
Benefit of a Short Chain Peptide as a Targeting Ligand of Nanocarriers for a Brain-Driven Purpose

Abstract
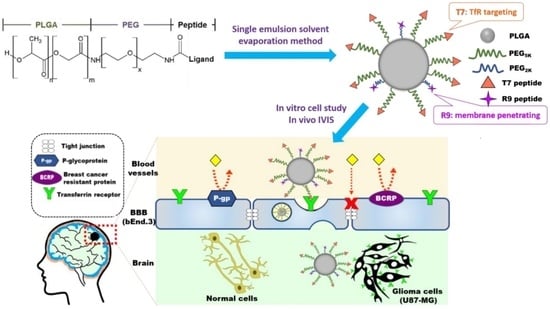
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Peptide-Conjugated Copolymers
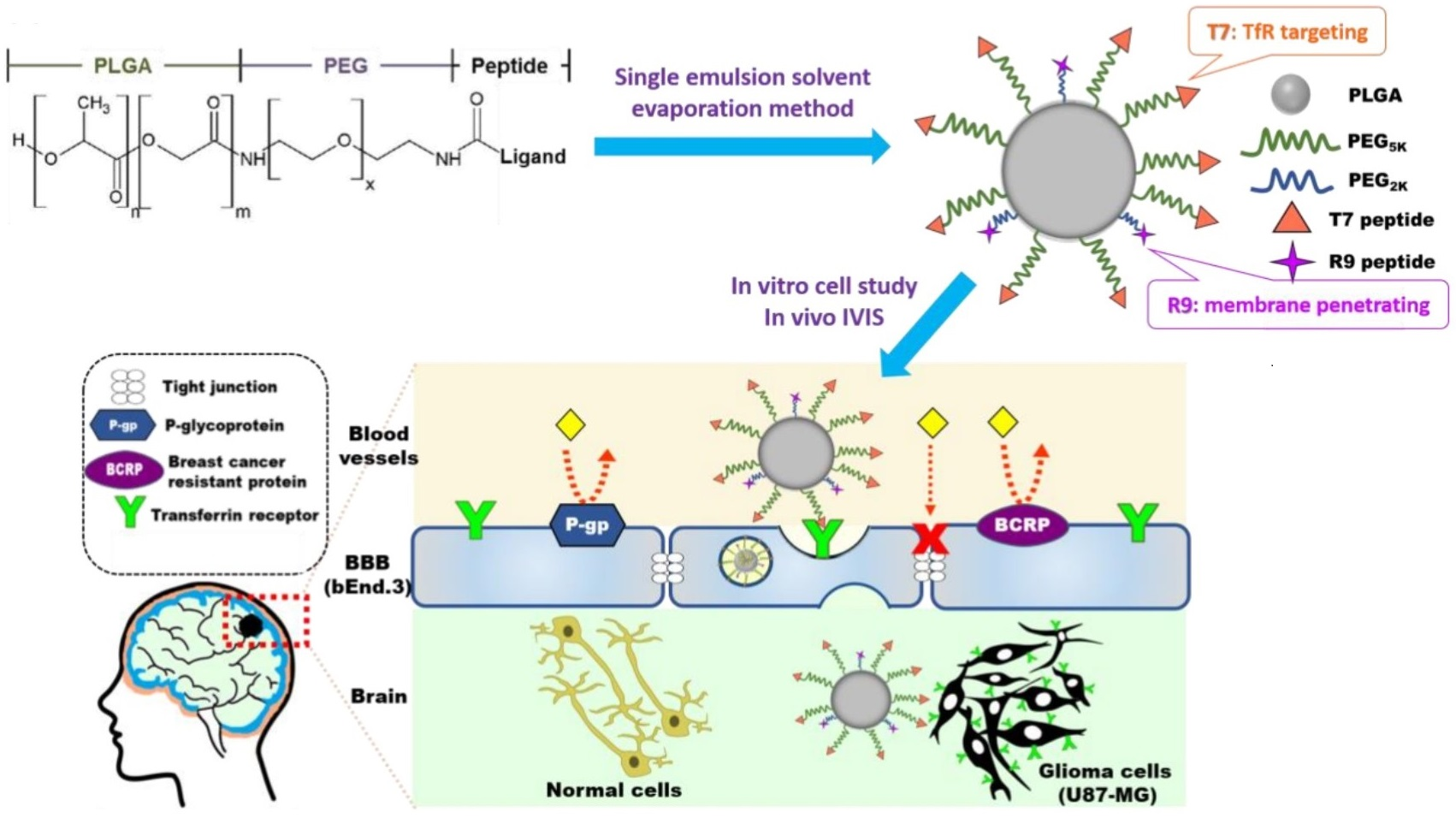
2.3. Preparation and Characterization of NPs
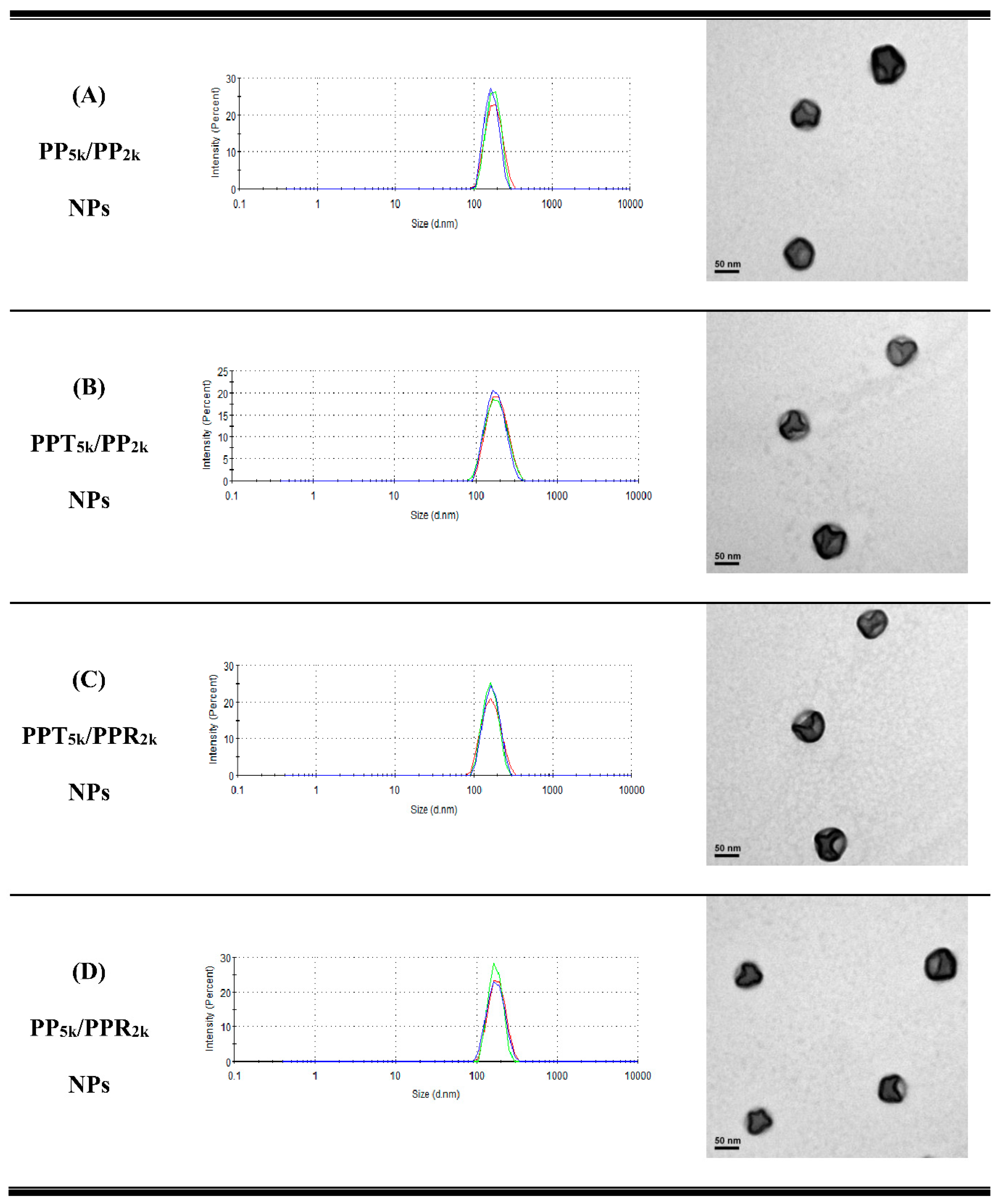
2.4. Cellular Uptake of T7-Conjugated NPs
2.5. Cellular Uptake of Single and Dual Peptide Conjugated NPs
2.6. Transport Study in Cocultured BBB Model
2.7. Endocytosis Mechanism Study
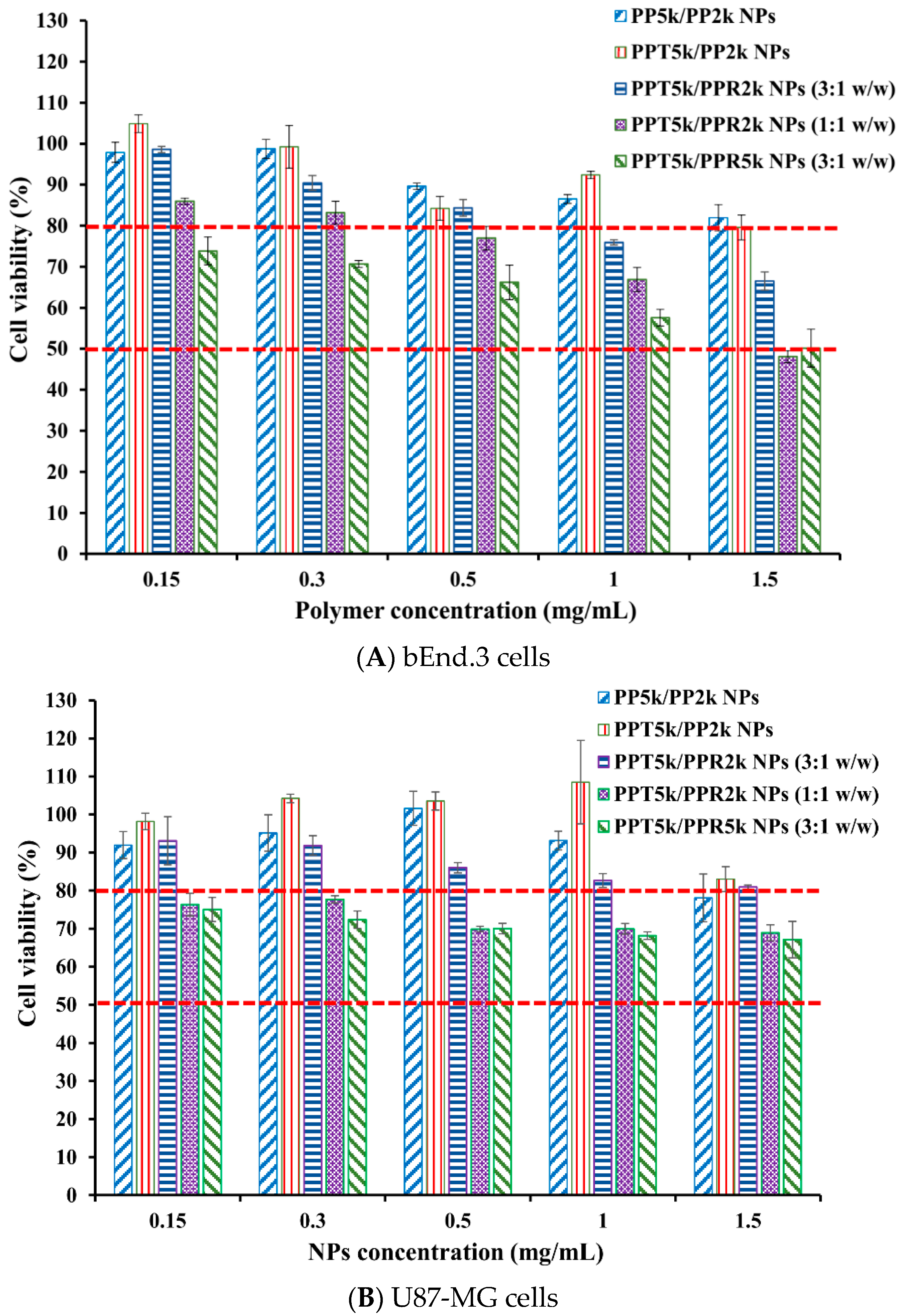
2.8. Cytotoxicity of Blended NPs
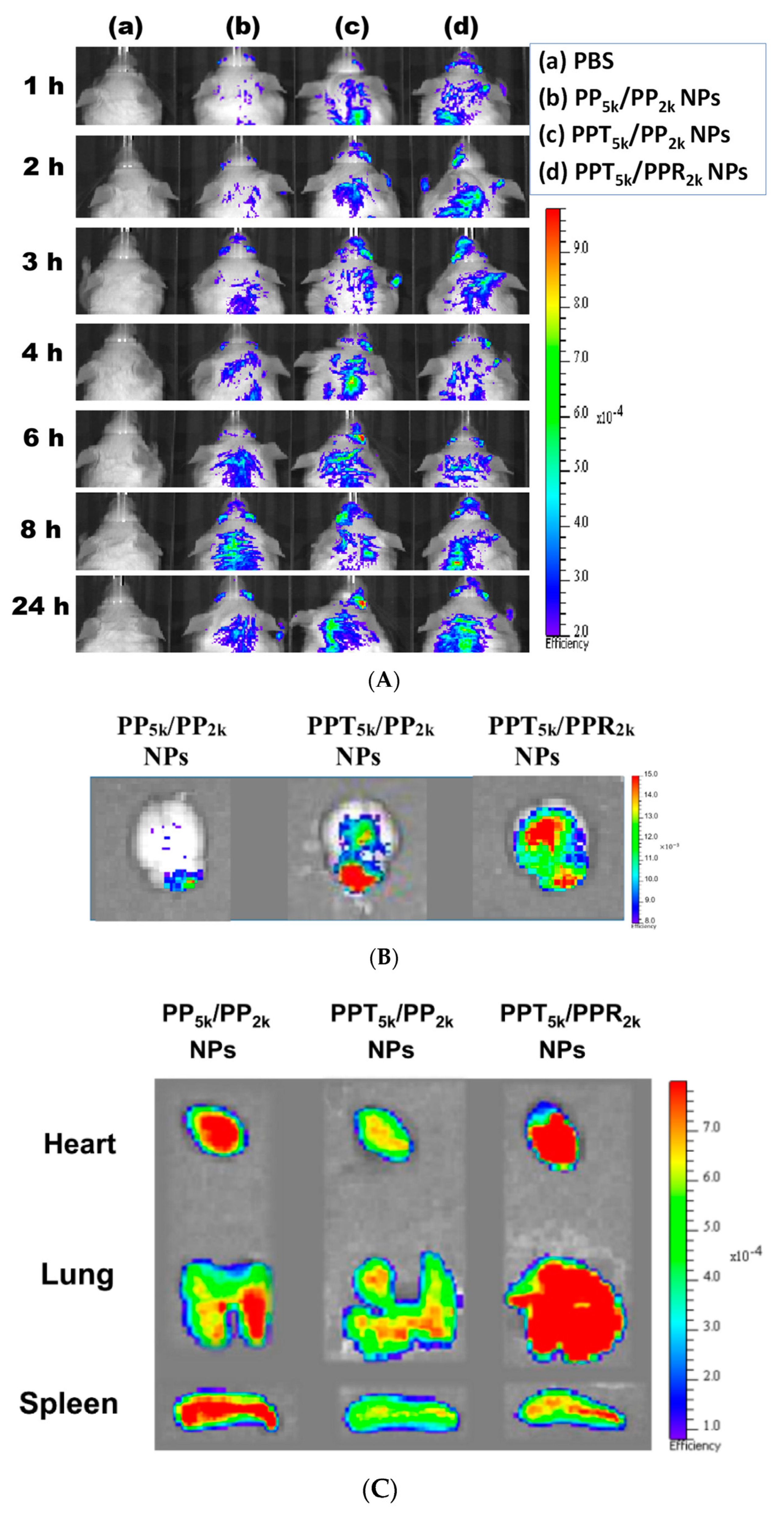
2.9. In Vivo Biodistribution Study
2.10. Statistics
3. Results and Discussion
3.1. Characterization of Peptide-Conjugated Copolymer
3.2. Characterization of NPs
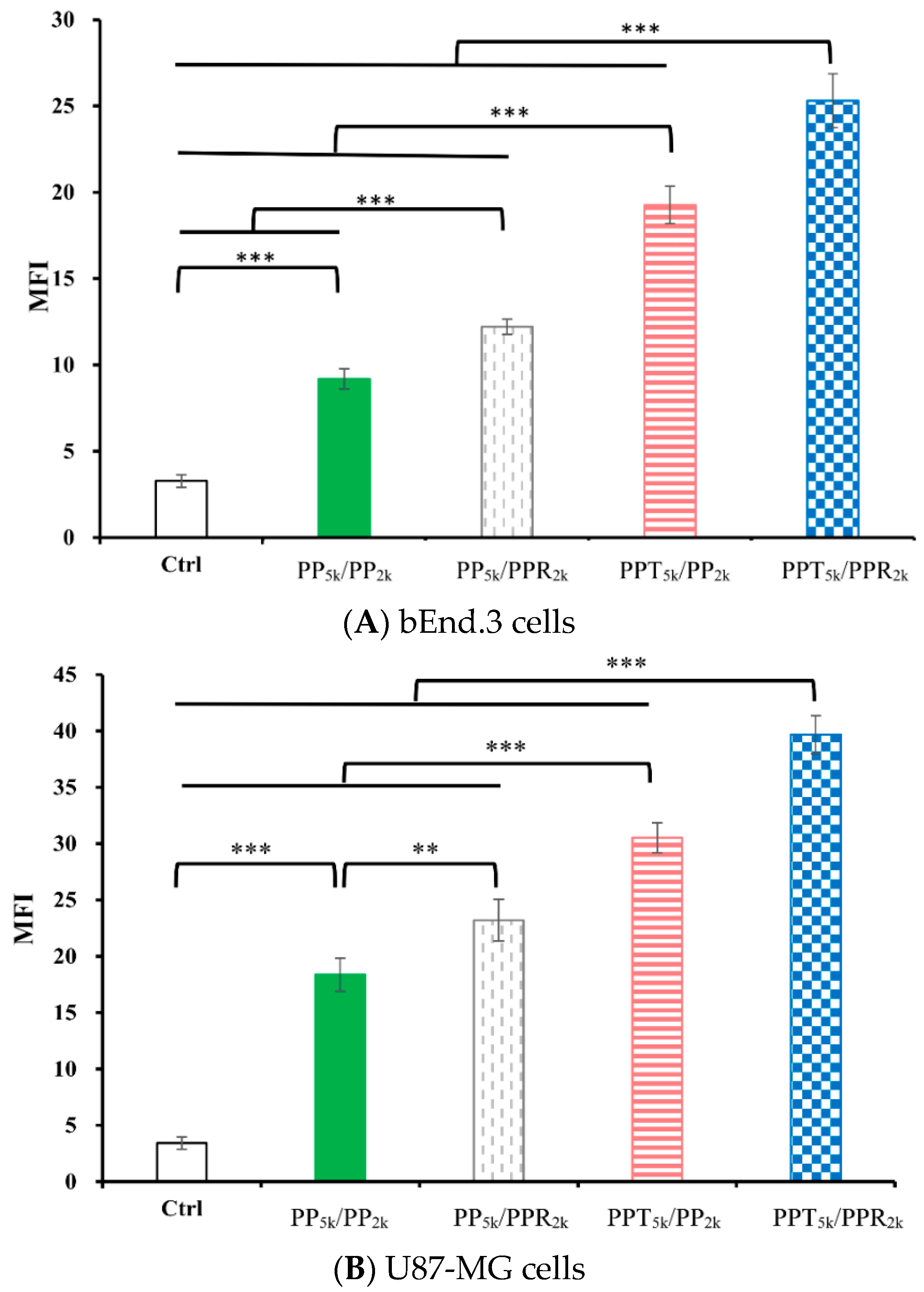
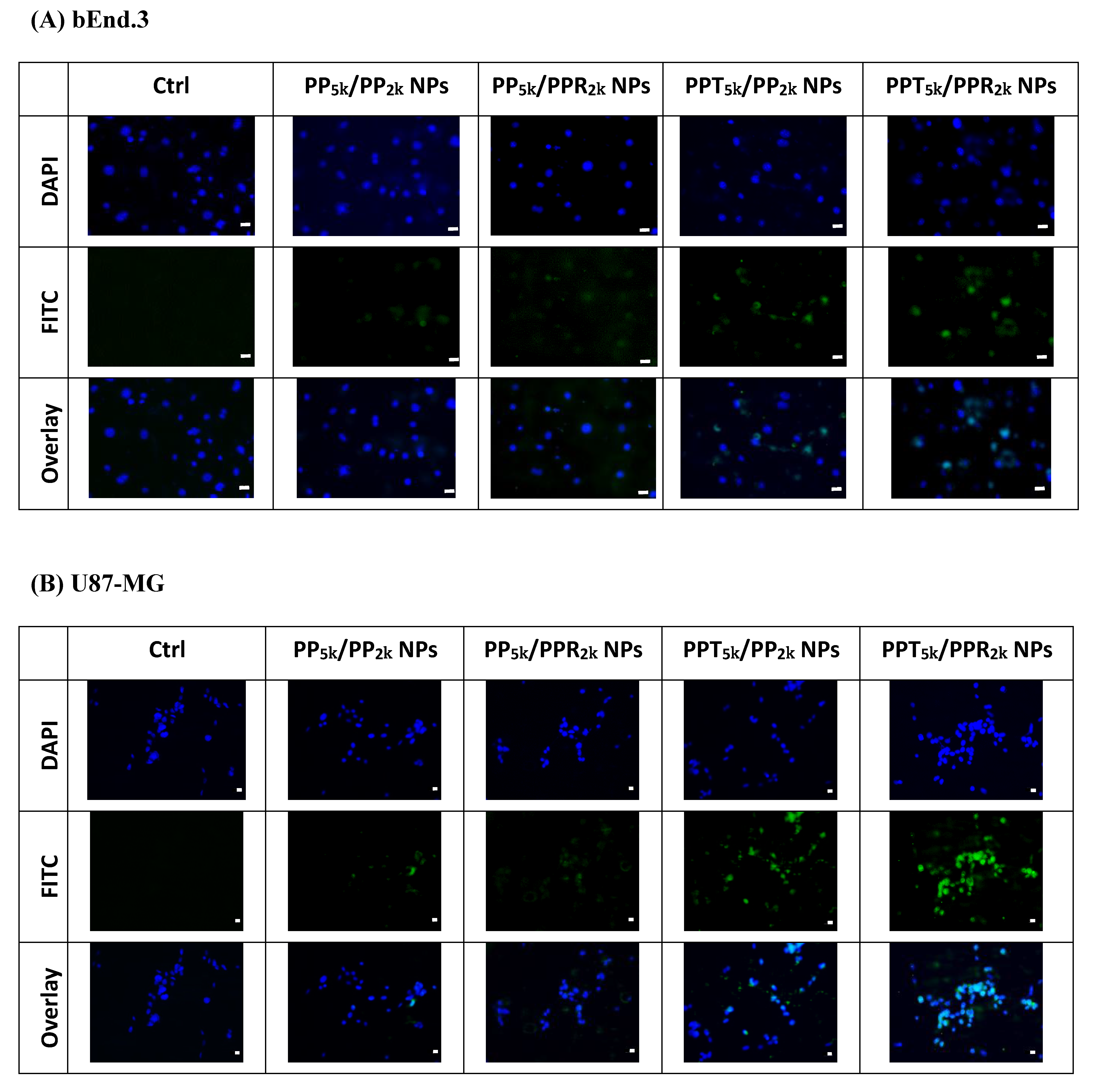
3.3. Cellular Uptake of Peptide-Conjugated NPs
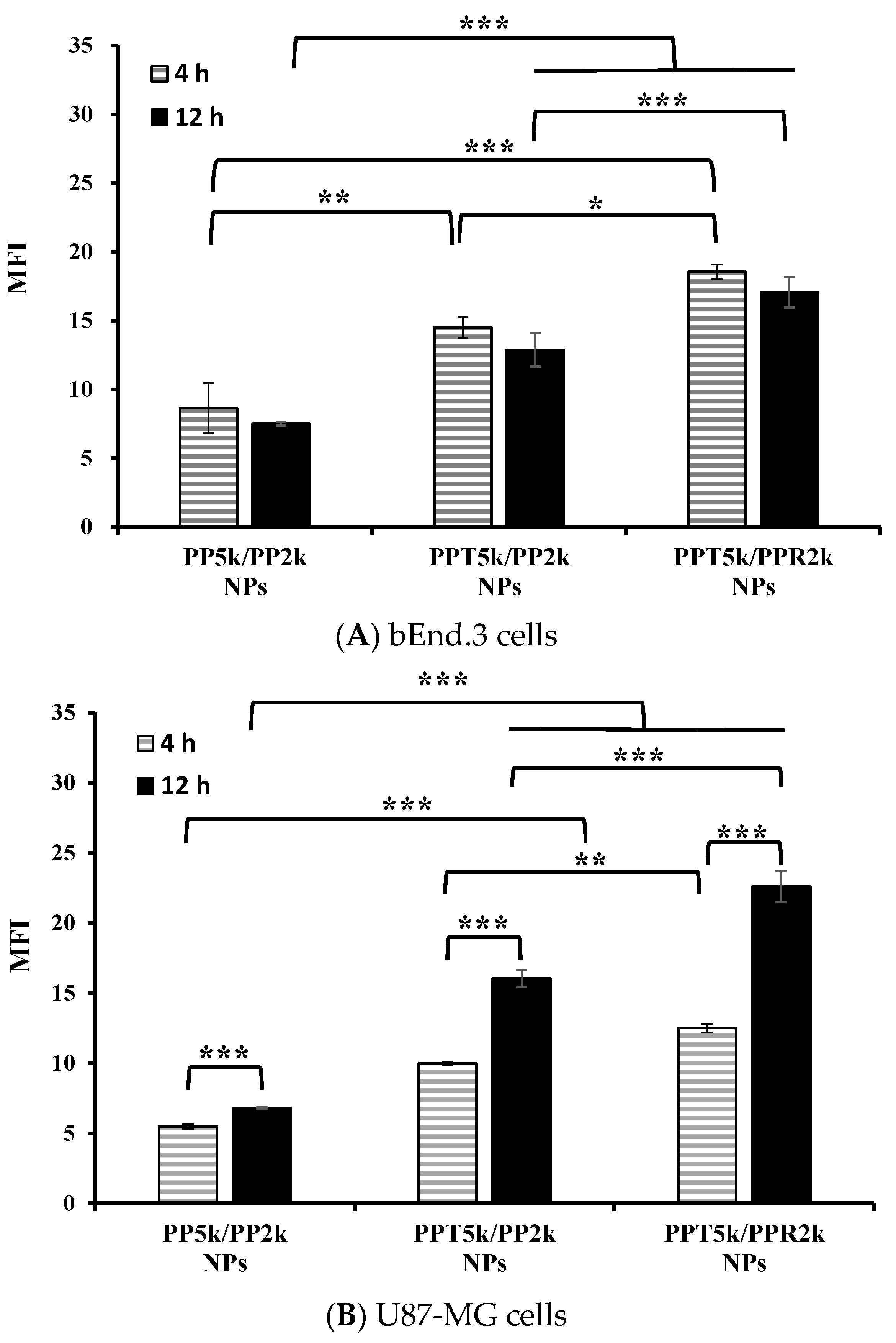
3.4. Cellular Uptake in the Cocultured BBB Model
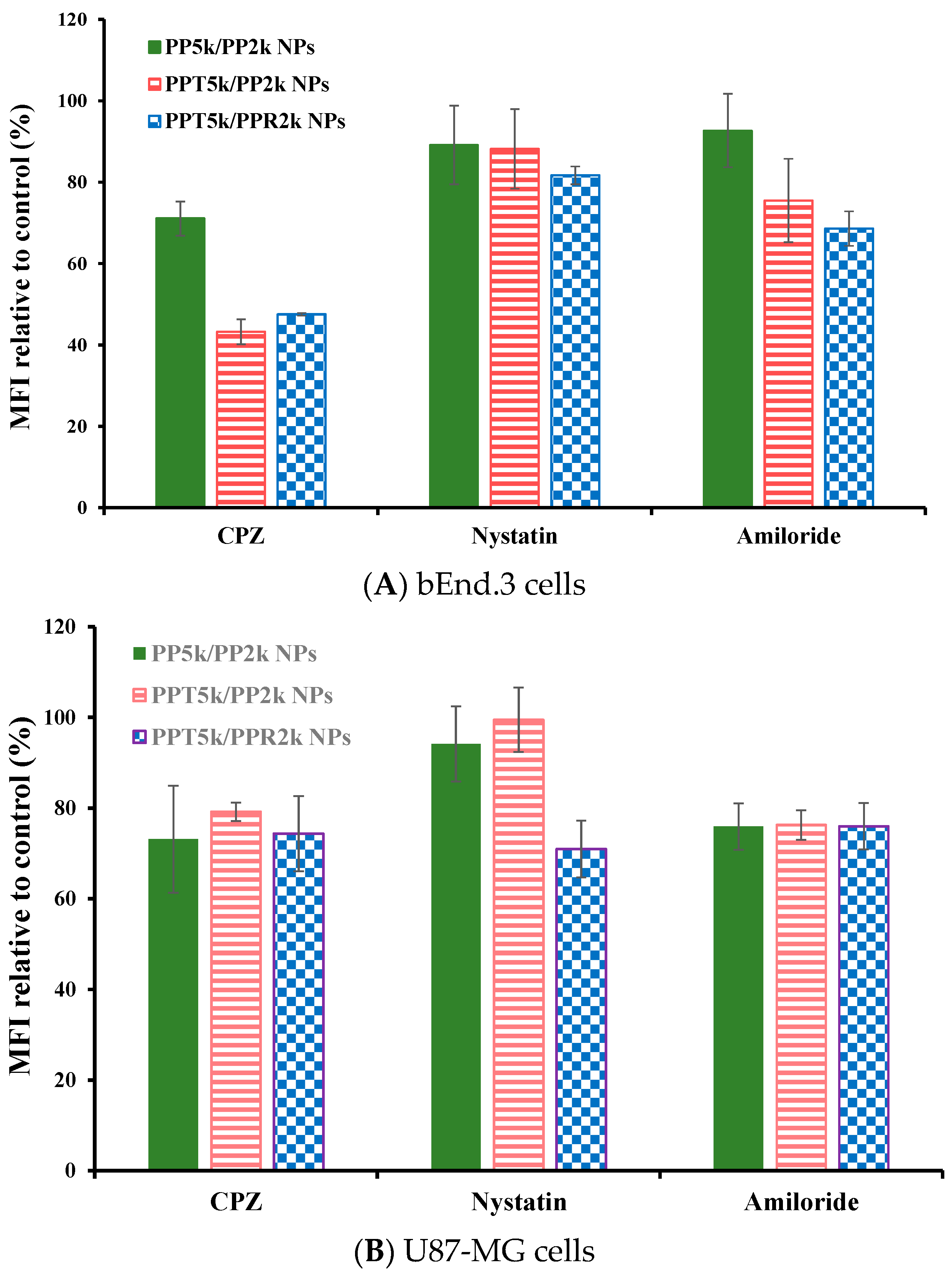
3.5. Endocytosis Mechanism
3.6. Cytotoxicity of Blended NPs
3.7. In Vivo Biodistribution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar]
- Oh, S.; Kim, B.J.; Singh, N.P.; Lai, H.; Sasaki, T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009, 274, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Liu, L.; Lu, Y.; Sun, T.; Shen, C.; Chen, X.; Chen, Q.; An, S.; He, X.; Ruan, C.; et al. T7 peptide-functionalized PEG-PLGA micelles loaded with carmustine for targeting therapy of glioma. ACS Appl. Mater. Interfaces 2016, 8, 27465–27473. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Zeng, F.; Jin, H.; Xu, Q.; Huang, Y. Dual-targeting magnetic PLGA nanoparticles for codelivery of paclitaxel and curcumin for brain tumor therapy. ACS Appl. Mater. Interfaces 2016, 8, 32159–32169. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Huang, R.; Liu, S.; Huang, S.; Jiang, C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol. Pharm. 2010, 7, 2156–2165. [Google Scholar] [CrossRef]
- Thakur, A.; Sidu, R.K.; Zou, H.; Alam, M.K.; Yang, M.; Lee, Y. Inhibition of glioma cells’ proliferation by doxorubicin-loaded exosomes via microfluidics. Int. J. Nanomed. 2020, 15, 8331–8343. [Google Scholar] [CrossRef] [PubMed]
