
Nanofibrous Formulation of Cyclodextrin Stabilized Lipases for Efficient Pancreatin Replacement Therapies

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
1.1. Enzyme Replacement Therapies (ERT)
1.2. Pancreatic Enzyme Replacement Therapy (PERT)
1.3. Lipases in PERT
1.4. Cyclodextrins as Additives
2. Materials and Methods
2.1. Materials
2.2. Standard Lipase Activity Assay
2.3. Lipase Activity Assay in Fed-State Simulated Intestinal Fluid (FeSSIF) System
2.4. Determination of Lipase Activity and Conversion of p-NPP Hydrolysis
2.5. Rheological Analysis
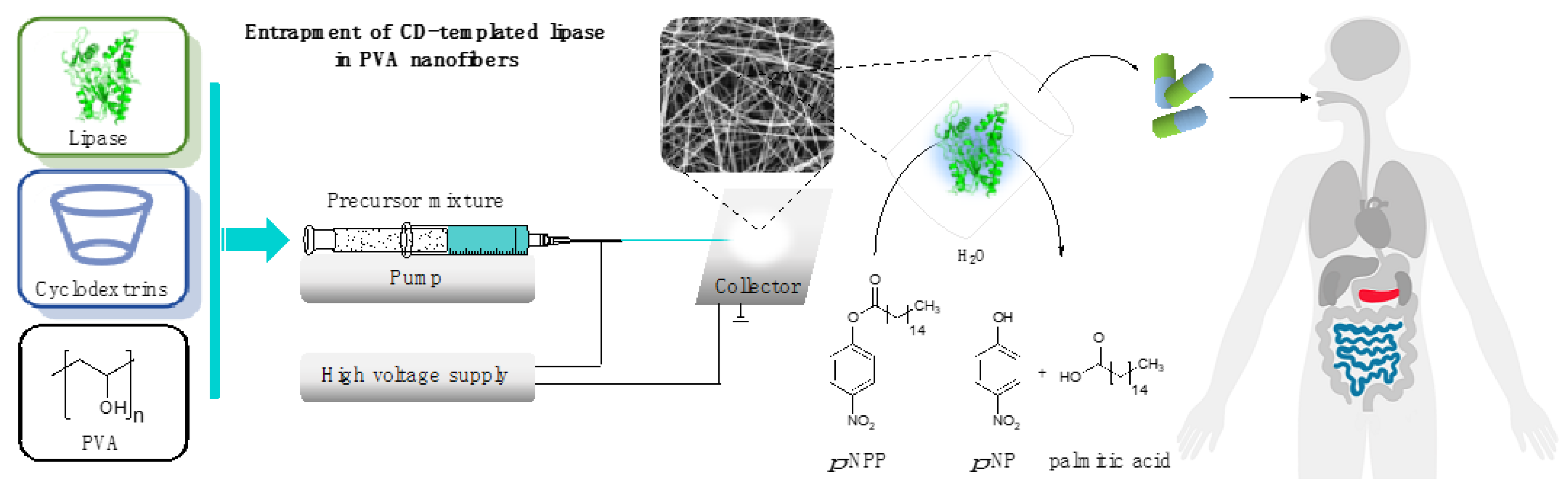
2.6. Entrapment of CD-Templated Lipases in Poly(vinyl alcohol) Nanofibers
2.7. Morphological Analysis
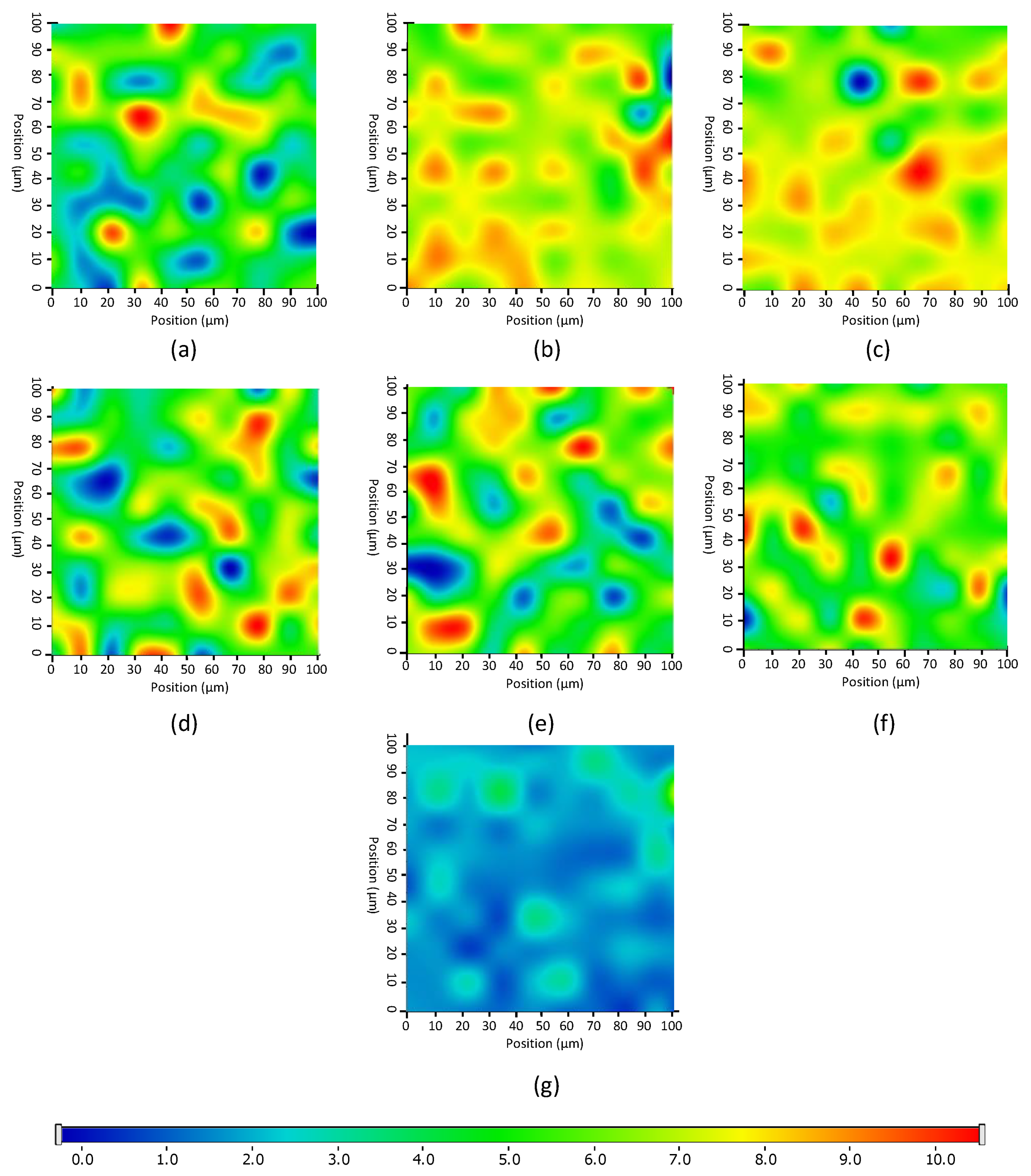
2.8. Raman Mapping Analysis of Nanofibrous Products
2.9. Investigation of Nanofibrous Lipase and Commercially Available Formulas
3. Results and Discussion
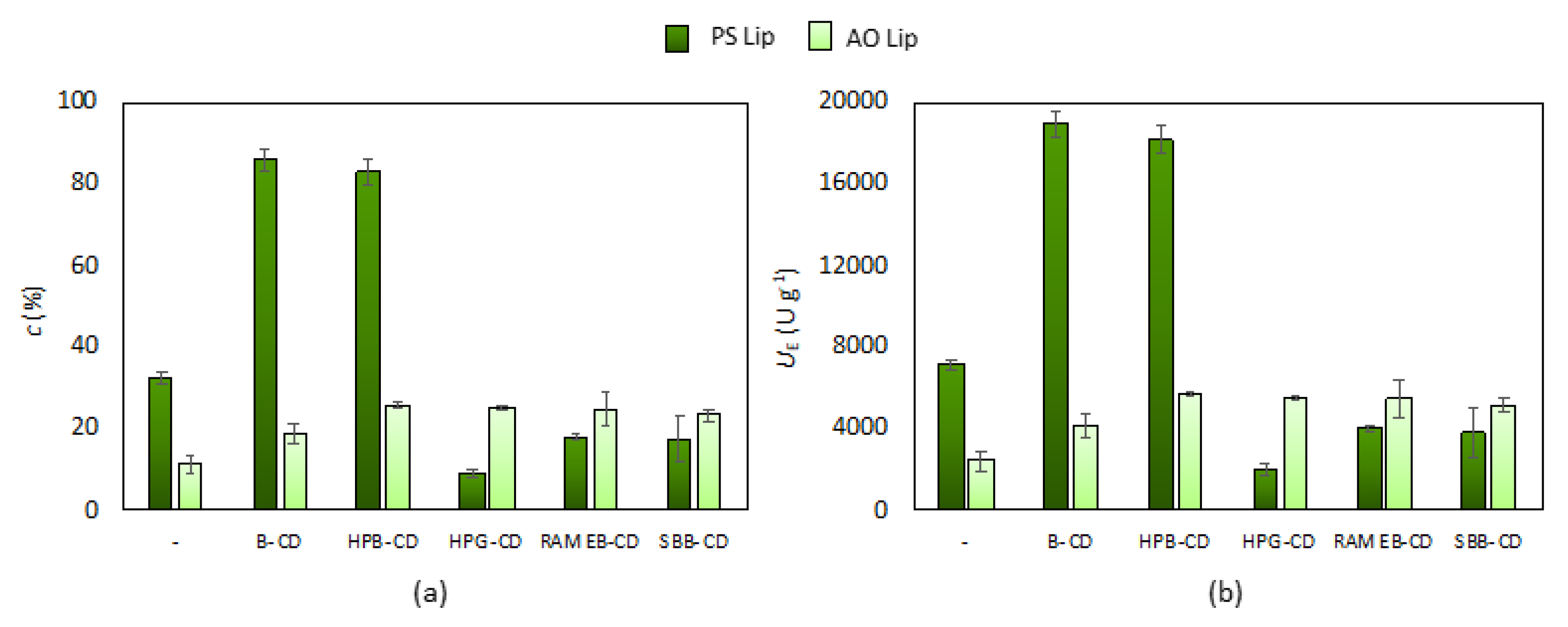
3.1. Effect of Cyclodextrins on the Native Lipase Activity
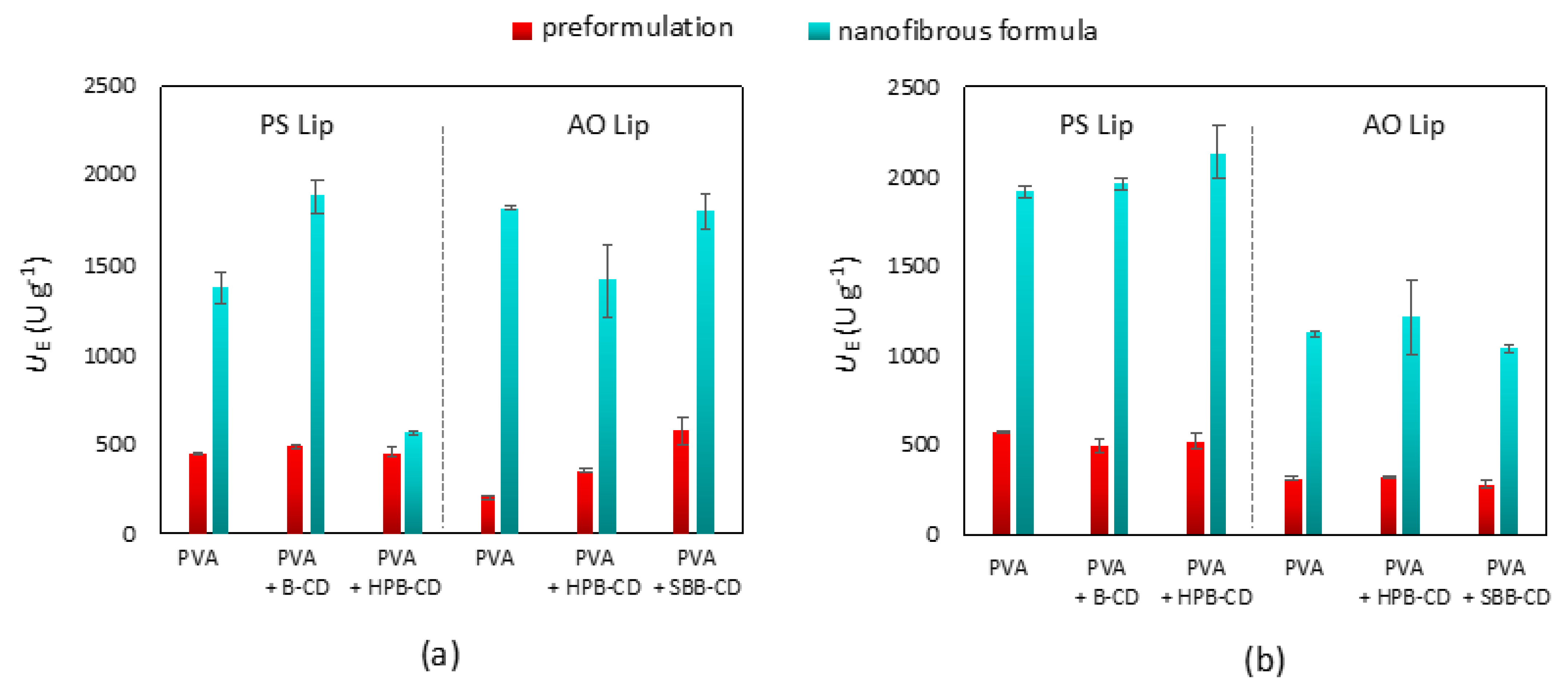
3.2. Solid Formulation of Lipases by Entrapment in Poly(vinyl alcohol) Nanofibers
3.3. Comparison of Lipase Activity of Cyclodextrin-Templated Nanofibers with Commercially Available Medicines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilcox, W.R. Lysosomal Storage Disorders: The Need for Better Pediatric Recognition and Comprehensive Care. J. Pediatr. 2004, 144, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. From Cytases to Lysosomes. Fed. Proc. 1964, 23, 1045–1049. [Google Scholar]
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of Glucocerebrosides II. Evidence of an Enzymatic Deficiency in Gaucher’s Disease. Biochem. Biophys. Res. Commun. 1965, 18, 221–225. [Google Scholar] [CrossRef]
- Ries, M. Enzyme Replacement Therapy and Beyond—In Memoriam Roscoe O. Brady, Md (1923–2016). J. Inherit. Metab. Dis. 2017, 40, 343–356. [Google Scholar] [CrossRef]
- Mechler, K.; Mountford, W.K.; Hoffmann, G.F.; Ries, M. Pressure for Drug Development in Lysosomal Storage Disorders–A Quantitative Analysis Thirty Years Beyond the Us Orphan Drug Act. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M. Treatment Strategies for Lysosomal Storage Disorders. Dev. Med. Child Neurol. 2018, 60, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr. Ann. 2018, 47, e191–e197. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme Replacement Therapy: Efficacy and Limitations. Ital. J. Pediatr. 2018, 44, 117–126. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Sikkens, E.C.; Cahen, D.L.; van Eijck, C.; Kuipers, E.J.; Bruno, M.J. The Daily Practice of Pancreatic Enzyme Replacement Therapy After Pancreatic Surgery: A Northern European Survey. J. Gastrointest. Surg. 2012, 16, 1487–1492. [Google Scholar] [CrossRef] [Green Version]
- Sikkens, E.C.; Cahen, D.L.; van Eijck, C.; Kuipers, E.J.; Bruno, M.J. Patients with Exocrine Insufficiency Due to Chronic Pancreatitis Are Undertreated: A Dutch National Survey. Pancreatology 2012, 12, 71–73. [Google Scholar] [CrossRef]
- Vellard, M. The Enzyme as Drug: Application of Enzymes as Pharmaceuticals. Curr. Opin. Biotechnol. 2003, 14, 444–450. [Google Scholar] [CrossRef]
- Boman, H.G.; Björk, W. General aspects of enzyme purification and characterization. In Modern Methods of Plant Analysis/Moderne Methoden der Pflanzenanalyse; Springer: Cham, Switzerland, 1963; pp. 374–392. [Google Scholar]
- Ferrone, M.; Raimondo, M.; Scolapio, J.S. Pancreatic Enzyme Pharmacotherapy. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2007, 27, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Kazi, Z.B.; Desai, A.K.; Berrier, K.L.; Troxler, R.B.; Wang, R.Y.; Abdul-Rahman, O.A.; Tanpaiboon, P.; Mendelsohn, N.J.; Herskovitz, E.; Kronn, D.; et al. Sustained Immune Tolerance Induction in Enzyme Replacement Therapy–Treated Crim-Negative Patients with Infantile Pompe Disease. JCI Insight 2017, 2, e94328. [Google Scholar] [CrossRef]
- Torchilin, V. Immobilised Enzymes as Drugs. Adv. Drug Deliv. Rev. 1987, 1, 41–86. [Google Scholar] [CrossRef]
- Kim, W.; Erlandsen, H.; Surendran, S.; Stevens, R.C.; Tyring, S.K.; Matalon, R.; Gamez, A.; Michols-Matalon, K. Trends in Enzyme Therapy for Phenylketonuria. Mol. Ther. 2004, 10, 220–224. [Google Scholar] [CrossRef]
- Galliani, M.; Santi, M.; Del Grosso, A.; Cecchettini, A.; Santorelli, F.M.; Hofmann, S.L.; Lu, J.-Y.; Angella, L.; Cecchini, M.; Signore, G. Cross-Linked Enzyme Aggregates as Versatile Tool for Enzyme Delivery: Application to Polymeric Nanoparticles. Bioconjug. Chem. 2018, 29, 2225–2231. [Google Scholar] [CrossRef]
- Nilsson, H.; Mosbach, R.; Mosbach, K. The Use of Bead Polymerization of Acrylic Monomers for Immobilization of Enzymes. Biochim. Biophys. Acta (BBA) Enzymol. 1972, 268, 253–256. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, H.S.; Kwon, T.-W.; Han, Y.-M.; Kang, N.-W.; Lee, M.Y.; Kim, D.-D.; Kim, M.G.; Lee, J.-Y. Single Enzyme Nanoparticle, an Effective Tool for Enzyme Replacement Therapy. Arch. Pharmacal Res. 2020, 43, 1–21. [Google Scholar] [CrossRef]
- Ketwaroo, G.A.; Graham, D.Y. Rational Use of Pancreatic Enzymes for Pancreatic Insufficiency and Pancreatic Pain. Ther. Enzym. Funct. Clin. Implic. 2019, 1148, 323–343. [Google Scholar]
- Majumder, S.; Chari, S.T. Chronic Pancreatitis. Lancet 2016, 387, 1957–1966. [Google Scholar] [CrossRef]
- Struyvenberg, M.R.; Martin, C.R.; Freedman, S.D. Practical Guide to Exocrine Pancreatic Insufficiency—Breaking the Myths. BMC Med. 2017, 15, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layer, P.; Go, V.; DiMagno, E.P. Fate of Pancreatic Enzymes During Small Intestinal Aboral Transit in Humans. Am. J. Physiol. Gastrointest. Liver Physiol. 1986, 251, G475–G480. [Google Scholar] [CrossRef]
- Granger, M.; Abadie, B.; Marchis-Mouren, G. Limited Action of Trypsin on Porcine Pancreatic Amylase: Characterization of the Fragments. FEBS Lett. 1975, 56, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Goebell, H.; Klotz, U.; Nehlsen, B.; Layer, P. Oroileal Transit of Slow Release 5-Aminosalicylic Acid. Gut 1993, 34, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Lederer, P.C.H. Optimal Size Range for Enteric-Coated Pancreatin Preparations. Pancreas 2011, 40, 310–311. [Google Scholar] [CrossRef]
- Naikwade, S.R.; Meshram, R.N.; Bajaj, A.N. Preparation and in Vivo Efficacy Study of Pancreatin Microparticles as an Enzyme Replacement Therapy for Pancreatitis. Drug Dev. Ind. Pharm. 2009, 35, 417–432. [Google Scholar] [CrossRef]
- Rosa-e-Silva, L.; Troncon, L.E.A.; Gallo, L.; Foss, M.C.; Passos, A.D.C.; Perdoná, G.C.; Achcar, J.A.; Oliveira, R.B. Determinants of Accelerated Small Intestinal Transit in Alcohol-Related Chronic Pancreatitis. Dig. Dis. Sci. 2010, 55, 1017–1025. [Google Scholar] [CrossRef]
- Domínguez-Muñoz, J.E. Pancreatic Enzyme Therapy for Pancreatic Exocrine Insufficiency. Curr. Gastroenterol. Rep. 2007, 9, 116–122. [Google Scholar] [CrossRef]
- Wang, H.; Hagedorn, J.; Svendsen, A.; Borch, K.; Otzen, D. Variant of the Thermomyces Lanuginosus Lipase with Improved Kinetic Stability: A Candidate for Enzyme Replacement Therapy. Biophys. Chem. 2013, 172, 43–52. [Google Scholar] [CrossRef]
- Zentler-Munro, P.; Assoufi, B.; Balasubramanian, K.; Cornell, S.; Benoliel, D.; Northfield, T.; Hodson, M. Therapeutic Potential and Clinical Efficacy of Acid-Resistant Fungal Lipase in the Treatment of Pancreatic Steatorrhoea Due to Cystic Fibrosis. Pancreas 1992, 7, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Münch, A.; Bührer, C.; Longardt, A.C. Digestive Enzyme Replacement Relieves Growth Failure in Preterm Infants with Poor Exocrine Pancreatic Function: A Retrospective Case Series. Eur. J. Pediatr. 2021, 1–8. [Google Scholar] [CrossRef]
- Hetrick, E.M.; Sperry, D.C.; Nguyen, H.K.; Strege, M.A. Characterization of a Novel Cross-Linked Lipase: Impact of Cross-Linking on Solubility and Release from Drug Product. Mol. Pharm. 2014, 11, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Somaraju, U.R.R.; Solis-Moya, A. Pancreatic Enzyme Replacement Therapy for People with Cystic Fibrosis. Cochrane Database Syst. Rev. 2020, CD008227. [Google Scholar] [CrossRef]
- Anbu, P.; Gopinath, S.C.B.; Cihan, A.C.; Chaulagain, B.P. Microbial Enzymes and Their Applications in Industries and Medicine. BioMed. Res. Int. 2013, 2195808. [Google Scholar] [CrossRef]
- Sikkens, E.C.M.; Cahen, D.L.; Kuipers, E.J.; Bruno, M.J. Pancreatic Enzyme Replacement Therapy in Chronic Pancreatitis. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 337–347. [Google Scholar] [CrossRef]
- Raimondo, M.; DiMagno, E.P. Lipolytic Activity of Bacterial Lipase Survives Better Than That of Porcine Lipase in Human Gastric and Duodenal Content. Gastroenterology 1994, 107, 231–235. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, Physicochemical Properties and Pharmaceutical Applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Crini, G.; Fenyvesi, É.; Szente, L. Professor József Szejtli: The godfather of cyclodextrins. In The History of Cyclodextrins; Crini, G., Fourmentin, S., Lichtfouse, E., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 95–155. ISBN 978-3-030-49308-0. [Google Scholar]
- Pang, Y.; Ritter, H.; Tabatabai, M. Cyclodextrins in Polymer Chemistry: Enzymatically Catalyzed Oxidative Polymerization of Para-Functionalized Phenol Derivatives in Aqueous Medium by Use of Horseradish Peroxidase. Macromolecules 2003, 36, 7090–7093. [Google Scholar] [CrossRef]
- Ghanem, A.; Schurig, V. Peracetylated β-Cyclodextrin as Additive in Enzymatic Reactions: Enhanced Reaction Rate and Enantiomeric Ratio in Lipase-Catalyzed Transesterifications in Organic Solvents. Tetrahedron Asymmetry 2001, 12, 2761–2766. [Google Scholar] [CrossRef]
- Ghanem, A. The Utility of Cyclodextrins in Lipase-Catalyzed Transesterification in Organic Solvents: Enhanced Reaction Rate and Enantioselectivity. Org. Biomol. Chem. 2003, 1, 1282–1291. [Google Scholar] [CrossRef]
- Gökmen, C.; Yasemin, A.; Doğaçb, I.; Devecic, I.; Teke, M. Synthesis and characterization of electrospun PVA/Zn2+ metal composite nanofibers for lipase immobilization with effective thermal, pH stabilities and reusability. Mater. Sci. Eng. C 2019, 99, 1226–1235. [Google Scholar]
- Wang, Y.; Hsieg, Y.L. Immobilization of lipase enzyme in polyvinyl alcohol (PVA) nanofibrous membranes. J. Membr. Sci. 2008, 309, 73–81. [Google Scholar] [CrossRef]
- Kordel, M.; Hofmann, B.; Schomburg, D.; Schmid, R.D. Extracellular lipase of Psudomonas sp. starin ATCC 21808: Purficitaion, characterization, crystallization, and preliminary X-ray data. J. Bacteriol. 1991, 173, 4836–4841. [Google Scholar] [CrossRef] [Green Version]
- Ozmen, E.Y.; Sezgin, M.; Yilmaz, M. Synthesis and characterization of cyclodextrin-based polymers as a support for immobilization of Candida rugosa lipase. J. Mol. Catal. B Enzym. 2009, 57, 109–114. [Google Scholar] [CrossRef]
- Ozyilmaz, E.; Sayin, S.; Arslan, M.; Yilmaz, M. Improving catalytic hydrolysis reaction efficiency of sol-gel-encapsulated Candida rugosa lipase with magnetic B-cyclodextrin nanoparticles. Coll. Surf. B Biointerface 2014, 113, 182–189. [Google Scholar] [CrossRef]
- Balogh-Weiser, D.; Németh, C.; Ender, F.; Gyarmati, B.; Szilágyi, A.; Poppe, L. Electrospun nanofibers for entrapment of biomolecules. In Electrospinning Method Used to Create Functional Nanocomposite Films; IntechOpen: London, UK, 2018; pp. 136–147. [Google Scholar]
- Weiser, D.; Sóti, P.L.; Bánóczi, G.; Bódai, V.; Kiss, B.; Gellért, Á.; Nagy, Z.K.; Koczka, B.; Szilágyi, A.; Marosi, G.; et al. Bioimprinted lipase in PVA nanofibers as efficient immobilized biocatalysts. Tetrahedron 2016, 72, 7335–7342. [Google Scholar] [CrossRef] [Green Version]
- Sóti, P.L.; Weiser, D.; Vigh, T.; Nagy, Z.K.; Poppe, L.; Marosi, G. Electrospun polylactic acid and polyvinyl alcohol fibers as efficient and stable nanomaterials immobilization of lipases. Bioprocess Biosyst. Eng. 2018, 39, 449–459. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Mahadevan, G.D.; Neelagund, S.E. Thermostable lipase from Geobacillus sp. Iso5: Bioseparation, characterization and native structural studies. J. Basic Microbiol. 2014, 54, 386–396. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipase | Cyclodextrin | c (%) | UE (U × g−1) |
|---|---|---|---|
| PS Lip | - | 18.2 ± 0.7 | 10,827 ± 429 |
| B-CD | 78.1 ± 2.5 | 47,178 ± 125 | |
| HPB-CD | 79.3 ± 0.9 | 47,045 ± 537 | |
| AO Lip | - | 12.0 ± 0.4 | 7120 ± 222 |
| HPB-CD | 13.7 ± 0.2 | 8135 ± 111 | |
| SBB-CD | 12.5 ± 0.3 | 7400 ± 178 |
| Lipase | Cyclodextrin | Precursor Viscosity (mPas) | Fiber Diameter (nm) |
|---|---|---|---|
| - | - | 426 ± 8 | 241 ± 48 |
| PS Lip | - | 356 ± 23 | 335 ± 51 |
| B-CD 1 | 446 ± 41 | 316 ± 49 | |
| HPB-CD 2 | 479 ± 63 | 368 ± 48 | |
| AO Lip | - | 354 ± 14 | 368 ± 67 |
| HPB-CD 2 | 399 ± 2 | 412 ± 117 | |
| SBB-CD 3 | 384 ± 14 | 371 ± 76 |
| Lipase | Cyclodextrin | Standard Assay 1 | FeSSIF Assay 2 | ||
|---|---|---|---|---|---|
| c (%) | UE (U × g−1) | c (%) | UE (U × g−1) | ||
| PS Lip | - | 34.2 ± 1.3 | 7530 ± 296 | 17.6 ± 0.5 | 104,515 ± 309 |
| B-CD | 55.7 ± 4.3 | 12,249 ± 957 | 15.7 ± 0.3 | 9297 ± 155 | |
| HPB-CD | 4.5 ± 0.1 | 983 ± 27 | 11.2 ± 0.4 | 6664 ± 234 | |
| AO Lip | - | 22.4 ± 0.7 | 4921 ± 164 | 7.3 ± 0.4 | 4340 ± 255 |
| HPB-CD | 18.4 ± 1.2 | 4048 ± 271 | 11.4 ± 0.2 | 6752 ± 117 | |
| SBB-CD | 21.6 ± 1.4 | 4760 ± 315 | 12.0 ± 0.6 | 7120 ± 331 | |
| