
A Bidirectional Permeability Assay for beyond Rule of 5 Compounds

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Determination of LogP Values
2.2. In Vivo Pharmacokinetics and Tissue Distribution Studies in Mice
2.3. Determination of Bidirectional Permeability in Caco-2 Cells and MDCK-MDR1 Cells
2.4. In Vitro Binding Assays
2.5. Dynamic Light Scattering Measurement
- G(2)(τ): Measured amplitude autocorrelation function
- B: So-called baseline
- f: Instrument constant
- Dm: Particle diffusion coefficient
- q: Scattering vector given by (4πn/λ)sin(θ/2)
- τ: Delay time
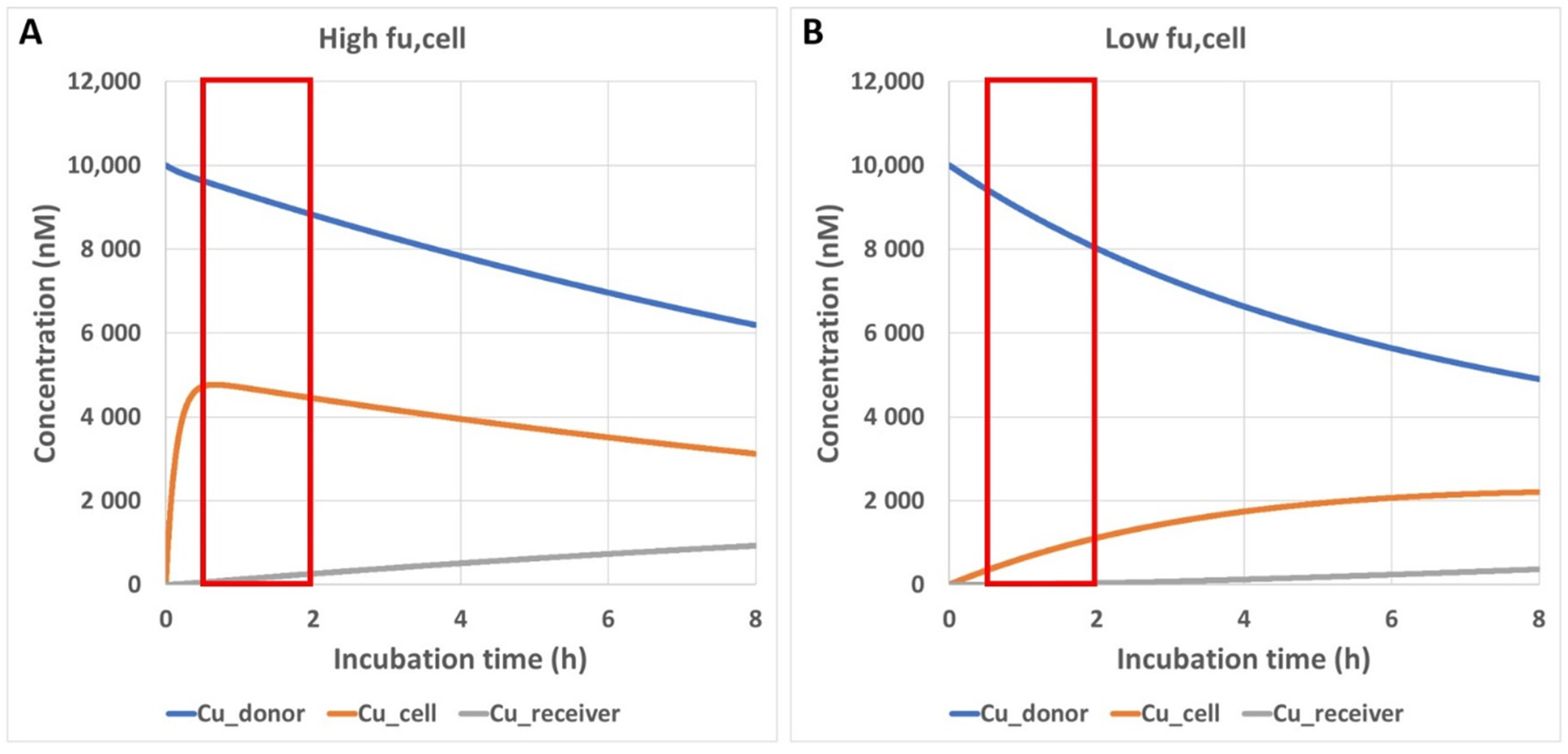
2.6. Simulation of Unbound Intracellular Drug Concentrations in a Transwell Permeability Experiment
3. Results
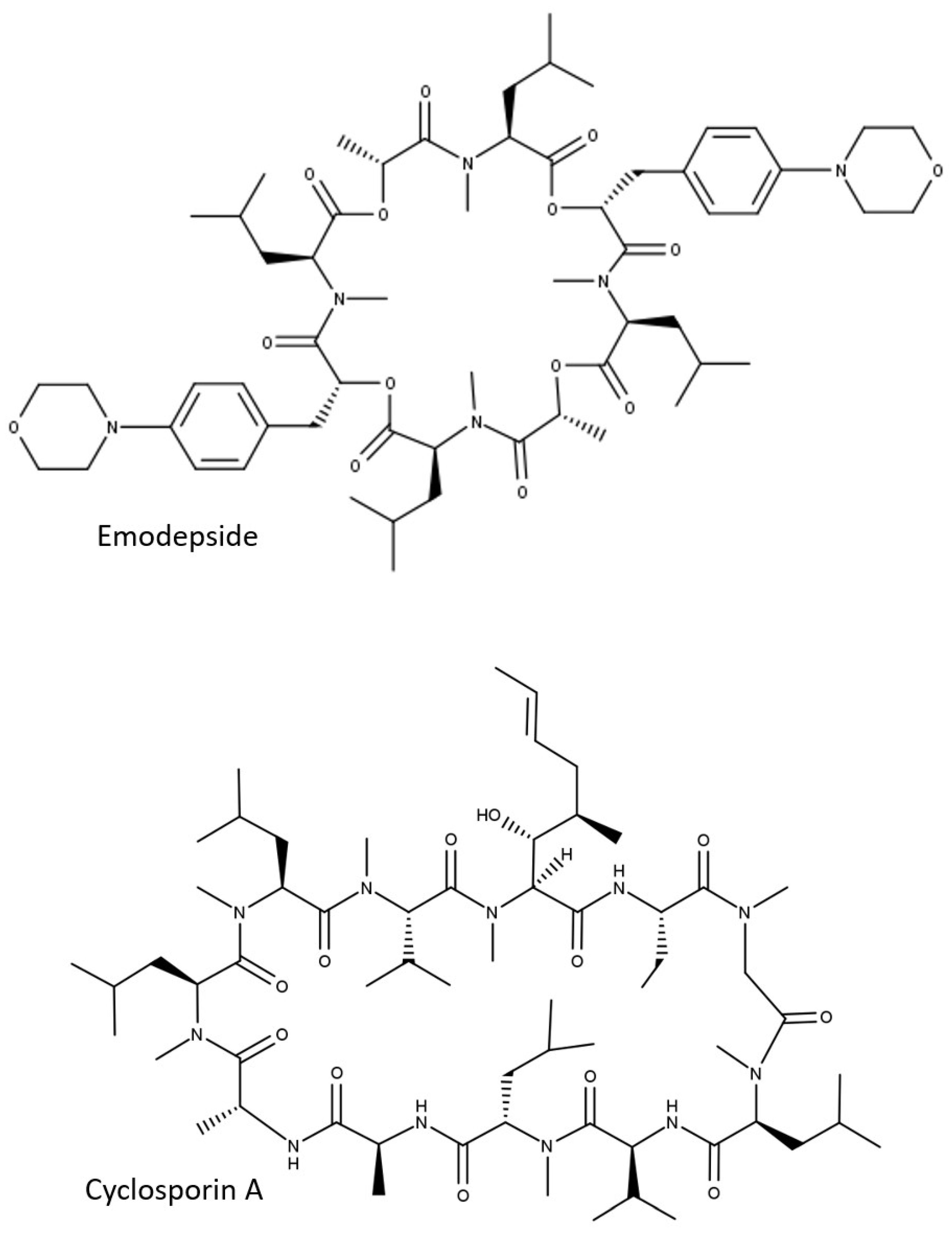
3.1. Depsipeptides Used in This Study
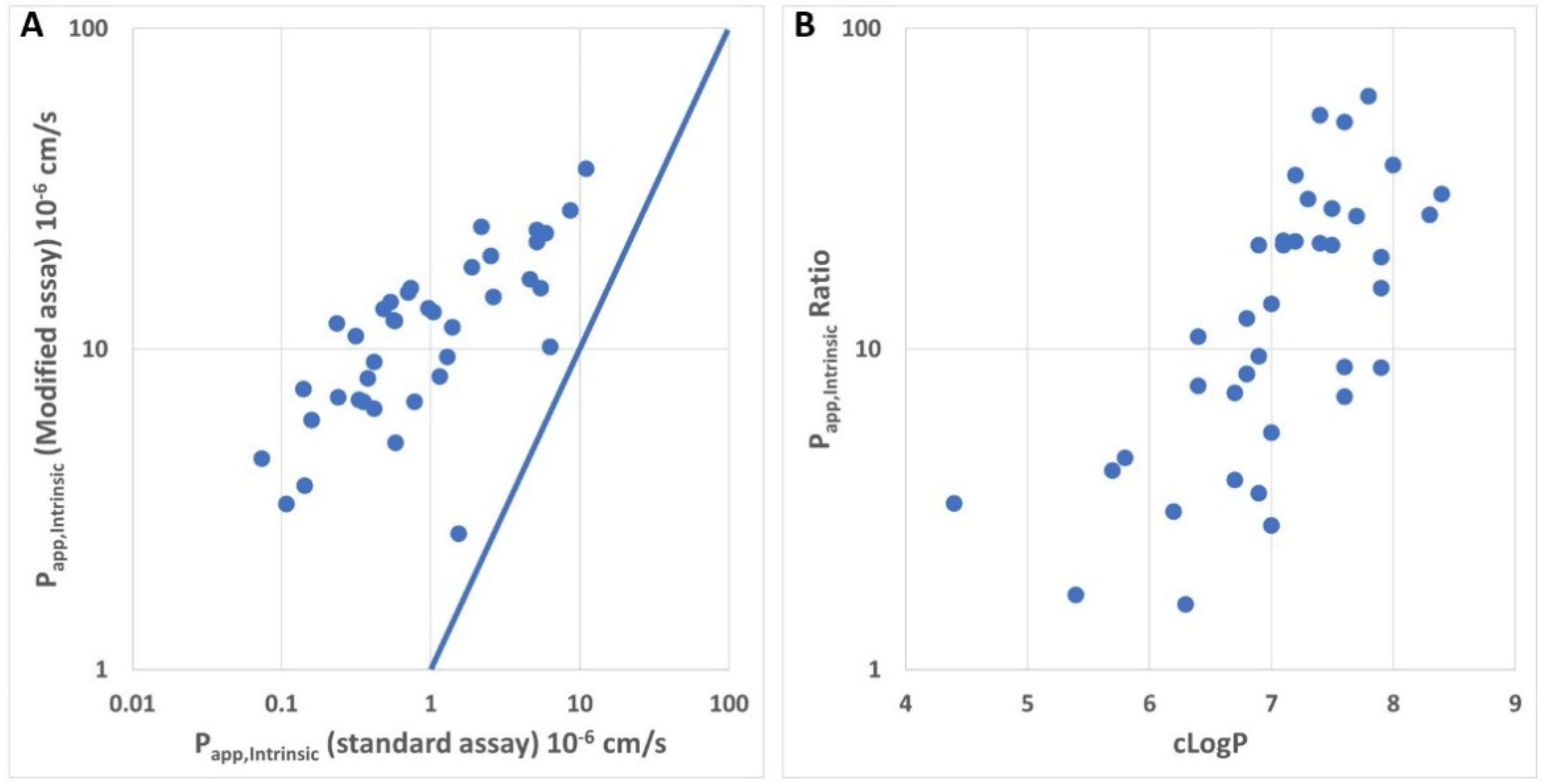
3.2. In Vitro Permeability and Efflux Measured for Depsipeptides in Standard Assays
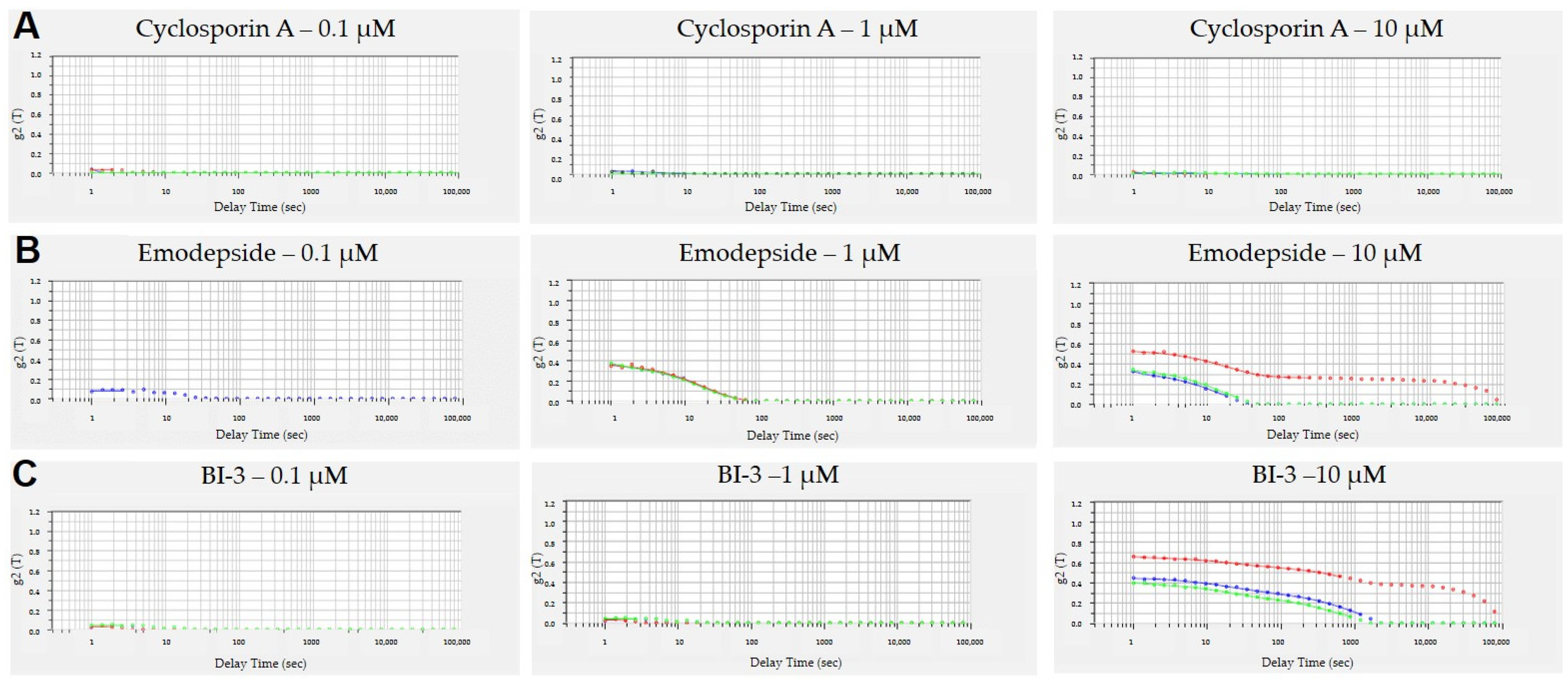
3.3. Aggregation of Peptides in Aqueous Solutions
3.4. Impact of Preincubation Time on Apparent In Vitro Permeability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lennernäs, H.; Palm, K.; Fagerholm, U.; Artursson, P. Comparison between Active and Passive Drug Transport in Human Intestinal Epithelial (Caco-2) Cells in Vitro and Human Jejunum in Vivo. Int. J. Pharm. 1996, 127, 103–107. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L.; Petusky, S.; Farris, M.; Ley, R.; Jupp, P. Combined Application of Parallel Artificial Membrane Permeability Assay and Caco-2 Permeability Assays in Drug Discovery. J. Pharm. Sci. 2004, 93, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Mills, J.B.; Davidson, R.E.; Mireles, R.J.; Janiszewski, J.S.; Troutman, M.D.; de Morais, S.M. In Vitro P-Glycoprotein Assays to Predict the in Vivo Interactions of P-Glycoprotein with Drugs in the Central Nervous System. Drug Metab. Dispos. 2008, 36, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, L.; Li, Y.; Fu, T.; Sun, X.; Zhang, Y.Y.; Gao, R.; Chen, Q.; Zhang, W.; Sahi, J.; et al. Correlation between Membrane Protein Expression Levels and Transcellular Transport Activity for Breast Cancer Resistance Protein. Drug Metab. Dispos. 2017, 45, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Lotz, R.; Rapp, H.; Klinder, K.; Himstedt, A.; Sauer, A. Muscle to Brain Partitioning as Measure of Transporter-Mediated Efflux at the Rat Blood-Brain Barrier and Its Implementation into Compound Optimization in Drug Discovery. Pharmaceutics 2019, 11, 595. [Google Scholar] [CrossRef] [Green Version]
- Sieger, P.; Cui, Y.; Scheuerer, S. PH-Dependent Solubility and Permeability Profiles: A Useful Tool for Prediction of Oral Bioavailability. Eur. J. Pharm. Sci. 2017, 105, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell Permeability beyond the Rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Takagi, M.; Yaguchi, T.; Miyadoh, S.; Okada, T.; Koyama, M. A new anthelmintic cyclodepsipeptide, PF1022A. J. Antibiot. 1992, 45, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, A.; Samson-Himmelstjerna, G. Activity of the Cyclic Depsipeptide Emodepside (BAY 44–4400) against Larval and Adult Stages of Nematodes in Rodents and the Influence on Worm Survival. Parasitol. Res. 2001, 87, 924–928. [Google Scholar] [CrossRef]
