
Neurovascular Unit: A New Target for Treating Early Stages of Diabetic Retinopathy


Abstract
:1. Introduction
2. The Neurovascular Unit
3. Current Treatments for Early Stages of Diabetic Retinopathy
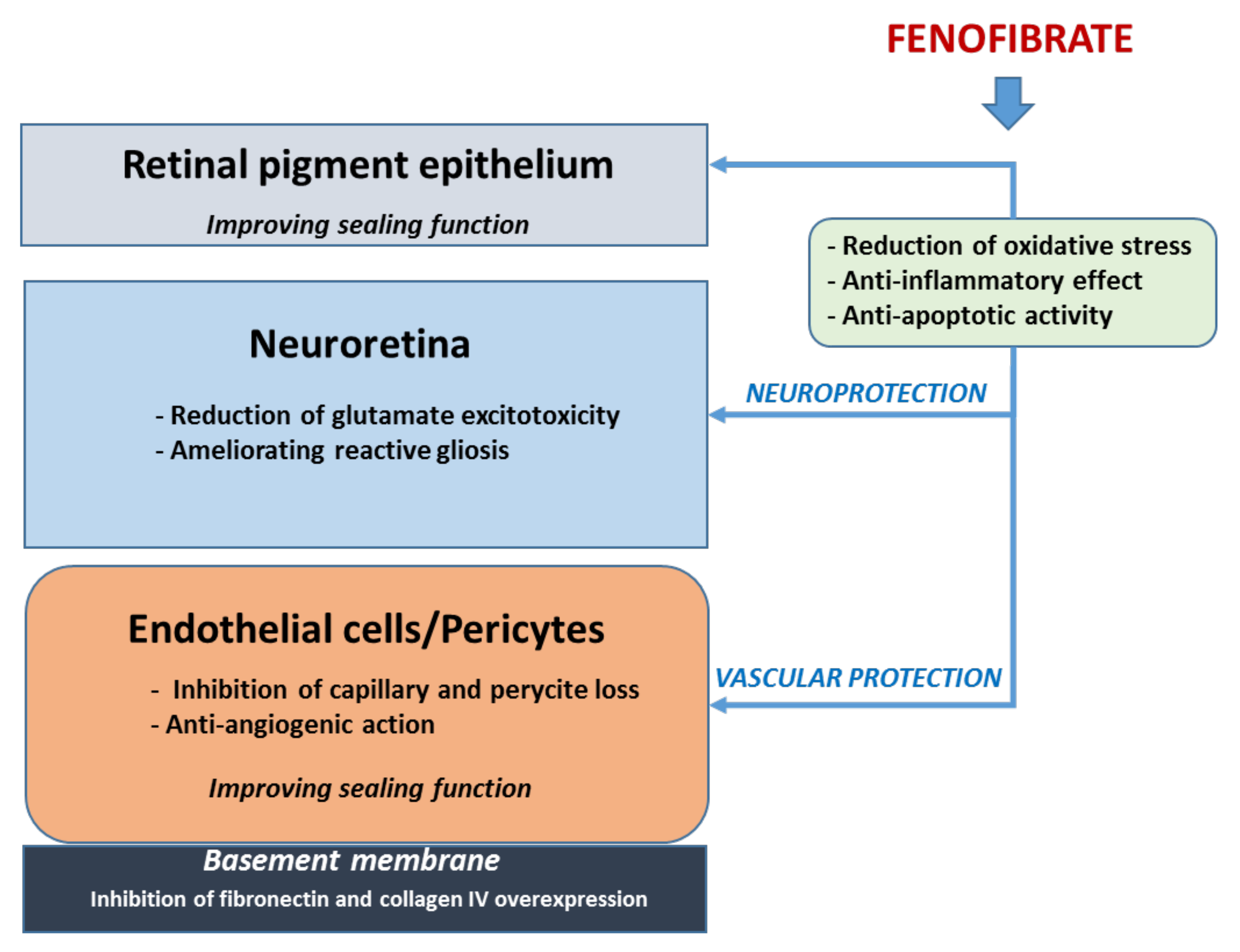
3.1. Fenofibrate
3.2. Calcium Dobesilate (CaD)
4. Administration Route When Treating Early Stages of DR
5. Treatment Based on Neuroprotection
5.1. Proteins with Neurotrophic and Angiogenic Properties
5.2. Peptides/Proteins with Neurotrophic and Anti-Angiogenic or Vasculotropic Properties
5.3. Blocking ET-1
5.4. Neurotransmitters
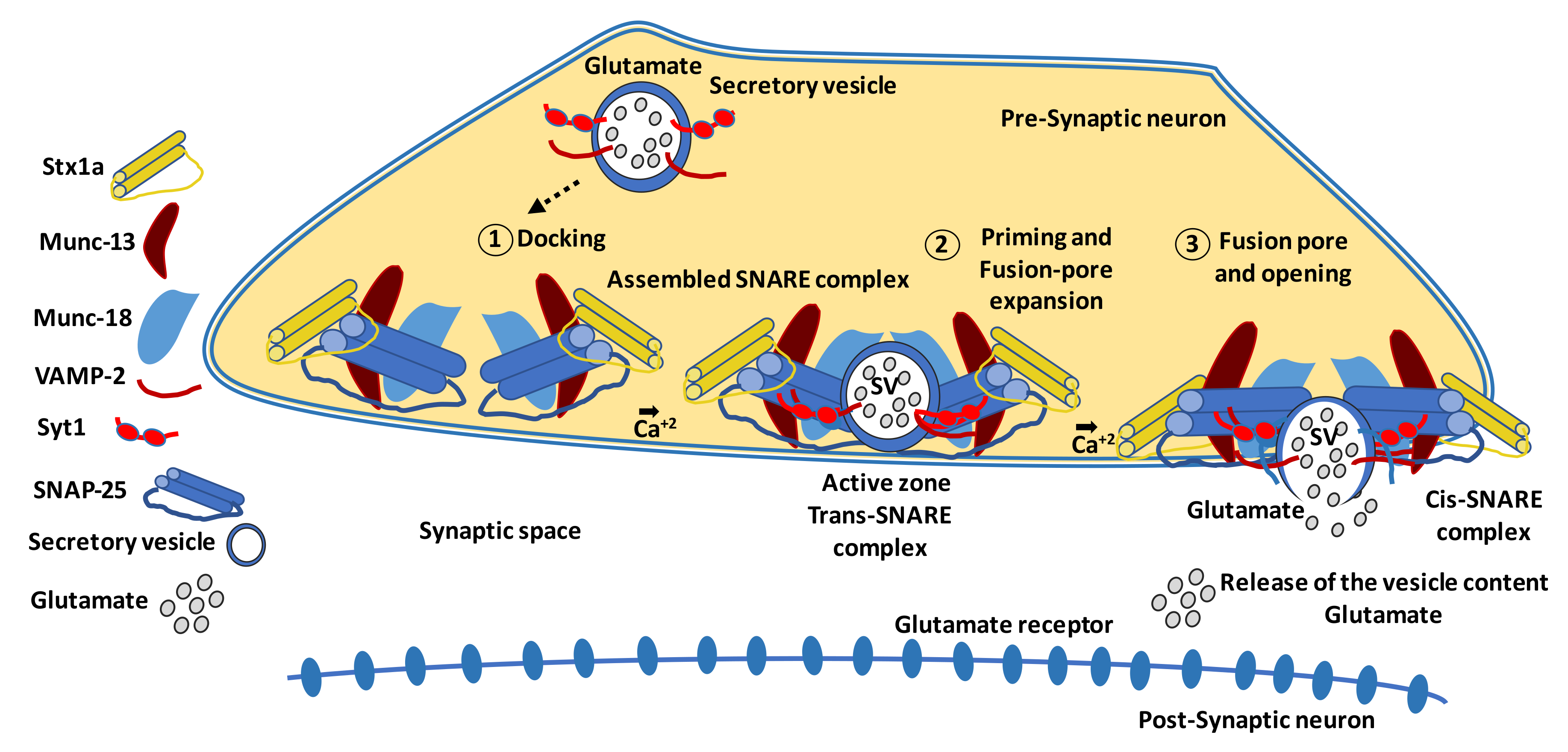
5.4.1. Proteins Involved in Synaptic Connectivity
5.4.2. Proteins Involved in Axonal Transport
6. Treatment Based on Targeting Inflammation
6.1. General Considerations
6.2. Targeting Pro-Inflammatory Cytokines
7. Concluding Remarks and New Perspectives in Clinical Practice
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yau, J.W.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Meta-Analysis for Eye Disease (META-EYE) Study Group. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.Y.; Cheung, C.M.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Primers 2016, 2, 16012. [Google Scholar] [CrossRef]
- Wong, T.Y.; Sabanayagam, C. Strategies to tackle the global burden of diabetic retinopathy: From epidemiology to artificial intelligence. Ophthalmologica 2020, 243, 9–20. [Google Scholar] [CrossRef]
- International Diabetes Federation. Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: http://www.idf.org/diabtesatlas (accessed on 12 June 2021).
- Sun, J.K.; Aiello, L.P.; Abràmoff, M.D.; Antonetti, D.A.; Dutta, S.; Pragnell, M.; Levine, S.R.; Gardner, T.W. Updating the Staging System for Diabetic Retinal Disease. Ophthalmology 2021, 128, 490–493. [Google Scholar] [CrossRef]
- Vujosevic, S.; Aldington, S.J.; Silva, P.; Hernández, C.; Scanlon, P.; Peto, T.; Simó, R. Screening for diabetic retinopathy: New perspectives and challenges. Lancet Diabetes Endocrinol. 2020, 8, 337–347. [Google Scholar] [CrossRef]
- Abcouwer, S.F.; Gardner, T.W. Diabetic retinopathy: Loss of neuroretinal adaptation to the diabetic metabolic environment. Ann. N. Y. Acad. Sci. 2014, 1311, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simó, R.; Hernández, C. Neurodegeneration in the diabetic eye: New insights and therapeutic perspectives. Trends Endocrinol. Metab. 2014, 25, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Hernández, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Retin. Eye Res. 2015, 48, 160–180. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; Mckay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammens, H.P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Chew, E.; Duh, E.J.; Sobrin, L.; Sun, J.K.; VanderBeek, B.L.; Wykoff, C.C.; Gardner, T.W. Diabetic retinopathy: A position statement by the American Diabetes Association. Diabetes Care 2017, 40, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Gardner, T.W.; Davila, J.R. The neurovascular unit and the pathophysiologic basis of diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1–6. [Google Scholar] [CrossRef]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.A. Functional hyperemia and mechanisms of neurovascular coupling in the retinal vasculature. J. Cereb. Blood Flow Metab. 2013, 33, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metea, M.R.; Newmman, E.A. Signalling within the neurovascular unit in the retina. Exp. Physiol. 2007, 92, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, E.; Hernández, C.; Miralles, A.; Huguet, P.; Farrés, J.; Simó, R. Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care 2007, 30, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ramírez, M.; Hernández, C.; Villarroel, M.; Canals, F.; Alonso, M.A.; Fortuny, R.; Masmiquel, L.; Navarro, A.; García-Arumí, J.; Simó, R. Interphotoreceptor retinoid-binding protein (IRBP) is downregulated at early stages of diabetic retinopathy. Diabetologia 2009, 52, 2633–2641. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.J.; Gardner, T.W.; Abcouwer, S.F. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1156–1163. [Google Scholar] [CrossRef]
- Barber, A.J. A new view of diabetic retinopathy: A neurodegenerative disease of the eye. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 283–290. [Google Scholar] [CrossRef]
- Ramsey, D.J.; Ripps, H.; Qian, H. An electrophysiological study of retinal function in the diabetic female rat. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5116–5124. [Google Scholar] [CrossRef]
- Bogdanov., P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; García-Arumí, J.; Ramos, D.; Ruberte, J.; Simó, R.; Hernández, C. The db/db mouse: A useful model for the study of diabetic retinal neurodegeneration. PLoS ONE 2014, 9, e97302. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.S.; Gunkel, R.D.; Podgor, M.J. Color vision defects in early diabetic retinopathy. Arch. Ophthalmol. 1986, 104, 225–228. [Google Scholar] [CrossRef]
- Trick, G.L.; Burde, R.M.; Gordon, M.O.; Santiago, J.V.; Kilo, C. The relationship between hue discrimination and contrast sensitivity deficits in patients with diabetes mellitus. Ophthalmology 1988, 95, 693–698. [Google Scholar] [CrossRef]
- Tonade, D.; Kern, T.S. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog. Retin. Eye Res. 2020, 83, 100919. [Google Scholar] [CrossRef]
- Picconi, F.; Parravano, M.; Ylli, D.; Pasqualetti, P.; Coluzzi, S.; Giordani, I.; Malandrucco, I.; Lauro, D.; Scarinci, F.; Giorno, P.; et al. Retinal neurodegeneration in patients with type 1 diabetes mellitus: The role of glycemic variability. Acta Diabetol. 2017, 54, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.R.; Ribeiro, L.; Bandello, F.; Lattanzio, R.; Egan, C.; Frydkjaer-Olsen, U.; García-Arumí, J.; Gibson, J.; Grauslund, J.; Harding, S.P.; et al. Functional and structural findings of neurodegeneration in early stages of diabetic retinopathy: Cross-sectional analyses of baseline data of the EUROCONDOR project. Diabetes 2017, 66, 2503–2510. [Google Scholar] [CrossRef] [Green Version]
- Simó, R.; Hernández, C.; Porta, M.; Bandello, F.; Grauslund, J.; Harding, S.P.; Aldington, S.J.; Egan, C.; Frydkjaer-Olsen, U.; García-Arumí, J.; et al. Effects of Topically Administered Neuroprotective Drugs in Early Stages of Diabetic Retinopathy: Results of the EUROCONDOR Clinical Trial. Diabetes 2019, 68, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keech, A.C.; Mitchell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- ACCORD Study Group; ACCORD Eye Study Group; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; Esser, B.A.; et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233–244. [Google Scholar]
- Simó, R.; Hernández, C. Fenofibrate for diabetic retinopathy. Lancet 2007, 370, 1667–1668. [Google Scholar] [CrossRef]
- Ciudin, A.; Hernández, C.; Simó, R. Molecular implications of the PPARs in the diabetic eye. PPAR Res. 2013, 2013, 686525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simó, R.; Roy, S.; Behar-Cohen, F.; Keech, A.; Mitchell, P.; Wong, T.Y. Fenofibrate: A new treatment for diabetic retinopathy. Molecular mechanisms and future perspectives. Curr. Med. Chem. 2013, 20, 3258–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasongko, M.B.; Wong, T.Y.; Nguyen, T.T.; Kawasaki, R.; Jenkins, A.J.; Shaw, J.; Robinson, C.; Wang, J.J. Serum apolipoproteins are associated with systemic and retinal microvascular function in people with diabetes. Diabetes 2012, 61, 1785–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.Y.; Simó, R.; Mitchell, P. Fenofibrate—A potential systemic treatment for diabetic retinopathy? Am. J. Ophthalmol. 2012, 154, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Ballarini, S.; Cunha-Vaz, J.; Ji, L.; Haller, H.; Zimmet, P.; Wong, T.Y. Non-traditional systemic treatments for diabetic retinopathy: An evidence-based review. Curr. Med. Chem. 2015, 22, 2580–2589. [Google Scholar] [CrossRef] [Green Version]
- Villarroel, M.; Garcia-Ramírez, M.; Corraliza, L.; Hernández, C.; Simó, R. Fenofibric acid prevents retinal pigment epithelium disruption induced by interleukin-1β by suppressing AMP-activated protein kinase (AMPK) activation. Diabetologia 2011, 54, 1543–1553. [Google Scholar] [CrossRef] [Green Version]
- Miranda, S.; González-Rodríguez, Á.; García-Ramírez, M.; Revuelta-Cervantes, J.; Hernández, C.; Simó, R.; Valverde, A.M. Beneficial effects of fenofibrate in retinal pigment epithelium by the modulation of stress and survival signaling under diabetic conditions. J. Cell. Physiol. 2012, 227, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Roy, S.; Guo, W.; Hernández, C.; Villarroel, M.; Simó, R.; Roy, S. Fenofibric acid reduces fibronectin and collagen type IV overexpression in human retinal pigment epithelial cells grown in conditions mimicking the diabetic milieu: Functional implications in retinal permeability. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6348–6354. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.X. Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am. J. Pathol. 2009, 175, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, A.C.; Mott, R.; Lyons, T.J.; Ma, J.X. Therapeutic effects of PPARα agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013, 62, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Ahn, J.H.; Kim, H.S.; Yu, Y.S.; Kim, H.S.; Ha, J.; Shinn, S.H.; Oh, Y.S. Fenofibrate regulates retinal endothelial cell survival through the AMPK signal transduction pathway. Exp. Eye Res. 2007, 84, 886–893. [Google Scholar] [CrossRef]
- Bogdanov, P.; Hernández, C.; Corraliza, L.; Carvalho, A.R.; Simó, R. Effect of fenofibrate on retinal neurodegeneration in an experimental model of type 2 diabetes. Acta Diabetol. 2015, 52, 113–122. [Google Scholar] [CrossRef]
- Roy, S.; Kim, D.; Hernández, C.; Simó, R.; Roy, S. Beneficial effects of fenofibric acid on overexpression of extracellular matrix components, COX-2, and impairment of endothelial permeability associated with diabetic retinopathy. Exp. Eye Res. 2015, 140, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ramírez, M.; Hernández, C.; Palomer, X.; Vázquez-Carrera, M.; Simó, R. Fenofibrate prevents the disruption of the outer blood retinal barrier through downregulation of NF-κB activity. Acta Diabetol. 2016, 53, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Ozawa, N.; Miwa, Y.; Ishida, A.; Ohta, M.; Tsubota, K.; Kurihara, T. Pemafibrate Prevents Retinal Pathological Neovascularization by Increasing FGF21 Level in a Murine Oxygen-Induced Retinopathy Model. Int. J. Mol. Sci. 2019, 20, 5878. [Google Scholar] [CrossRef] [Green Version]
- Leite, E.B.; Mota, M.C.; de Abreu, J.R.; Cunha-Vaz, J.G. Effect of calcium dobesilate on the blood-retinal barrier in early diabetic retinopathy. Int. Ophthalmol. 1990, 14, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.L.; Seres, A.I.; Carneiro, A.M.; Stur, M.; Zourdani, A.; Caillon, P.; Cunha-Vaz, J.G. DX-Retinopathy Study Group. Effect of calcium dobesilate on progression of early diabetic retinopathy: A randomised double-blind study. Graefe’s Arch. Clin. Exp. Ophthalmol. 2006, 244, 1591–1600. [Google Scholar] [CrossRef]
- Haritoglou, C.; Gerss, J.; Sauerland, C.; Kampik, A.; Ulbig, M.W.; CALDIRET study group. Effect of calcium dobesilate on occurrence of diabetic macular oedema (CALDIRET study): Randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2009, 373, 1364–1371. [Google Scholar] [CrossRef]
- Simó-Servat, O.; Solà-Adell, C.; Bogdanov, P.; Hernández, C.; Simó, R. Mechanisms of retinal neuroprotection of calcium dobesilate: Therapeutic implications. Neural Regen. Res. 2017, 12, 1620–1622. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.C.; Martins, J.; Voabil, P.; Liberal, J.; Chiavaroli, C.; Bauer, J.; Cunha-Vaz, J.; Ambrosio, A.F. Calcium dobesilate inhibits the alterations in tight junction proteins and leukocyte adhesion to retinal endothelial cells induced by diabetes. Diabetes 2010, 59, 2637–2645. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, P.; Solà –Adell, C.; Hernández, C.; García-Ramírez, M.; Sampedro, J.; Simó-Servat, O.; Valeri, M.; Pasquali, C.; Simó, R. Calcium dobesilate prevents the oxidative stress and inflammation induced by diabetes in the retina of db/db mice. J. Diabetes Complicat. 2017, 31, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Voabil, P.; Liberal, J.; Leal, E.C.; Bauer, J.; Cunha-Vaz, J.; Santiago, A.R.; Ambrósio, A.F. Calcium Dobesilate Is Protective against Inflammation and Oxidative/Nitrosative Stress in the Retina of a Type 1 Diabetic Rat Model. Ophthalmic Res. 2017, 58, 150–161. [Google Scholar] [CrossRef]
- Solà-Adell, C.; Bogdanov, P.; Hernández, C.; Sampedro, J.; Valeri, M.; Garcia-Ramirez, M.; Pasquali, C.; Simó, R. Calcium Dobesilate Prevents Neurodegeneration and Vascular Leakage in Experimental Diabetes. Curr. Eye Res. 2017, 42, 1273–1286. [Google Scholar] [CrossRef]
- Park, J.Y.; Takahara, N.; Gabriele, A.; Chou, E.; Naruse, K.; Suzuma, K.; Yamauchi, T.; Ha, S.W.; Meier, M.; Rhodes, C.J.; et al. Induction of endothelin-1 expression by glucose: An effect of protein kinase C activation. Diabetes 2000, 49, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Liu, J.; Wang, D.; Qiu, S.; Yuan, Y.; Wang, F.; Wen, L.; Song, Q.; Sun, Z.L. Efficacy of calcium dobesilate in treating Chinese patients with mild-to-moderate non-proliferative diabetic retinopathy (CALM-DR): Protocol for a single-blind, multicentre, 24-armed cluster-randomised, controlled trial. BMJ Open 2021, 11, e045256. [Google Scholar] [CrossRef]
- Thagaard, M.S.; Vergmann, A.S.; Grauslund, J. Topical treatment of diabetic retinopathy: A systematic review. Acta Ophthalmol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; García-Ramírez, M.; Corraliza, L.; Fernández-Carneado, J.; Farrera-Sinfreu, J.; Ponsati, B.; González-Rodríguez, A.; Valverde, A.M.; Simó, R. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes 2013, 62, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Bogdanov, P.; Corraliza, L.; García-Ramírez, M.; Solà-Adell, C.; Arranz, J.A.; Arroba, A.I.; Valverde, A.M.; Simó, R. Topical Administration of GLP-1 Receptor Agonists Prevents Retinal Neurodegeneration in Experimental Diabetes. Diabetes 2016, 65, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, P.; Simó-Servat, O.; Sampedro, J.; Solà-Adell, C.; Garcia-Ramírez, M.; Ramos, H.; Guerrero, M.; Suñé-Negre, J.M.; Ticó, J.R.; Montoro, B.; et al. Topical Administration of Bosentan Prevents Retinal Neurodegeneration in Experimental Diabetes. Int. J. Mol. Sci. 2018, 19, 3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parravano, M.; Scarinci, F.; Parisi, V.; Giorno, P.; Giannini, D.; Oddone, F.; Varano, M. Citicoline and vitamin B12 eye drops in type 1 diabetes: Results of a 3-year pilot study evaluating mopho-functional retinal changes. Adv. Ther. 2020, 37, 1646–1663. [Google Scholar] [CrossRef] [Green Version]