- Zong, T.; Mei, L.; Gao, H.; Cai, W.; Zhu, P.; Shi, K.; Chen, J.; Wang, Y.; Gao, F.; He, Q. Synergistic dual-ligand doxorubicin liposomes improve targeting and therapeutic efficacy of brain glioma in animals. Mol. Pharm. 2014, 11, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; An, S.; Guo, Y.; Huang, S.; Shao, K.; Liu, Y.; Ma, L.H.; Jiang, C. T7 peptide-functionalized nanoparticles utilizing RNA interference for glioma dual targeting. Int. J. Pharm. 2013, 454, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Guo, X.Y.; Yang, T.; Yu, M.Z.; Chen, D.W.; Wang, J.C. Brain tumor-targeted therapy by systemic delivery of siRNA with Transferrin receptor-mediated core-shell nanoparticles. Int. J. Pharm. 2016, 510, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Chen, X.; Ying, M.; Lu, W. Brain tumor-targeted drug delivery strategies. Acta Pharm. Sin. B 2014, 4, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.W.; Lin, W.J. Using doxorubicin and siRNA-loaded heptapeptide-conjugated nanoparticles to enhance chemosensitization in epidermal growth factor receptor high-expressed breast cancer cells. J. Drug Target. 2013, 21, 776–786. [Google Scholar] [CrossRef]