- Krücken, J.; Harder, A.; Jeschke, P.; Holden-Dye, L.; O’Connor, V.; Welz, C.; von Samson-Himmelstjerna, G. Anthelmintic Cyclooctadepsipeptides: Complex in Structure and Mode of Action. Trends Parasitol. 2012, 28, 385–394. [Google Scholar] [CrossRef]
- Harder, A.; von Samson-Himmelstjerna, G. Cyclooctadepsipeptides—A New Class of Anthelmintically Active Compounds. Parasitol. Res. 2002, 88, 481–488. [Google Scholar] [CrossRef]
- Gillon, J.; Dennison, J.; Berg, F.; Delhomme, S.; Cheeseman, K.D.; Rossi, C.P.; Wourgaft, N.S.; Specht, S.; Pedrique, B.; Monnot, F.; et al. Safety, Tolerability and Pharmacokinetics of Emodepside, a Potential Novel Treatment for Onchocerciasis (River Blindness), in Healthy Male Subjects. Br. J. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Elmshäuser, S.; Straehle, L.C.; Kranz, J.; Krebber, R.; Geyer, J. Brain Penetration of Emodepside Is Increased in P-glycoprotein-deficient Mice and Leads to Neurotoxicosis. J. Vet. Pharmacol. Ther. 2015, 38, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, T.; Subramanian, G.; Menon, S.; Townsend, C.E.; Lokey, R.S.; Ebert, M.-O.; Riniker, S. Connecting the Conformational Behavior of Cyclic Octadepsipeptides with Their Ionophoric Property and Membrane Permeability. Org. Biomol. Chem. 2020, 18, 7110–7126. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.F.; Pescatore, M.C. Method for Measuring the Logarithm of the Octanol–Water Partition Coefficient by Using Short Octadecyl–Poly(Vinyl Alcohol) High-Performance Liquid Chromatography Columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Kalvass, J.C.; Pollack, G.M. Kinetic Considerations for the Quantitative Assessment of Efflux Activity and Inhibition: Implications for Understanding and Predicting the Effects of Efflux Inhibition. Pharm. Res. 2007, 24, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.; Sakamoto, A.; Fuchs, H.; Ishiguro, N.; Suzuki, S.; Cui, Y.; Klinder, K.; Watanabe, M.; Terasaki, T.; Sauer, A. Quantification of Transporter and Receptor Proteins in Dog Brain Capillaries and Choroid Plexus: Relevance for the Distribution in Brain and CSF of Selected BCRP and P-Gp Substrates. Mol. Pharm. 2017, 14, 3436–3447. [Google Scholar] [CrossRef]
- Beyer, S.; Xie, L.; Schmidt, M.; de Bruin, N.; Ashtikar, M.; Rüschenbaum, S.; Lange, C.M.; Vogel, V.; Mäntele, W.; Parnham, M.J.; et al. Optimizing Novel Implant Formulations for the Prolonged Release of Biopharmaceuticals Using in Vitro and in Vivo Imaging Techniques. J. Control. Release 2016, 235, 352–364. [Google Scholar] [CrossRef]
- Ueda, C.T.; Lemaire, M.; Gsell, G.; Misslin, P.; Nussbaumer, K. Apparent Dose-dependent Oral Absorption of Cyclosporin a in Rats. Biopharm. Drug Dispos. 1984, 5, 141–151. [Google Scholar] [CrossRef]
- Faulds, D.; Goa, K.L.; Benfield, P. Cyclosporin. Drugs 1993, 45, 953–1040. [Google Scholar] [CrossRef] [PubMed]