- Simó, R.; Carrasco, E.; García-Ramírez, M.; Hernández, C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr. Diabetes Rev. 2006, 2, 71–98. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Sundstrom, J.M.; Antonetti, D.A. Ocular Anti-VEGF therapy for diabetic retinopathy: The role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care 2014, 37, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef] [Green Version]
- Saint-Geniez, M.; Maharaj, A.S.R.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous VEGF is required for visual function: Evidence for a survival role on Müller cells and photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, K.; Ng, Y.S.; Zhong, L.; Bradley, J.; Schubert, W.; Jo, N.; Akita, J.; Samuelsson, S.J.; Robinson, G.S.; Adamis, A.P.; et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptative response ischemic injury. Am. J. Pathol. 2007, 171, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Seigel, G.M.; Chiu, L.; Paxhia, A. Inhibition of neuroretinal cell death by insulin-like growth factor-1 and its analogs. Mol. Vis. 2000, 31, 157–163. [Google Scholar]
- Wilson, S.H.; Davis, M.I.; Caballero, S.; Grant, M.B. Modulation of retinal endothelial cell behaviour by insulin-like growth factor I and somatostatin analogues: Implications for diabetic retinopathy. Growth Horm. IGF Res. 2001, 11, S53–S59. [Google Scholar] [CrossRef]
- Hellström, A.; Svenssonm, E.; Carlssonm, B.; Niklassonm, A.; Albertsson-Wikland, K. Reduced retinal vascularization in children with growth hormone deficiency. J. Clin. Endocrinol. Metab. 1999, 84, 795–798. [Google Scholar] [PubMed] [Green Version]
- García-Ramírez, M.; Hernández, C.; Simó, R. Expression of erythropoietin and its receptor in the human retina: A comparative study of diabetic and nondiabetic subjects. Diabetes Care 2008, 31, 1189–1194. [Google Scholar] [CrossRef] [Green Version]
- Becerra, S.P.; Amaral, J. Erythropoietin: An endogenous retinal survival factor. N. Engl. J. Med. 2002, 347, 1968–1970. [Google Scholar] [CrossRef]
- Rex, T.S.; Wong, Y.; Kodali, K.; Merry, S. Neuroprotection of photoreceptors by direct delivery of erythropoietin to the retina of the retinal degeneration slow mouse. Exp. Eye Res. 2009, 89, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Wu, Y.; Xu, J.Y.; Zhang, J.; Sinclair, S.H.; Yanoff, M.; Xu, G.; Li, W.; Xu, G.T. ERK- and Akt-dependent neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via modulation of Bcl-xL, Bax, and BAD. Investig. Ophthalmol. Vis. Sci. 2010, 5, 35–46. [Google Scholar] [CrossRef]
- Hernández, C.; Fonollosa, A.; García-Ramírez, M.; Higuera, M.; Catalán, R.; Miralles, A.; García-Arumí, J.; Simó, R. Erythropoietin is expressed in the human retina and it is highly elevated in the vitreous fluid of patients with diabetic macular edema. Diabetes Care 2006, 29, 2028–2033. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cinnor, K.M.; Aderman, C.M.; Smith, L.E.H. Erythropoietin deficiency decreases vascular stability in mice. J. Clin. Investig. 2008, 118, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Feng, Y.; Pfister, F.; Brownlee, M. Diabetic retinopathy: Targeting vasoregression. Diabetes 2011, 60, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizu, Y.; Nakao, S.; Yoshida, S.; Hayami, T.; Arima, M.; Yamaguchi, M.; Wada, I.; Hisatomi, T.; Ikeda, Y.; Ishibashi, T.; et al. Optical Coherence Tomography Angiography Reveals Spatial Bias of Macular Capillary Dropout in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4889–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, M.E.; Wang, W.; Chen, X.; Caberoy, N.B.; Guo, F.; Shen, C.; Ji, Y.; Tian, H.; Wang, H.; Chen, R.; et al. Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J. Exp. Med. 2017, 214, 1029–1047. [Google Scholar] [CrossRef]
- Li, W.; Webster, K.A.; LeBlanc, M.E.; Tian, H. Secretogranin III: A diabetic retinopathy-selective angiogenic factor. Cell. Mol. Life Sci. 2018, 75, 635–647. [Google Scholar] [CrossRef]
- Rong, X.; Tian, H.; Yang, L.; Li, W. Function-first ligandomics for ocular vascular research and drug target discovery. Exp. Eye Res. 2019, 182, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; LeBlanc, M.E.; Wang, W.; Liang, D.; Chen, P.; Chou, T.-H.; Tian, H.; Li, W. Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis 2019, 22, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Barnstable, C.J.; Tombran-Tink, J. Neuroprotective and antiangiogenic actions of PEDF in the eye: Molecular targets and therapeutic potential. Prog. Retin. Eye Res. 2004, 23, 561–577. [Google Scholar] [CrossRef]
- Polato, F.; Becerra, S.P. Pigment epithelium-derived factor, a protective factor for photoreceptors in Vivo. Adv. Exp. Med. Biol. 2016, 854, 699–706. [Google Scholar] [PubMed]
- Elahy, M.; Baindur-Hudson, S.; Cruzat, V.F.; Newsholme, P.; Dass, C.R. Mechanisms of PEDF-mediated protection against reactive oxygen species damage in diabetic retinopathy and Neuropathy. J. Endocrinol. 2014, 222, R129–R139. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhong, Y.; Xie, B.; Cheng, Y.; Jiao, Q. Pigment epithelium derived factor as an anti-inflammatory factor against decrease of glutamine synthetase expression in retinal Müller cells under high glucose conditions. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Jiao, Q.; Cheng, Y.; Zhong, Y.; Shen, X. Effect of pigment epithelium-derived factor on glutamate uptake in retinal Müller cells under high-glucose conditions. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lu, Q.; Gao, S.; Zhu, Y.; Gao, Y.; Xier, B.; Shen, X. Pigment epithelium-derived factor regulates glutamine synthetase and l-glutamate/l-aspartate transporter in retinas with oxygen-induced retinopathy. Curr. Eye Res. 2015, 40, 1232–1244. [Google Scholar] [CrossRef]
- Liu, Y.; Leo, L.F.; McGregor, C.; Grivitishvili, A.; Barnstable, C.J.; Tombran-Tink, J. Pigment epithelium-derived factor (PEDF) peptide eye drops reduce inflammation, cell death and vascular leakage in diabetic retinopathy in Ins2Akita mice. Mol. Med. 2012, 18, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Esmaeili, M.; Deer, L.; Berry, M.; Logan, A.; Ahmed, Z. Eye drop delivery of pigment epithelium-derived factor-34 promotes retinal ganglion cell neuroprotection and axon regeneration. Mol. Cell. Neurosci. 2015, 68, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Haurigot, V.; Villacampa, P.; Ribera, A.; Bosch, A.; Ramos, D.; Ruberte, J.; Bosch, F. Long-term retinal PEDF overexpression prevents neovascularization in a murine adult model of retinopathy. PLoS ONE 2012, 7, e41511. [Google Scholar] [CrossRef] [Green Version]
- Calado, S.M.; Diaz-Corrales, F.; Silva, G.A. pEPito-Driven PEDF Expression Ameliorates Diabetic Retinopathy Hallmarks. Hum. Gene Ther. Methods 2016, 27, 79–86. [Google Scholar] [CrossRef]
- Araújo, R.S.; Silva, G.A. PlGF silencing combined with PEDF overexpression: Modeling RPE secretion as potential therapy for retinal neovascularization. Mol. Biol. Rep. 2020, 47, 4413–4425. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Carrasco, E.; Casamitjana, R. Somatostatin molecular variants in the vitreous fluid: A comparative study between diabetic patients with proliferative diabetic retinopathy and nondiabetic control subjects. Diabetes Care 2005, 28, 1941–1947. [Google Scholar] [CrossRef] [Green Version]
- Cervia, D.; Casini, G.; Bagnoli, P. Physiology and pathology of somatostatin in the mammalian retina: A current view. Mol. Cell. Endocrinol. 2008, 286, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Lambooij, A.C.; Kuijpers, R.W.; van Lichtenauer-Kaligis, E.G.; Kliffen, M.; Baarsma, G.S.; van Hagen, P.M.; Mooy, C.M. Somatostatin receptor 2A expression in choroidal neovascularization secondary to age related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2329–2335. [Google Scholar]
- Simó, R.; Carrasco, E.; Fonollosa, A.; García-Arumí, J.; Casamitjana, R.; Hernández, C. Deficit of somatostatin in the vitreous fluid of patients with diabetic macular edema. Diabetes Care 2007, 30, 725–727. [Google Scholar] [CrossRef] [Green Version]
- Simó, R.; Lecube, A.; Sararols, L.; García-Arumi, J.; Segura, R.M.; Casamitjana, R.; Hernández, C. Deficit of somatostatin-like immunoreactivity in the vitreous fluid of diabetic patients: Possible role in the development of proliferative diabetic retinopathy. Diabetes Care 2002, 25, 2282–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, M.B.; Mames, R.N.; Fitzgeral, C.; Hazariwala, K.M.; Cooper-DeHoff, R.; Caballero, S.; Estes, K.S. The efficacy of octreotide in the therapy of severe non-proliferative and early proliferative diabetic retinopathy: A randomized controlled study. Diabetes Care 2000, 23, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, B.O.; Lang, G.K.; Jehle, P.M.; Feldman, B.; Lang, G.E. Octreotide reduces vitreous hemorrhage and loss of visual acuity in patients with high-risk proliferative diabetic retinopathy. Horm. Metab. Res. 2001, 33, 300–306. [Google Scholar] [CrossRef]
- Simó-Servat, O.; Hernández, C.; Simó, R. Somatostatin and diabetic retinopathy: An evolving story. Endocrine 2018, 60, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, C.; Arroba, A.I.; Bogdanov, P.; Ramos, H.; Simó-Servat, O.; Simó, R.; Valverde, A.M. Effect of Topical Administration of Somatostatin on Retinal Inflammation and Neurodegeneration in an Experimental Model of Diabetes. J. Clin. Med. 2020, 9, 2579. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Mazzeo, A.; Cazzoni, D.; Beltramo, E.; Hernández, C.; Porta, M.; Simó, R.; Valverde, Á.M. Somatostatin protects photoreceptor cells against high glucose-induced apoptosis. Mol. Vis. 2016, 22, 1522–1531. [Google Scholar]
- Grauslund, J.; Frydkjaer-Olsen, U.; Peto, T.; Fernández-Carneado, J.; Ponsati, B.; Hernández, C.; Cunha-Vaz, J.; Simó, R.; EUROCONDOR. Topical Treatment with Brimonidine and Somatostatin Causes Retinal Vascular Dilation in Patients with Early Diabetic Retinopathy from the EUROCONDOR. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2257–2262. [Google Scholar] [CrossRef]
- Hölscher, C. Potential role of glucagon-like peptide-1 (GLP-1) in neuroprotection. CNS Drugs 2012, 26, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Arnold, S.E. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 2014, 63, 2253–2261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Q.; Zhang, J.; Lei, X.; Xu, G.T.; Ye, W. Protection of exendin-4 analogue in early experimental diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 699–706. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Wang, Q.; Lei, X.; Chu, Q.; Xu, G.T.; Ye, W. Intravitreal injection of exendin-4 analogue protects retinal cells in early diabetic rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Liu, K.; Wang, Q.; Ruan, Y.; Zhang, Y.; Ye, W. Exendin-4 protects retinal cells from early diabetes in Goto-Kakizaki rats by increasing the Bcl-2/Bax and Bcl-xL/Bax ratios and reducing reactive gliosis. Mol. Vis. 2014, 20, 1557–1568. [Google Scholar]
- Sampedro, J.; Bogdanov, P.; Ramos, H.; Solà-Adell, C.; Turch, M.; Valeri, M.; Simó-Servat, O.; Lagunas, C.; Simó, R.; Hernández, C. New Insights into the Mechanisms of Action of Topical Administration of GLP-1 in an Experimental Model of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, H.; Bogdanov, P.; Sampedro, J.; Huerta, J.; Simó, R.; Hernández, C. Beneficial Effects of Glucagon-Like Peptide-1 (GLP-1) in Diabetes-Induced Retinal Abnormalities: Involvement of Oxidative Stress. Antioxidants 2020, 9, 846. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, K.; Wang, F.; Zhou, L.; Hu, Y.; Tang, M.; Zhang, S.; Jin, S.; Zhang, J.; Wang, J.; et al. The glucagon like peptide 1 analogue, exendin-4, attenuates oxidative stress-induced retinal cell death in early diabetic rats through promoting Sirt1 and Sirt3 expression. Exp. Eye Res. 2016, 151, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, K.; Wang, Q.; Ruan, Y.; Ye, W.; Zhang, Y. Exendin-4 alleviates retinal vascular leakage by protecting the blood-retinal barrier and reducing retinal vascular permeability in diabetic Goto-Kakizaki rats. Exp. Eye Res. 2014, 127, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Vilsbøll, T.; Bain, S.C.; Leiter, L.A.; Lingvay, I.; Matthews, D.; Simó, R.; Helmark, I.C.; Wijayasinghe, N.; Larsen, M. Semaglutide, reduction in glycated haemoglobin and the risk of diabetic retinopathy. Diabetes Obes. Metab. 2018, 20, 889. [Google Scholar] [CrossRef]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Bogdanov, P.; Solà-Adell, C.; Sampedro, J.; Valeri, M.; Genís, X.; Simó-Servat, O.; García-Ramírez, M.; Simó, R. Topical administration of DPP-IV inhibitors prevents retinal neurodegeneration in experimental diabetes. Diabetologia 2017, 60, 2285–2298. [Google Scholar] [CrossRef]
- Breckler, M.; Berthouze, M.; Laurent, A.C.; Crozatier, B.; Morel, E.; Lezoualc’h, F. Rap-linked cAMP signaling Epac proteins: Compartmentation, functioning and disease implications. Cell. Signal. 2011, 23, 1257–1266. [Google Scholar] [CrossRef]
- Boddu, S.H.S.; Gupta, H.; Patel, S. Drug delivery to the back of the eye following topical administration: An update on research and patenting activity. Recent Pat. Drug Deliv. Formul. 2014, 8, 27–36. [Google Scholar] [CrossRef]
- Deng, D.; Evans, T.; Mukherjee, K.; Downey, D.; Chakrabarti, S. Diabetes-induced vascular dysfunction in the retina: Role of endothelins. Diabetologia 1999, 42, 1228–1234. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Gan, X.T.; Merry, A.; Karmazyn, M.; Sima, A.A. Augmented retinal endothelin-1, endothelin-2, endothelinA and endothelinB gene expression in chronic diabetes. Curr. Eye Res. 1998, 17, 301–307. [Google Scholar] [CrossRef]