- Liu, C.W.; Lin, W.J. Systemic co-delivery of doxorubicin and siRNA using nanoparticles conjugated with EGFR-specific targeting peptide to enhance chemotherapy in ovarian tumor bearing mice. J. Nanoparticle Res. 2013, 15, 1956. [Google Scholar] [CrossRef]
- Wang, H.; Chen, W.; Wu, G.; Kong, J.; Yuan, S.; Chen, L. A magnetic T7 peptide & AS1411 aptamer-modified microemulsion for triple glioma-targeted delivery of shikonin and docetaxel. J. Pharm. Sci. 2021, 110, 2946–2954. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, Y.; Wang, H.; Wang, J.; Shin, M.C.; Byun, Y.; He, H.; Liang, Y.; Yang, V.C. Curb challenges of the “Trojan Horse” approach: Smart strategies in achieving effective yet safe cell-penetrating peptide-based drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 1299–1315. [Google Scholar] [CrossRef] [Green Version]
- Tong, R.; Kohane, D.S. New strategies in cancer nanomedicine. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Almatroudi, A.; Alzohairy, M.A.; AlYahya, S.; Alomary, M.N.; Al-Dossary, H.A.; Alghamdi, S. Lipid-based nano delivery of Tat-peptide conjugated drug or vaccine-promising therapeutic strategy for SARS-CoV-2 treatment. Expert Opin. Drug Deliv. 2020, 17, 1671–1674. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiong, S.; Liu, P. Polymeric nanoparticles-based brain delivery with improved therapeutic efficacy of ginkgolide B in parkinson’s disease. Int. J. Nanomed. 2020, 24, 10453–10467. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Robison, A.D.; Sun, S.; Poyton, M.F.; Johnson, G.A.; Pellois, J.P.; Jungwirth, P.; Vazdar, M.; Cremer, P.S. Polyarginine interacts more strongly and cooperatively than polylysine with phospholipid bilayers. J. Phys. Chem. B 2016, 120, 9287–9296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhakamy, N.A.; Berkland, C.J. Polyarginine molecular weight determines transfection efficiency of calcium condensed complexes. Mol. Pharm. 2013, 10, 1940–1948. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhu, Z.; Wang, H.; Han, C.; Liu, D.; Tian, L.; Yang, X.; Pan, W. Surface density of polyarginine influence the size, zeta potential, cellular uptake and tissue distribution of the nanostructured lipid carrier. Drug Deliv. 2017, 24, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfram, J.; Ferrari, M. Clinical cancer nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, S.; De Giovanni, P.J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Huang, H.L.; Lin, W.J. Dual Peptide-modified nanoparticles improve combination chemotherapy of etoposide and siPIK3CA against drug-resistant small cell lung carcinoma. Pharmaceutics 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Yang, Z.; Zhang, S.; Cao, S.; Shen, S.; Pang, Z.; Jiang, X. Ligand modified nanoparticles increases cell uptake, alters endocytosis and elevates glioma distribution and internalization. Sci. Rep. 2013, 3, 2534. [Google Scholar] [CrossRef] [Green Version]
- Kou, L.; Sun, J.; Zhai, Y.; He, Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian J. Pharm. Sci. 2013, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Monteiro-Riviere, N.A. Mechanisms of cell uptake, inflammatory potential and protein corona effects with gold nanoparticles. Nanomedicine 2016, 11, 3185–3203. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Tang, W.; Luo, X.; Zhang, X.; Zhang, C.; Li, H.; Gao, D.; Luo, H.; Jiang, Q.; Liu, J. Dual-ligand modified polymer-lipid hybrid nanoparticles for docetaxel targeting delivery to her2/neu overexpressed human breast cancer cells. J. Biomed. Nanotechnol. 2015, 11, 1401–1417. [Google Scholar] [CrossRef] [PubMed]
- Farkhani, S.M.; Shirani, A.; Mohammadi, S.; Zakeri-Milani, P.; Mojarrad, J.S.; Valizadeh, H. Effect of poly-glutamate on uptake efficiency and cytotoxicity of cell penetrating peptides. IET Nanobiotechnol. 2016, 10, 87–95. [Google Scholar] [CrossRef]
- Liu, B.R.; Huang, Y.W.; Koriv, M.; Lo, S.Y.; Aronstam, R.S.; Lee, H.J. The primary mechanism of cellular internalization for a short cell-penetrating peptide as a nano-scale delivery system. Curr. Pharm. Biotechnol. 2017, 18, 569–584. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs | Yield (%) | Size (nm) | PdI | Zeta Potential (mV) |
|---|---|---|---|---|
| PP5k NPs | 75.3 ± 6.1 | 166.9 ± 2.4 | 0.14 ± 0.04 | −17.0 ± 1.6 |
| PPT5k NPs | 73.0 ± 1.7 | 167.4 ± 7.6 | 0.15 ± 0.02 | −16.6 ± 1.9 |
| PPR2k NPs | 80.0 ± 3.5 | 162.6 ± 8.5 | 0.12 ± 0.04 | +7.9 ± 0.6 |
| PP5k/PP2k NPs | 70.7 ± 4.2 | 173.5 ± 3.6 | 0.07 ± 0.04 | −18.6 ± 1.2 |
| PPT5k/PP2k NPs | 79.3 ± 3.1 | 170.1 ± 4.2 | 0.13 ± 0.02 | −15.6 ± 1.4 |
| PPT5k/PPR2k NPs | 80.3 ± 3.8 | 160.9 ± 3.3 | 0.06 ± 0.07 | −7.2 ± 1.0 |
| PP5k/PPR2k NPs | 70.7 ± 4.2 | 168.8 ± 2.0 | 0.10 ± 0.07 | −4.7 ± 0.8 |
| Inhibitors | CPZ | Nystatin | Amiloride | |||
|---|---|---|---|---|---|---|
| Endocytosis Pathway | Clathrin | Caveolin | Macropinocytosis | |||
| Cell Line | bEnd.3 | U87-MG | bEnd.3 | U87-MG | bEnd.3 | U87-MG |
| PP5k/PP2k NPs | ++ | ++ | + | + | + | ++ |
| PPT5k/PP2k NPs | +++ | ++ | + | + | ++ | ++ |
| PPT5k/PPR2k NPs | +++ | ++ | + | ++ | ++ | ++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo, Y.-C.; Lin, W.-J. Benefit of a Short Chain Peptide as a Targeting Ligand of Nanocarriers for a Brain-Driven Purpose. Pharmaceutics 2021, 13, 1249. https://doi.org/10.3390/pharmaceutics13081249
Lo Y-C, Lin W-J. Benefit of a Short Chain Peptide as a Targeting Ligand of Nanocarriers for a Brain-Driven Purpose. Pharmaceutics. 2021; 13(8):1249. https://doi.org/10.3390/pharmaceutics13081249
Chicago/Turabian StyleLo, Yu-Chen, and Wen-Jen Lin. 2021. "Benefit of a Short Chain Peptide as a Targeting Ligand of Nanocarriers for a Brain-Driven Purpose" Pharmaceutics 13, no. 8: 1249. https://doi.org/10.3390/pharmaceutics13081249