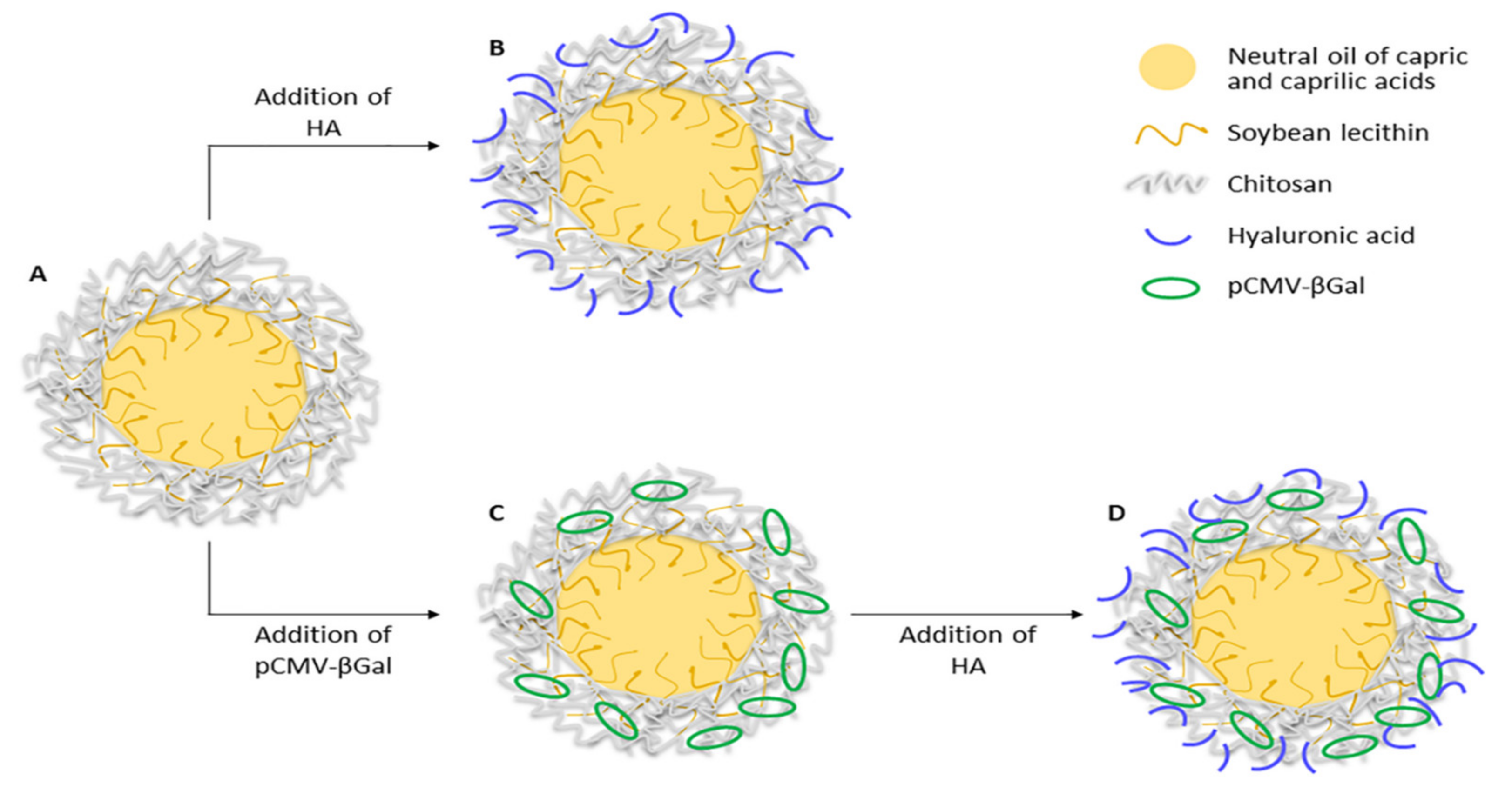
2.3. Preparation of CS-Based NCs
2.4. Determination of NCs Production Yield
2.5. Characterization of NCs
2.6. Preliminary Study of the pCMV-βGal Association to the NCs
2.7. Determination of the pCMV-βGal Association to the NCs
2.8. Preparation of Dry Powders and Determination of Spray-Drying Process Yield
2.9. Characterization of the Dry Powders
2.10. Release of NCs from Dry Powders
2.11. In Vivo Studies
2.11.1. Lung Distribution of Microencapsulated NCs
2.11.2. In Vivo Gene Expression Study
3. Results and Discussion
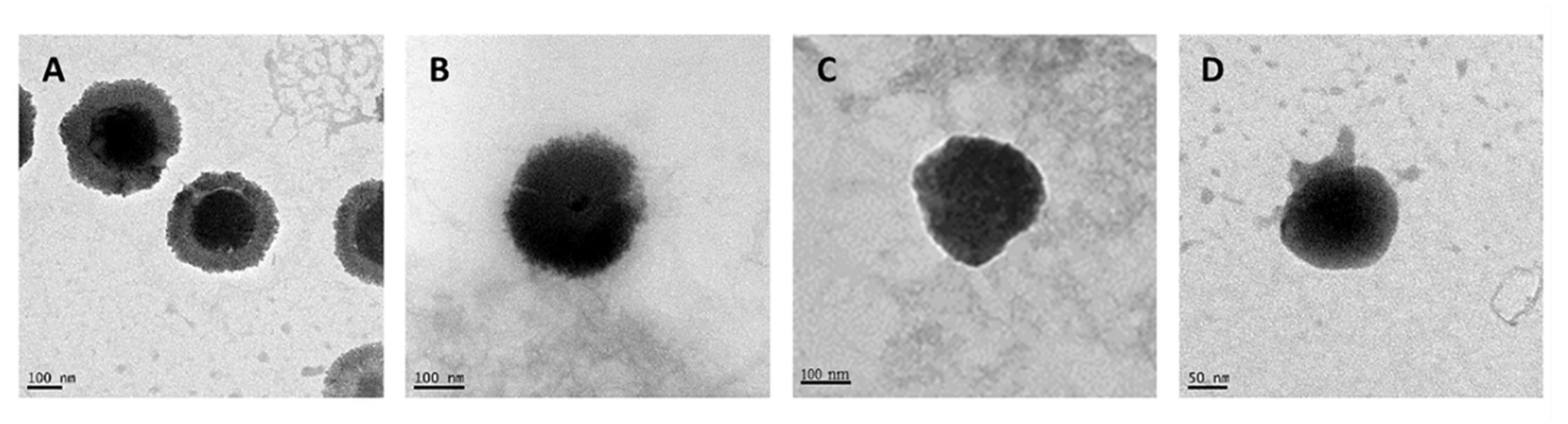
3.1. Preparation and Characterization of CS-Based NCs
3.2. Determination of the pCMV-βGal Association to the NCs
3.3. Preparation and Characterization of the Dry Powders
3.4. Release of NCs from Dry Powders
3.5. In Vivo Studies
3.5.1. Lung Distribution of Microencapsulated NCs
3.5.2. In Vivo Gene Expression Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geiger, J.; Aneja, M.K.; Rudolph, C. Vectors for Pulmonary Gene Therapy. Int. J. Pharm. 2010, 390, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Villate-Beitia, I.; Zarate, J.; Puras, G.; Pedraz, J.L. Gene Delivery to the Lungs: Pulmonary Gene Therapy for Cystic Fibrosis. Drug Dev. Ind. Pharm. 2017, 47, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-P.; Zhang, W.-T.; Qiu, Y.; Ju, M.-J.; Tu, G.-W.; Luo, Z. Understanding Gene Therapy in Acute Respiratory Distress Syndrome. Curr. Gene Ther. 2019, 19, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.J.; King, J.A.; Thorpe, P.H.; McLachlan, G.; Sallenave, J.-M. Towards Gene Therapy for Inflammatory and Infective Pulmonary Diseases. Curr. Genom. 2004, 5, 365–383. [Google Scholar] [CrossRef]
- Skvortsov, T.A.; Ignatov, D.V.; Majorov, K.B.; Apt, A.S.; Azhikina, T.L. Mycobacterium Tuberculosis Transcriptome Profiling in Mice with Genetically Different Susceptibility to Tuberculosis. Acta Nat. 2013, 5, 62–69. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Grenha, A.; Remuñán-López, C. Chitosan and its Derivatives as Nanocarriers for siRNA Delivery. J. Drug Deliv. Sci. Technol. 2012, 22, 29–42. [Google Scholar] [CrossRef]
- Morille, M.; Passirani, C.; Vonarbourg, A.; Clavreul, A.; Benoit, J.P. Progress in Developing Cationic Vectors for Non-Viral Systemic Gene Therapy Against Cancer. Biomaterials 2008, 29, 3477–3496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, S.; Lavigne, P.; Corsi, K.; Benderdour, M.; Beaumont, E.; Fernandes, J.C. Chitosan–DNA Nanoparticles as Non-Viral Vectors in Gene Therapy: Strategies to Improve Transfection Efficacy. Eur. J. Pharm. Biopharm. 2004, 57, 1–8. [Google Scholar] [CrossRef]
- Liu, X.; Howard, K.A.; Dong, M.; Andersen, M.O.; Rahbek, U.L.; Johnsen, M.G.; Hansen, O.C.; Besenbacher, F.; Kjems, J. The Influence of Polymeric Properties on Chitosan/siRNA Nanoparticle Formulation and Gene Silencing. Biomaterials 2006, 28, 1280–1288. [Google Scholar] [CrossRef]
- Islam, N.; Wang, H.; Maqbool, F.; Ferro, V. In Vitro Enzymatic Digestibility of Glutaraldehyde-Crosslinked Chitosan Nanoparticles in Lysozyme Solution and their Applicability in Pulmonary Drug Delivery. Molecules 2019, 24, 1271. [Google Scholar] [CrossRef] [Green Version]
- Portero, A.; Remuñán-López, C.; Nielsen, H.M. The Potential of Chitosan in Enhancing Peptide and Protein Absortion Across the TR146 Cell Culture Model-An In Vitro Model of the Buccal Epithelium. Pharm. Res. 2002, 19, 169–174. [Google Scholar] [CrossRef]
- Prego, C.; García Fuentes, M.; Torres, D.; Alonso, M.J. Transmucosal Macromolecular Drug Delivery. J. Control. Release 2005, 101, 151–162. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Grenha, A.; Carrión-Recio, D.; Seijo, B.; Remuñán-López, C. Microencapsulated Chitosan Nanoparticles for Pulmonary Protein Delivery: In Vivo Evaluation of Insulin-Loaded Formulations. J. Control. Release 2012, 157, 383–390. [Google Scholar] [CrossRef]
- De Jesús Valle, M.J.; Dinis-Oliveira, R.J.; Carvalho, F.; Bastos, M.L.; Sánchez Navarro, A. Toxicological Evaluation of Lactose and Chitosan Delivered by Inhalation. J. Biomater. Sci. 2008, 19, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenha, A.; Al-Qadi, S.; Seijo, B.; Remuñán-López, C. The Potential of Chitosan for Pulmonary Drug Delivery. J. Drug Deliv. Sci. Technol. 2010, 20, 33–43. [Google Scholar] [CrossRef]
- Dua, K.; Bebawy, M.; Awasthi, R.; Tekade, R.K.; Tekade, M.; Gupta, G.; de Jesús Andreoli Pinto, T.; Hansbro, P.M. Application of Chitosan and its Derivatives in Nanocarrier Based Pulmonary Drug Delivery Systems. Pharm. Nanotechnol. 2017, 5, 243–249. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non Viral Vectors in Gene Therapy-An Overview. J. Clin. Diagn. Res. 2015, 9, 1–6. [Google Scholar] [CrossRef]
- Csaba, N.; Köping-Höggård, M.; Alonso, M.J. Ionically Crosslinked Chitosan/Tripolyphosphate Nanoparticles for Oligonucleotide and Plasmid DNA Delivery. Int. J. Pharm. 2009, 382, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, W.E.; Aminabhavi, T.M. Chitosan as a Carrier for Targeted Delivery of Small Interfering RNA. Int. J. Pharm. 2010, 399, 1–11. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Alatorre-Meda, M.; Zaghloul, E.M.; Taboada, P.; Remunán-López, C. Chitosan–Hyaluronic Acid Nanoparticles for Gene Silencing: The Role of Hyaluronic Acid on the Nanoparticles’ Formation and Activity. Colloids Surf. B Biointerfaces 2013, 103, 615–623. [Google Scholar] [CrossRef]
- Bastow, E.R.; Byers, S.; Goluba, S.B.; Clarkinc, C.E.; Pitsillides, A.A.; Fosang, A.J. Hyaluronan Synthesis and Degradation in Cartilage and Bone. Cell. Mol. Life Sci. 2008, 65, 395–413. [Google Scholar] [CrossRef]
- Vasvani, S.; Kulkarni, P.; Rawtani, D. Hyaluronic Acid: A Review on its Biology, Aspects of Drug Delivery, Route of Administrations and a Special Emphasis on its Approved Marketed Products and Recent Clinical Studies. Int. J. Biol. Macromol. 2020, 151, 1012–1029. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan in Tissue Injury and Repair. Annu. Rev. Cell Dev. Biol. 2007, 23, 435–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, A.; Guo, L.; Tang, L. Effect of an Intrathoracic Injection of Sodium Hyaluronic Acid on the Prevention of Pleural Thickening in Excess Fluid of Tuberculous Thoracic Cavity. Clin. Exp. Pharmacol. Physiol. 2003, 30, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Sevilla, I.; Artiga, A.; Mitchell, S.G.; de Matteis, L.; de la Fuente, J.M. Natural Polysaccharides for siRNA Delivery: Nanocarriers Based on Chitosan, Hyaluronic Acid, and their Derivatives. Molecules 2019, 24, 2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.; Chawla, S.; Gadeval, A.; Reddy, G.; Maheshwari, R.; Kalia, K.; Tekade, R.K. Nanostructured Hyaluronic Acid-Based Materials for the Delivery of siRNA. Curr. Pharm. Des. 2018, 24, 2678–2691. [Google Scholar] [CrossRef]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Novel Hyaluronic Acid-Chitosan Nanoparticles for Ocular Gene Therapy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2016–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, J.M.; Goycoolea, F.M.; Higuera-Ciapara, I. Chitosan-Polysaccharide Blended Nanoparticles for Controlled Drug Delivery. Nat.-Based Polym. Biomed. Appl. 2008, 25, 644–679. [Google Scholar] [CrossRef]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Novel Hyaluronan-Based Nanocarriers for Transmucosal Delivery of Macromolecules. Macromol. Biosci. 2008, 8, 441–450. [Google Scholar] [CrossRef]
- Goycoolea, F.M.; Valle-Gallego, A.; Stefani, R.; Menchicchi, B.; David, L.; Rochas, C.; Santander-Ortega, M.J.; Alonso, M.J. Chitosan-Based Nanocapsules: Physical Characterization, Stability in Biological Media and Capsaicin Encapsulation. Colloid Polym. Sci. 2012, 290, 1423–1434. [Google Scholar] [CrossRef]
- Cadete, A.; Olivera, A.; Besev, M.; Dhal, P.K.; Gonçalves, L.; Almeida, A.J.; Bastiat, G.; Benoit, J.-P.; de la Fuente, M.; García-Fuentes, M.; et al. Self-Assembled Hyaluronan Nanocapsules for the Intracelular Delivery of Anticancer Drugs. Sci. Rep. 2019, 9, 11565. [Google Scholar] [CrossRef] [PubMed]
- Lollo, G.; Hervella, P.; Calvo, P.; Avilés, P.; Guillén, M.J.; García-Fuentes, M.; Alonso, M.J.; Torres, D. Enhanced In Vivo Therapeutic Efficacy of Plitidepsin-Loaded Nanocapsules Decorated with a New Poly-Aminoacid-PEG Derivative. Int. J. Pharm. 2015, 483, 212–219. [Google Scholar] [CrossRef]
- Peleteiro, M.; Presas, E.; González-Aramundiz, J.V.; Sánchez-Correa, B.; Simón-Vázquez, R.; Csaba, N.; Alonso, M.J.; González-Fernández, Á. Polymeric Nanocapsules for Vaccine Delivery: Influence of the Polymeric Shell on the Interaction with the Immune System. Front. Immunol. 2018, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Abellan-Pose, R.; Teijeiro-Valiño, C.; Santander-Ortega, M.J.; Borrajo, E.; Vidal, A.; García-Fuentes, M.; Csaba, N.; Alonso, M.J. Polyaminoacid Nanocapsules for Drug Delivery to the Lymphatic System: Effect of the Particle Size. Int. J. Pharm. 2016, 509, 107–117. [Google Scholar] [CrossRef]
- Calvo, P.; Remuñán-López, C.; Vila-Jato, J.L.; Alonso, M.J. Development of Positively Charged Colloidal Drug Carriers: Chitosan-Coated Polyester Nanocapsules and Submicron-Emulsions. Colloid Polym. Sci. 1997, 275, 46–53. [Google Scholar] [CrossRef]
- Crecente-Campo, J.; Alonso, M.J. Engineering, On-Demand Manufacturing, and Scaling-Up of Polymeric Nanocapsules. Bioeng. Transl. Med. 2019, 4, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Kolonko, A.K.; Efing, J.; González-Espinosa, Y.; Bangel-Ruland, N.; van Driessche, W.; Goycoolea, F.M.; Weber, W.-M. Capsaicin-Loaded Chitosan Nanocapsules for wtCFTR-mRNA Delivery to a Cystic Fibrosis Cell Line. Biomedicines 2020, 8, 364. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Sogali, B.S. Inhalation Therapy–Approaches and Challenges. Asian J. Pharm. Clin. Res. 2018, 11, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Grenha, A.; Carrión-Recio, D.; Teijeiro-Osorio, D.; Seijo, B.; Remuñán-López, C. Nano-and Microparticulate Carriers for Pulmonary Drug Delivery. In Handbook of Particulate Drug Delivery; Kumar, M.N.V., Ed.; American Scientific Publishers: Valencia, CA, USA, 2008; Volume 2, pp. 165–192. [Google Scholar]
- Newman, S.P. Delivering Drugs to the Lungs: The History of Repurposing in the Treatment of Respiratory Diseases. Adv. Drug Deliv. Rev. 2018, 133, 5–18. [Google Scholar] [CrossRef]
- Newman, S.P. Drug Delivery to the Lungs: Challenges and Opportunities. Ther. Deliv. 2017, 8, 647–661. [Google Scholar] [CrossRef]
- Sung, J.; Pulliam, B.; Edwards, D. Nanoparticles for Drug Delivery to the Lungs. Trends Biotechnol. 2007, 25, 563–570. [Google Scholar] [CrossRef]
- Hussain, A.; Arnold, J.J.; Khan, M.A.; Ahsan, F. Absorption Enhancers in Pulmonary Protein Delivery. J. Control. Release 2004, 94, 15–24. [Google Scholar] [CrossRef] [PubMed]
- FDA Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm (accessed on 13 April 2021).
- WHO Model Lists of Essential Medicines and Health Products. Available online: https://www.who.int/medicines/publications/essentialmedicines/en/ (accessed on 13 April 2021).
- Ohrem, H.L.; Schornick, E.; Kalivoda, A.; Ognibene, R. Why Is Mannitol Becoming More and More Popular as a Pharmaceutical Excipient in Solid Dosage Forms? Pharm. Dev. Technol. 2013, 19, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.G.; Schaldach, G.; Littringer, E.M.; Mescher, A.; Griesser, U.J.; Braun, D.E.; Walzel, P.E.; Urbanetz, N.A. The Impact of Spray Drying Outlet Temperature on the Particle Morphology of Mannitol. Powder Technol. 2011, 213, 27–35. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Taboada, P.; Remuñán-López, C. Micro/Nanostructured Inhalable Formulation Based on Polysaccharides: Effect of a Thermoprotectant on Powder Properties and Protein Integrity. Int. J. Pharm. 2018, 551, 23–33. [Google Scholar] [CrossRef]
- Li, X.; Vogt, F.G.; Hayes, D., Jr.; Mansour, H.M. Design, Characterization, and Aerosol Dispersion Performance Modeling of Advanced Spray-Dried Microparticulate/Nanoparticulate Mannitol Powders for Targeted Pulmonary Delivery as Dry Powder Inhalers. J. Aerosol Med. Pulm. Drug Deliv. 2014, 27, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Teper, A.; Jaques, A.; Charlton, B. Inhaled Mannitol in Patients with Cystic Fibrosis: A Randomised Open-Label Dose Response Trial. J. Cyst. Fibros. 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, D.P.; Gaspar, M.M.; Eleutério, C.V.; Grenha, A.; Blanco, M.; Gonçalves, L.M.D.; Taboada, P.; Almeida, A.J.; Remuñán-López, C. Microencapsulated Solid Lipid Nanoparticles as a Hybrid Platform for Pulmonary Antibiotic delivery. Mol. Pharm. 2017, 14, 2977–2990. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Faria, V.; Gonçalves, L.M.D.; Taboada, P.; Remuñán-López, C.; Almeida, A.J. Rifabutin-Loaded Solid Lipid Nanoparticles for Inhaled Antitubercular Therapy: Physicochemical and In Vitro Studies. Int. J. Pharm. 2016, 497, 199–209. [Google Scholar] [CrossRef]
- Grenha, A.; Remuñán-López, C.; Carvalho, E.L.S.; Seijo, B. Microspheres Containing Lipid/Chitosan Nanoparticles Complexes for Pulmonary Delivery of Therapeutic Proteins. Eur. J. Pharm. Biopharm. 2008, 69, 83–93. [Google Scholar] [CrossRef]
- Fernández-Paz, C.; Rojas, S.; Salcedo-Abraira, P.; Simón-Yarza, T.; Remuñán-López, C.; Horcajada, P. Metal-Organic Framework Microsphere Formulation for Pulmonary Administration. ACS Appl. Mater. Interfaces 2020, 12, 25676–25682. [Google Scholar] [CrossRef] [PubMed]
- El-Gibaly, I. Development and In Vitro Evaluation of Novel Floating Chitosan Microcapsules for Oral Use: Comparison with Non-Floating Chitosan Microspheres. Int. J. Pharm. 2002, 294, 7–21. [Google Scholar] [CrossRef]
- Alves, A.D.; Cavaco, J.S.; Guerreiro, F.; Lourenço, J.P.; da Costa, A.M.R.; Grenha, A. Inhalable Antitubercular Therapy Mediated by Locust Bean Gum Microparticles. Molecules 2016, 21, 702. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Pant, G.; Mitra, K.; Madan, J.; Chourasia, M.K.; Misra, A. Inhalable Particles Containing Rapamycin for Induction of Autophagy in Macrophages Infected with Mycobacterium Tuberculosis. Mol. Pharm. 2014, 11, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.; Limberis, M.; Gao, G.; Wu, D.; Bove, M.S.; Sanmiguel, J.C.; Wilson, J.M. An Optimized Protocol for Detection of E. Coli β-Galactosidase in Lung Tissue Following Gene Transfer. Histochem. Cell Biol. 2005, 124, 77–85. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Grenha, A.; Remuñán-López, C. Microspheres Loaded with Polysaccharide Nanoparticles for Pulmonary Delivery: Preparation, Structure and Surface Analysis. Carbohydr. Polym. 2011, 86, 25–34. [Google Scholar] [CrossRef]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Design of Novel Polysaccharidic Nanostructures for Gene Delivery. Nanotechnology 2008, 19, 075105. [Google Scholar] [CrossRef]
- Prego, C.; Torres, D.; Alonso, M.J. Chitosan Nanocapsules: A New Carrier for Nasal Peptide Delivery. J. Drug Del. Sci. Technol. 2006, 16, 331–337. [Google Scholar] [CrossRef]
- Prego, C.; Torres, D.; Fernandez-Megia, E.; Novoa-Carballal, R.; Quiñoá, E.; Alonso, M.J. Chitosan–PEG Nanocapsules as New Carriers for Oral Peptide Delivery Effect of Chitosan Pegylation Degree. J. Control. Release 2006, 111, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Prego, C.; Fabre, M.; Torres, D.; Alonso, M.J. Efficacy and Mechanism of Action of Chitosan Nanocapsules for Oral Peptide Delivery. Pharm. Res. 2006, 23, 549–556. [Google Scholar] [CrossRef]
- Pilcer, G.; Amighi, K. Formulation Strategy and Use of Excipients in Pulmonary Drug Delivery. Int. J. Pharm. 2010, 392, 1–19. [Google Scholar] [CrossRef]
- Broadhead, J.; Edmond-Rouan, S.K.; Rhodes, C.T. The Spray Drying of Pharmaceuticals. Drug Dev. Ind. Pharm. 1992, 18, 1169–1206. [Google Scholar] [CrossRef]
- Grenha, A.; Seijo, B.; Remuñán-López, C. Microencapsulated Chitosan Nanoparticles for Lung Protein Delivery. Eur. J. Pharm. Sci. 2005, 25, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Al-Qadi, S.; Remuñán-López, C. A Micro-and Nano-Structured Drug Carrier Based on Biocompatible, Hybrid Polymeric Nanoparticles for Potential Application in Dry Powder Inhalation Therapy. Polymer 2014, 55, 4012–4021. [Google Scholar] [CrossRef]
- Khan, I.; Apostolou, M.; Bnyan, R.; Houacine, C.; Elhissi, A.; Yousaf, S.S. Paclitaxel-Loaded Micro or Nano Transfersome Formulation into Novel Tablets for Pulmonary Drug Delivery Via Nebulization. Int. J. Pharm. 2020, 575, 118919. [Google Scholar] [CrossRef]
- Bosquillon, C.; Lombry, C.; Préat, V.; Vanbever, R. Influence of Formulation Excipients and Physical Characteristics of Inhalation Dry Powders on their Aerosolization Performance. J. Control. Release 2001, 70, 329–339. [Google Scholar] [CrossRef]
- Mejias, J.C.; Roy, K. In-Vitro and In-Vivo Characterization of a Multi-Stage Enzyme-Responsive Nanoparticle-in-Microgel Pulmonary Drug Delivery System. J. Control. Release 2019, 316, 393–403. [Google Scholar] [CrossRef]
- Grenha, A.; Seijo, B.; Serra, C.; Remuñán-López, C. Chitosan Nanoparticle-Loaded Mannitol Microspheres: Structure and Surface Characterization. Biomacromolecules 2007, 8, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Sinsuebpol, C.; Chatchawalsaisin, J.; Kulvanich, P. Preparation and In Vivo Absorption Evaluation of Spray Dried Powders Containing Salmon Calcitonin Loaded Chitosan Nanoparticles for Pulmonary Delivery. Drug Des. Dev. Ther. 2013, 7, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.M.; Gorman, E.M.; Munson, E.J.; Berkland, C. Pure Insulin Nanoparticle Agglomerates for Pulmonary Delivery. Langmuir 2008, 24, 13614–13620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paranjpe, M.; Müller-Goymann, C.C. Nanoparticle-Mediated Pulmonary Drug Delivery: A Review. Int. J. Mol. Sci. 2014, 15, 5852–5873. [Google Scholar] [CrossRef]
- Berkebile, A.R.; McCray, P.B. Effects of Airway Surface Liquid pH on Host Defense in Cystic Fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Gupta, N.; Ahsan, F. Particle Engineering to Enhance or Lessen Uptake by Alveolar Macrophages and to Influence the Therapeutic Outcome. Eur. J. Pharm. Biopharm. 2015, 89, 163–174. [Google Scholar] [CrossRef]
- Weiss, D.J.; Liggitt, D.; Clark, J.G. Histochemical Discrimination of Endogenous Mammalian β-Galactosidase Activity From that Resulting from Lac-Z Gene Expression. Histochem. J. 1999, 31, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Iida-Tanaka, N.; Niidome, T.; Kawano, T.; Kubo, K.; Yoshikawa, K.; Sato, T.; Yang, Z.; Koyama, Y. Hyaluronic Acid and its Derivative as a Multi-Functional Gene Expression Enhancer: Protection from Non-Specific Interactions, Adhesion to Targeted Cells, and Transcriptional Activation. J. Control. Release 2006, 112, 382–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evanko, S.P.; Wight, T.N. Intracellular Localization of Hyaluronan in Proliferating Cells. J. Histochem. Cytochem. 1999, 47, 1331–1341. [Google Scholar] [CrossRef]
- Ito, T.; Okuda, T.; Takashima, Y.; Okamoto, H. Naked pDNA Inhalation Powder Composed of Hyaluronic Acid Exhibits High Gene Expression in the Lungs. Mol. Pharm. 2019, 16, 489–497. [Google Scholar] [CrossRef]
- Frydas, S.; Varvara, G.; Murmura, G.; Saggini, A.; Caraffa, A.; Antinolfi, P.; Teté, S.; Tripodi, D.; Conti, F.; Cianchetti, E.; et al. Impact of Capsaicin on Mast Cell Inflammation. Int. J. Immunopathol. Pharmacol. 2013, 26, 597–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiobara, T.; Usui, T.; Han, J.; Isoda, H.; Nagumo, Y. The Reversible Increase in Tight Junction Permeability Induced by Capsaicin is Mediated via Cofilin-Actin Cytoskeletal Dynamics and Decreased Level of Occluding. PLoS ONE 2013, 8, e79954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, M.; Pereira, S.; Pohl, L.; Ketelhut, S.; Kemper, B.; Gorzelanny, C.; Galla, H.-J.; Moerschbacher, B.M.; Goycoolea, F.M. Chitosan Encapsulation Modulates the Effect of Capsaicin on the Tight Junctions of MDCK Cells. Sci. Rep. 2015, 5, 10048. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | MilliQ Water (µL) | pCMV-βGal (628 µg/mL) (µL) | CS NCs Suspension (34.3 mg/mL) (µL) | Hyaluronic Acid (HA) Solution (625 µg/mL) (µL) |
|---|---|---|---|---|
| pCMV-βGal-CS NCs | 125 | 125 | 250 | - |
| pCMV-βGal-HA/CS NCs | 125 | 93.80 | 250 | 31.20 |
| Formulation | Size Range (nm) | PdI | ζ-Potential Range (mV) | Production Yield (%) |
|---|---|---|---|---|
| CS NCs | 160 ± 3 | 0.19 | +56.5 ± 1.4 | 83 ± 4 |
| HA/CS NCs | 154 ± 2 | 0.15 | +34.7 ± 0.8 | 67 ± 7 |
| pCMV-βGal-CS NCs | 165 ± 4 | 0.25 | +54.0 ± 1.6 | 79 ± 6 |
| pCMV-βGal-HA/CS NCs | 162 ± 3 | 0.21 | +29.2 ± 1.2 | 65 ± 8 |
| Pre-dialyzed Cu6-CS NCs | 161 ± 2 | 0.21 | +57.4 ± 0.6 | - |
| Pre-dialyzed Cu6-HA/CS NCs | 155 ± 1 | 0.16 | +37.5 ± 0.4 | - |
| Dialyzed Cu6-CS NCs | 162 ± 1 | 0.20 | +39.6 ± 0.1 | - |
| Dialyzed Cu6-HA/CS NCs | 156 ± 2 | 0.15 | +26.2 ± 0.1 | - |
| Formulation | E.E. (%) | D.L. (%) |
|---|---|---|
| pCMV-βGal-CS NCs | 90.6 ± 0.9 | 36.2 ± 0.4 |
| pCMV-βGal-HA/CS NCs | 89.0 ± 4.8 | 35.6 ± 1.9 |
| Dry Powder | TOutlet (°C) | P.Y. (w/w, %) |
|---|---|---|
| Mannitol (Ma) microspheres (MS) | 56 | 64 ± 3 |
| CS NCs-loaded Ma MS | 60 | 67 ± 5 |
| HA/CS NCs-loaded Ma MS | 58 | 67 ± 6 |
| pCMV-βGal-Ma MS | 57 | 70 ± 5 |
| pCMV-βGal-CS NCs-loaded Ma MS | 55 | 69 ± 8 |
| pCMV-βGal-HA/CS NCs-loaded Ma MS | 58 | 71 ± 8 |
| Dry Powder Samples | Geometric Diameter (µm) | Apparent Density (g/cm3) | Real Density (g/cm3) | Theoretical Aerodynamic Diameter (µm) |
|---|---|---|---|---|
| Ma MS | 3.75 ± 1.56 | 0.50 ± 0.01 | 1.29 ± 0.01 | 4.21 ± 0.01 |
| pCMV-βGal-Ma MS | 2.85 ± 1.49 | - | - | - |
| CS NCs-loaded Ma MS | 2.10 ± 0.86 | 0.44 ± 0.02 | 1.43 ± 0.01 | 2.51 ± 0.02 |
| pCMV-βGal-CS NCs-loaded Ma MS | 1.98 ± 0.70 | - | - | - |
| HA/CS NCs-loaded Ma MS | 2.77 ± 1.35 | 0.42 ± 0.02 | 1.43 ± 0.02 | 3.35 ± 0.02 |
| pCMV-βGal-HA/CS NCs-loaded Ma MS | 2.43 ± 1.32 | - | - | - |
| Types of CS-Based NCs | Count Rate (kcps) |
|---|---|
| CS NCs | 266–281 |
| CS NCs released in MilliQ water | 262–359 |
| CS NCs released in simulated pulmonary medium | 280–399 |
| HA/CS NCs | 196–200 |
| HA/CS NCs released in MilliQ water | 102–216 |
| HA/CS NCs released in simulated pulmonary medium | 185–226 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Paz, E.; Feijoo-Siota, L.; Gaspar, M.M.; Csaba, N.; Remuñán-López, C. Microencapsulated Chitosan-Based Nanocapsules: A New Platform for Pulmonary Gene Delivery. Pharmaceutics 2021, 13, 1377. https://doi.org/10.3390/pharmaceutics13091377
Fernández-Paz E, Feijoo-Siota L, Gaspar MM, Csaba N, Remuñán-López C. Microencapsulated Chitosan-Based Nanocapsules: A New Platform for Pulmonary Gene Delivery. Pharmaceutics. 2021; 13(9):1377. https://doi.org/10.3390/pharmaceutics13091377
Chicago/Turabian StyleFernández-Paz, Estefanía, Lucía Feijoo-Siota, Maria Manuela Gaspar, Noemi Csaba, and Carmen Remuñán-López. 2021. "Microencapsulated Chitosan-Based Nanocapsules: A New Platform for Pulmonary Gene Delivery" Pharmaceutics 13, no. 9: 1377. https://doi.org/10.3390/pharmaceutics13091377