- Chou, J.C.; Rollins, S.D.; Ye, M.; Batlle, D.; Fawzi, A.A. Endothelin receptor-A antagonist attenuates retinal vascular and neuroretinal pathology in diabetic mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2516–2525. [Google Scholar] [CrossRef] [Green Version]
- Minton, A.Z.; Phatak, N.R.; Stankowska, D.L.; He, S.; Ma, H.Y.; Mueller, B.H.; Jiang, M.; Luedtke, R.; Yang, S.; Brownlee, C.; et al. Endothelin B receptors contribute to retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e43199. [Google Scholar] [CrossRef]
- Tonari, M.; Kurimoto, T.; Horie, T.; Sugiyama, T.; Ikeda, T.; Oku, H. Blocking endothelin-B receptors rescues retinal ganglion cells from optic nerve injury through suppression of neuroinflammation. Investig. Ophthalmol. Vis. Sci. 2012, 8, 3490–3500. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Bogdanov, P.; Gómez-Guerrero, C.; Sampedro, J.; Solà-Adell, C.; Espejo, C.; García-Ramírez, M.; Prieto, I.; Egido, J.; Simó, R. SOCS1-Derived Peptide Administered by Eye Drops Prevents Retinal Neuroinflammation and Vascular Leakage in Experimental Diabetes. Int. J. Mol. Sci. 2019, 20, 3615. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.J.; Antonetti, D.A.; Kern, T.S.; Reiter, C.E.; Soans, R.S.; Krady, J.K.; Levison, S.W.; Gardner, T.W.; Bronson, S.K. The Ins2Akita mouse as a model of early retinal complications in diabetes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2210–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanGuilder, H.D.; Brucklacher, R.M.; Patel, K.; Ellis, R.W.; Freeman, W.M.; Barber, A.J. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur. J. Neurosci. 2008, 28, 1–11. [Google Scholar] [CrossRef]
- Gaspar, J.M.; Baptista, F.I.; Galvão, J.; Castilho, A.F.; Cunha, R.A.; Ambrósio, A.F. Diabetes differentially affects the content of exocytotic proteins in hippocampal and retinal nerve terminals. Neuroscience 2010, 169, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Scheerer, M.F.; Zukunft, C.S.; Merl, J.; Adamski, J.; Hrabe de Angelis, M.; Neschen, S.; Hauck, S.M.; Ueffing, M. Retinal proteome alterations in a mouse model of type 2 diabetes. Diabetologia 2014, 57, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhof, T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef] [Green Version]
- Saheki, Y.; De Camilli, P. Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005645. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O. Synaptic vesicle recycling: Steps and principles. EMBO J. 2014, 33, 788–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz, R.W.; Zimmerberg, J. Dynamic Relationship of the SNARE Complex with a Membrane. Biophys. J. 2019, 117, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhou, P.; Wang, A.L.; Wu, D.; Zhao, M.; Südhof, T.C.; Brunger, A.T. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 2017, 548, 420–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Rizo, J.; Südhof, T. Snares and munc18 in synaptic vesicle fusion. Nat. Rev. Neurosci. 2002, 3, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Calloway, N.; Gouzer, G.; Xue, M.; Ryan, T.A. The active-zone protein Munc13 controls the use-dependence of presynaptic voltage-gated calcium channels. Elife 2015, 4, e07728. [Google Scholar] [CrossRef]
- Robinson, W.F.; VanGuilder, H.D.; D’Cruz, T.S.; El-Remessy, A.B.; Barber, A.J. Synapsin 1 Protein Expression and Phosphorylation Are Compromised by Diabetes in Rodent and Human Retinas. Invest. Ophthalmol. Vis. Sci. 2008, 49, 4920. [Google Scholar]
- Masser, D.R.; VanGuilder, H.D.; Bixler, G.V.; Dunton, W.; Bronson, S.K.; Freeman, W.M. Insulin treatment normalizes retinal neuroinflammation but not markers of synapse loss in diabetic rats. Exp. Eye Res. 2014, 125, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Lai, J.; Yuan, Y.; Wang, L.; Wang, Q.; Yuan, F. Taurine Protects Retinal Cells and Improves Synaptic Connections in Early Diabetic Rats. Curr. Eye Res. 2020, 45, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Sasaki, M.; Takahashi, N.; Kamoshita, M.; Miyake, S.; Tsubota, K. Neuroprotective effects of lutein in the retina. Curr. Pharm. Des. 2012, 18, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3β-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, X.; Zhang, Y.; Li, M.; Huang, X.; Yang, Y.; Zeng, J.; Zhao, Y.; Wang, X.; Zhang, W.; Ying, Y. Topical ocular administration of the GLP-1 receptor agonist liraglutide arrests hyperphosphorylated tau-triggered diabetic retinal neurodegeneration via activation of GLP-1R/Akt/GSK3β signaling. Neuropharmacology 2019, 153, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.M.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 2016, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- García-Ramírez, M.; Canals, F.; Hernández, C.; Colomé, N.; Ferrer, C.; Carrasco, E.; García-Arumí, J.; Simó, R. Proteomic analysis of human vitreous fluid by fluorescence-based difference gel electrophoresis (DIGE): A new strategy for identifying potential candidates in the pathogenesis of proliferative diabetic retinopathy. Diabetologia 2007, 50, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Perlson, E.; Maday, S.; Fu, M.M.; Moughamian, A.J.; Holzbaur, E.L.F. Retrograde axonal transport: Pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Baptista, F.I.; Pinto, M.J.; Elvas, F.; Martins, T.; Almeida, R.D.; Ambrósio, A.F. Diabetes induces changes in KIF1A, KIF5B and dynein distribution in the rat retina: Implications for axonal transport. Exp. Eye Res. 2014, 127, 91–103. [Google Scholar] [CrossRef]
- Sundstrom, J.M.; Hernández, C.; Weber, S.R.; Zhao, Y.; Dunklebarger, M.; Tiberti, N.; Laremore, T.; Simó-Servat, O.; García-Ramirez, M.; Barber, A.J.; et al. Proteomic analysis of early diabetic retinopathy reveals mediators of neurodegennerative brain diseases. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2264–2274. [Google Scholar] [CrossRef] [Green Version]
- Simó-Servat, O.; Hernández, C.; Sundstrom, J.; García-Ramirez, M.; Gardner, T.W.; Simó, R. Mediators between Reactive gliosis and Vascular leakage in Diabetic Retinopathy: A Proteomic approach using Human Retinas. Diabetologia 2016, 59 (Suppl. 1), S478. [Google Scholar]
- Kevenaar, J.T.; Hoogenraad, C.C. The axonal cytoskeleton: From organization to function. Front. Mol. Neurosci. 2015, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Grubb, M.S.; Shu, Y.; Kuba, H.; Rasband, M.N.; Wimmer, V.C.; Bender, K.J. Short- and long-term plasticity at the axon initial segment. J. Neurosci. 2011, 31, 16049–16550. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.L.; Korobova, F.; Svitkina, T. Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J. Cell. Biol. 2014, 205, 67–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamis, A.P. Is diabetic retinopathy an inflammatory disease? Br. J. Ophthalmol. 2002, 86, 363–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, T.S. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 95103. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liu, H.; Rojas, M.; Caldwell, R.W.; Caldwell, R.B. Anti-inflammatory therapy for diabetic retinopathy. Immunotherapy 2011, 3, 609–628. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Segura, R.M.; Fonollosa, A.; Carrasco, E.; Francisco, G.; Simó, R. Interleukin-8, monocyte chemoattractant protein-1 and IL-10 in the vitreous fluid of patients with proliferative diabetic retinopathy. Diabet. Med. 2005, 22, 719–722. [Google Scholar] [CrossRef]
- El-Asrar, A.M.; Nawaz, M.I.; Kangave, D.; Geboes, K.; Ola, M.S.; Ahmad, S.; Al-Shabrawey, M. High-mobility group box-1 and biomarkers of inflammation in the vitreous from patients with proliferative diabetic retinopathy. Mol. Vis. 2011, 17, 1829–1838. [Google Scholar]
- Simó-Servat, O.; Hernández, C.; Simó, R. Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediat. Inflamm. 2012, 2012, 872978. [Google Scholar] [CrossRef]
- Sorrentino, F.S.; Allkabes, M.; Salsini, G.; Bonifazzi, C.; Perri, P. The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci. 2016, 162, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnalagu, M.; Subramani, M.; Jayadev, C.; Shetty, R.; Das, D. Retinal pigment epithelium-secretome: A diabetic retinopathy perspective. Cytokine 2017, 95, 126–135. [Google Scholar] [CrossRef]
- Rangasamy, S.; McGuire, P.G.; Franco, C.; Monickaraj, F.; Oruganti, S.R.; Das, A. Chemokine mediated monocyte trafficking into the retina: Role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS ONE 2014, 9, e108508. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Murata, T.; Tsujikawa, A.; Kirchhof, B.; Bursell, S.E.; Adamis, A.P. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am. J. Pathol. 2001, 158, 147–152. [Google Scholar] [CrossRef] [Green Version]
- McLeod, D.S.; Lefer, D.J.; Merges, C.; Lutty, G.A. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am. J. Pathol. 1995, 147, 642–653. [Google Scholar] [PubMed]
- Bringmann, A.; Wiedemann, P. Müller glial cells in retinal disease. Ophthalmologica 2012, 227, 1–19. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Stitt, A.W.; O’Neill, C.L.; O’Doherty, M.T.; Archer, D.B.; Gardiner, T.A.; Medina, R.J. Vascular stem cells and ischaemic retinopathies. Prog. Retin. Eye Res. 2011, 30, 149–166. [Google Scholar] [CrossRef]
- Franze, K.; Grosche, J.; Skatchkov, S.N.; Schinkinger, S.; Foja, C.; Schild, D.; Uckermann, O.; Travis, K.; Reichenbach, A.; Guck, J. Muller cells are living optical fibers in the vertebrate retina. Proc. Natl. Acad. Sci. UAS 2007, 104, 8287–8292. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Labin, A.M.; Safuri, S.K.; Ribak, E.N.; Perlman, I. Müller cells separate between wavelengths to improve day vision with minimal effect upon night vision. Nat. Commun. 2014, 5, 4319. [Google Scholar] [CrossRef] [Green Version]
- Lindenau, W.; Kuhrt, H.; Ulbricht, E.; Körner, K.; Bringmann, A.; Reichenbach, A. Cone-to-Müller cell ratio in the mammalian retina: A survey of seven mammals with different lifestyle. Exp. Eye Res. 2019, 181, 38–48. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.; Allen, D.M.; Cardona, A.E. The role of microglia in diabetic retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, P.; Kijlstra, A. Distribution, markers, and functions of retinal microglia. Ocul. Immunol. Inflamm. 2002, 10, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Jehle, T.; Dimitriu, C.; Auer, S.; Knoth, R.; Vidal-Sanz, M.; Gozes, I.; Lagrèze, W.A. The neuropeptide NAP provides neuroprotection against retinal ganglion cell damage after retinal ischemia and optic nerve crush. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 1255–1263. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polazzi, E.; Contestabile, A. Reciprocal interactions between microglia and neurons: From survival to neuropathology. Rev. Neurosci. 2002, 13, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Harada, T.; Quah, H.M.A.; Maekawa, F.; Yoshida, K.; Ohno, S.; Wada, K.; Parada, L.F.; Tanaka, K. Potential role of glial cell line-derived neurotrophic factor receptors in Müller glial cells during light-induced retinal degeneration. Neuroscience 2003, 122, 229–235. [Google Scholar] [CrossRef]
- Arai-Gaun, S.; Katai, N.; Kikuchi, T.; Kurokawa, T.; Ohta, K.; Yoshimura, N. Heme oxygenase-1 induced in Müller cells plays a protective role in retinal ischemia-reperfusion injury in rats. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4226–4232. [Google Scholar] [CrossRef] [Green Version]
- Hauck, S.M.; Kinkl, N.; Deeg, C.A.; Swiatek-de Lange, M.; Schöffmann, S.; Ueffing, M. GDNF family ligands trigger indirect neuroprotective signaling in retinal glial cells. Mol. Cell. Biol. 2006, 26, 2746–2757. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.A. The Multifaceted Profile of Activated Microglia. Mol. Neurobiol. 2009, 40, 139–156. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Odenbach, S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br. J. Ophthalmol. 2004, 88, 1343–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.H. Interleukin-1β induces pericyte apoptosis via the NF-κB pathway in diabetic retinopathy. Biochem. Biophys. Res. Commun. 2021, 546, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.A.; Mohr, S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 2007, 56, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Xu, C.; Zhong, Y.; Shen, X. Role of interleukin-1b in hypoxia-induced depression of glutamate uptake in retinal Müller cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2014, 252, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrósio, A.F.; Antonetti, D.A. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Wijk, A.E.; Vogels, I.M.C.; van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. TNFα-Induced Disruption of the Blood-Retinal Barrier In Vitro Is Regulated by Intracellular 3’,5’-Cyclic Adenosine Monophosphate Levels. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3496–3505. [Google Scholar] [CrossRef] [Green Version]
- Kitaoka, Y.; Kitaoka, Y.; Kwong, J.M.; Ross-Cisneros, F.N.; Wang, J.; Tsai, R.K.; Sadun, A.A.; Lam, T.T. TNF-α-induced optic nerve degeneration and nuclear factor-κB p65. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1448–1457. [Google Scholar] [CrossRef]
- Madigan, M.C.; Sadun, A.A.; Rao, N.S.; Dugel, P.U.; Tenhula, W.N.; Gill, P.S. Tumor necrosis factor-alpha (TNF-alpha)-induced optic neuropathy in rabbits. Neurol. Res. 1996, 18, 176–184. [Google Scholar] [CrossRef]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Kirchhof, B.; Koizumi, K.; Döhmen, S.; Adamis, A.P. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002, 16, 438–440. [Google Scholar] [CrossRef]
- Joussen, A.M.; Doehmen, S.; Le, M.L.; Koizumi, K.; Radetzky, S.; Krohne, T.U.; Poulaki, V.; Semkova, I.; Kociok, N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol. Vis. 2009, 15, 1418–1428. [Google Scholar]
- Behl, Y.; Krothapalli, P.; Desta, T.; DiPiazza, A.; Roy, S.; Graves, D.T. Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. Am. J. Pathol. 2008, 172, 1411–1418. [Google Scholar] [CrossRef] [Green Version]
- Behl, Y.; Krothapalli, P.; Desta, T.; Roy, S.; Graves, D.T. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 2009, 58, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Sama, D.M.; Mohmmad Abdul, H.; Furman, J.L.; Artiushin, I.A.; Szymkowski, D.E.; Scheff, S.W.; Norris, C.M. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and cA2+ dysregulation in aged rats. PLoS ONE 2012, 7, e38170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albensi, B.C.; Mattson, M.P. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse 2000, 35, 151–159. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Linossi, E.M.; Babon, J.J.; Hilton, D.J.; Nicholson, S.E. Suppression of cytokine signaling: The SOCS perspective. Cytokine Growth Factor Rev. 2013, 24, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Ohira, A.; Hara, K.; Jóhannesson, G.; Tanito, M.; Asgrimsdóttir, G.M.; Lund, S.H.; Loftsoon, T.; Stefánsson, E. Topical dexamethasone γ-cyclodextrin nanoparticle eye drops increase visual acuity and decrease macular thickness in diabetic macular oedema. Acta Ophthalmol. 2015, 93, 610–615. [Google Scholar] [CrossRef]
- Semeraro, F.; Russo, A.; Gambicorti, E.; Duse, S.; Morescalchi, F.; Vezzoli, S.; Costagliola, C. Efficacy and vitreous levels of topical NSAIDs. Expert Opin. Drug Deliv. 2015, 12, 1767–1782. [Google Scholar] [CrossRef]
- Friedman, S.M.; Almukhtar, T.H.; Baker, C.W.; Glassman, A.R.; Elman, M.J.; Bressler, N.M.; Maker, M.P.; Jampol, L.M.; Melia, M. Diabetic Retinopathy Clinical Research Network. Topical nepafenec in eyes with noncentral diabetic macular edema. Retina 2015, 35, 944–956. [Google Scholar] [CrossRef]
- Park, J.C.; Chen, Y.F.; Liu, M.; Liu, K.; McAnany, J.J. Structural and Functional Abnormalities in Early-stage Diabetic Retinopathy. Curr. Eye Res. 2020, 45, 975–985. [Google Scholar] [CrossRef]
- Jackson, G.R.; Barber, A.J. Visual dysfunction associated with diabetic retinopathy. Curr. Diabetes. Rep. 2010, 10, 380–384. [Google Scholar] [CrossRef]
- Wolff, B.E.; Bearse, M.A.; Schneck, M.E.; Dhamdhere, K.; Harrisson, W.W.; Barez, S.; Adams, A.J. Color vision and neuroretinal function in diabetes. Doc. Ophthalmol. 2015, 130, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Trento, M.; Durando, O.; Lavecchia, S.; Charrier, L.; Cavallo, F.; Costa, M.A.; Hernández, C.; Simó, R.; Porta, M.; EUROCONDOR trial investigators. Vision related quality of life in patients with type 2 diabetes in the EUROCONDOR trial. Endocrine 2017, 57, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Rohrschneider, K.; Bültmann, S.; Springer, C. Use of fundus perimetry (microperimetry) to quantify macular sensitivity. Prog. Retin. Eye Res. 2008, 27, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ayton, L.N.; Guymer, R.H.; Luu, C.D. Comparison between multifocal electroretinography and microperimetry in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6431–6439. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Kondo, M.; Sugimoto, M.; Ikesugi, K.; Matsubara, H. Effect of pupil size on flicker ERGs recorded with RETeval system: New mydriasis-free full-field ERG system. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3684–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garhofer, G.; Bek, T.; Boehm, A.G.; Gherghel, D.; Grunwald, J.; Jeppesen, P.; Kergoat, H.; Kotliar, K.; Lanzl, I.; Lovasik, J.V.; et al. Use of the retinal vessel analyzer in ocular blood flow research. Acta Ophthalmol. 2010, 88, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Mandecka, A.; Dawczynski, J.; Blum, M.; Müller, N.; Kloos, C.; Wolf, G.; Vilser, W.; Hoyer, H.; Müller, U.A. Influence of flickering light on the retinal vessels in diabetic patients. Diabetes Care 2007, 30, 3048–3052. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Kawasaki, R.; Wang, J.J.; Kreis, A.J.; Shaw, J.; Vilser, W.; Wong, T.Y. Flicker light-induced retinal vasodilation in diabetes and diabetic retinopathy. Diabetes Care 2009, 32, 2075–2080. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.S.; Ling, L.H.; Ong, P.G.; Foulds, W.; Tai, E.S.; Wong, E.; Lee, S.Y.; Wong, D.; Cheung, C.M.G.; Wong, T.Y. Dynamic responses in retinal vessel caliber with flicker light stimulation in eyes with diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5207–5213. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.S.; Ling, L.H.; Ong, P.G.; Foulds, W.; Tai, E.S.; Wong, T.Y. Dynamic Responses in Retinal Vessel Caliber with Flicker Light Stimulation and Risk of Diabetic Retinopathy and Its Progression. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Fujimoto, J.G.; Waheed, N.K.; Sadda, S.R.; Staurenghi, G. Optical coherence tomography angiography. Prog. Retin. Eye Res. 2018, 64, 1–55. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Treatment | Reference | Neuro- degeneration | Micro- angiopathy | Mechanisms of Action | |||
|---|---|---|---|---|---|---|---|
| Glial Inflammation | Neuronal Apoptosis | Oxidative Stress | Vascular Permeability | ||||
| PEDF | [91] | yes | yes | + | + | + | |
| SST | [59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104] | yes | + | + | |||
| GLP-1 | [60,112, 113] | yes | yes | + | + | + | + |
| DPP-IVi | [118] | yes | yes | + | + | + | + |
| Bosentan | [61] | yes | yes | + | + | + | |
| SOCS-1 | [126] | yes | yes | + | + | + | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simó, R.; Simó-Servat, O.; Bogdanov, P.; Hernández, C. Neurovascular Unit: A New Target for Treating Early Stages of Diabetic Retinopathy. Pharmaceutics 2021, 13, 1320. https://doi.org/10.3390/pharmaceutics13081320
Simó R, Simó-Servat O, Bogdanov P, Hernández C. Neurovascular Unit: A New Target for Treating Early Stages of Diabetic Retinopathy. Pharmaceutics. 2021; 13(8):1320. https://doi.org/10.3390/pharmaceutics13081320
Chicago/Turabian StyleSimó, Rafael, Olga Simó-Servat, Patricia Bogdanov, and Cristina Hernández. 2021. "Neurovascular Unit: A New Target for Treating Early Stages of Diabetic Retinopathy" Pharmaceutics 13, no. 8: 1320. https://doi.org/10.3390/pharmaceutics13081320