- Boland, J.; Atkinson, K.; Britton, K.; Darveniza, P.; Johnson, S.; Biggs, J. Tissue distribution and toxicity of cyclosporin a in the mouse. Pathology 1984, 16, 117–123. [Google Scholar] [CrossRef]
- Pye, C.R.; Hewitt, W.M.; Schwochert, J.; Haddad, T.D.; Townsend, C.E.; Etienne, L.; Lao, Y.; Limberakis, C.; Furukawa, A.; Mathiowetz, A.M.; et al. Nonclassical Size Dependence of Permeation Defines Bounds for Passive Adsorption of Large Drug Molecules. J. Med. Chem. 2017, 60, 1665–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaghloul, I.; Ptachcinski, R.J.; Burckart, G.J.; Thiel, D.V.; Starzel, T.E.; Venkataramanan, R. Blood Protein Binding of Cyclosporine in Transplant Patients. J. Clin. Pharmacol. 1987, 27, 240–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huth, F.; Domange, N.; Poller, B.; Vapurcuyan, A.; Durrwell, A.; Hanna, I.D.; Faller, B. Predicting Oral Absorption for Compounds Outside the Rule of Five Property Space. J. Pharm. Sci. 2021, 110, 2562–2569. [Google Scholar] [CrossRef]
- Tran, T.T.; Mittal, A.; Aldinger, T.; Polli, J.W.; Ayrton, A.; Ellens, H.; Bentz, J. The Elementary Mass Action Rate Constants of P-Gp Transport for a Confluent Monolayer of MDCKII-HMDR1 Cells. Biophys. J. 2005, 88, 715–738. [Google Scholar] [CrossRef] [Green Version]
- Abreu, M.S.C.; Estronca, L.M.B.B.; Moreno, M.J.; Vaz, W.L.C. Binding of a Fluorescent Lipid Amphiphile to Albumin and Its Transfer to Lipid Bilayer Membranes. Biophys. J. 2003, 84, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Helander, H.F.; Fändriks, L. Surface Area of the Digestive Tract—Revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW | cLogP 1 | LogP 2 | Oral Availability in Mice (%) 3 | Kp,br in WT Mice | Kp,br/mu in WT Mice |
|---|---|---|---|---|---|---|
| Cyclosporin A | 1202.6 | 6.6 | 5.5 | n.d. | n.d. | ~0.1 4 |
| Emodepside | 1119.4 | 4.3 | 5.6 | n.d. | 0.05 5 | 0.034 5 |
| BI-1 | 1181.5 | 6.5 | 6.4 | 33 | 0.020 5 | 0.038 5 |
| BI-2 | 1177.4 | 5.7 | 6.1 | 28 | 0.13 5 | 0.082 5 |
| BI-3 | 1121.2 | 6.4 | 6.1 | 14 | 0.011 5 | 0.025 5 |
| BI-4 | 1153.2 | 7.0 | 6.3 | 10 | 0.023 5 | n.d. |
| Compound | MDCK-MDR1 | Caco-2 | ||||
|---|---|---|---|---|---|---|
| Papp,AB (10−6 cm/s) | PappBA (10−6 cm/s) | Efflux | Papp,AB (10−6 cm/s) | PappBA (10−6 cm/s) | Efflux | |
| Metoprolol | 67.7 | 68.5 | 1.0 | 54.3 | 41.0 | 0.8 |
| Propranolol | 38.4 | 32.6 | 0.8 | 79.0 | 69.5 | 1.1 |
| Cyclosporin | 7.6 | 68.2 | 10.8 | 22.2 | 17.9 | 0.8 |
| Emodepside | 3.6 | 34.1 | 9.5 | 27.4 | 16.4 | 0.8 |
| BI-1 | <0.3 | 1.2 | n.c. | <0.12 | 0.12 | n.c. |
| BI-2 | Low recovery | n.c. | <0.47 | 1.73 | n.c. | |
| BI-3 | <0.02 | 0.06 | n.c. | <0.14 | 0.05 | n.c. |
| BI-4 | <0.1 | <0.06 | n.c. | <0.06 | 0.02 | n.c. |
| Compound | Caco-2 | ||
|---|---|---|---|
| Papp,AB (10−6 cm/s) | PappBA (10−6 cm/s) | Efflux | |
| Cyclosporin | 7.2 | 26.4 | 3.7 |
| Emodepside | 8.8 | 36 | 4.1 |
| BI-1 | <0.53 | <0.23 | n.c. |
| BI-2 | BLQ | BLQ | n.c. |
| BI-3 | <1.2 | 0.65 | n.c. |
| BI-4 | <0.036 | <0.014 | n.c. |
| Compound | Caco-2 | Caco-2 in the Presence of Zosuquidar | ||||
|---|---|---|---|---|---|---|
| Papp,AB (10−6 cm/s) | Papp,BA (10−6 cm/s) | Efflux | Papp,AB (10−6 cm/s) | Papp,BA (10−6 cm/s) | Efflux | |
| Cyclosporin | 10 | 44 | 4.4 | |||
| Emodepside | 48 | 110 | 2.3 | 60 | 35 | 0.7 |
| BI-1 | 5 | 29 | 5.8 | 4.8 | 5.0 | 1.0 |
| BI-2 | 13 | 43 | 3.3 | 26 | 23 | 0.9 |
| BI-3 | 0.9 | 8.2 | 9.3 | 2.0 | 2.4 | 1.2 |
| BI-4 | 0.2 | 4.2 | 23.3 | 1.8 | 1.3 | 0.7 |
| Compound | MDCK-MDR1 | MDCK-MDR1 in the Presence of Zosuquidar | ||||
|---|---|---|---|---|---|---|
| Papp,AB (10−6 cm/s) | Papp,BA (10−6 cm/s) | Efflux | Papp,AB (10−6 cm/s) | Papp,BA (10−6 cm/s) | Efflux | |
| Cyclosporin | 2.1 | 62 | 29.5 | 28 | 13 | 0.5 |
| Emodepside | 6.3 | 190 | 30.2 | 56 | 39 | 0.7 |
| BI-1 | 0.7 | 30 | 45.5 | 4.4 | 3.7 | 0.8 |
| BI-2 | 2.4 | 47 | 19.6 | 9.4 | 7.3 | 0.8 |
| BI-3 | <0.037 | 3.1 | n.c. | 1.8 | 1.6 | 0.9 |
| BI-4 | 0.3 | 12.4 | 47.5 | 1.6 | 1.4 | 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Desevaux, C.; Truebenbach, I.; Sieger, P.; Klinder, K.; Long, A.; Sauer, A. A Bidirectional Permeability Assay for beyond Rule of 5 Compounds. Pharmaceutics 2021, 13, 1146. https://doi.org/10.3390/pharmaceutics13081146
Cui Y, Desevaux C, Truebenbach I, Sieger P, Klinder K, Long A, Sauer A. A Bidirectional Permeability Assay for beyond Rule of 5 Compounds. Pharmaceutics. 2021; 13(8):1146. https://doi.org/10.3390/pharmaceutics13081146
Chicago/Turabian StyleCui, Yunhai, Cyril Desevaux, Ines Truebenbach, Peter Sieger, Klaus Klinder, Alan Long, and Achim Sauer. 2021. "A Bidirectional Permeability Assay for beyond Rule of 5 Compounds" Pharmaceutics 13, no. 8: 1146. https://doi.org/10.3390/pharmaceutics13081146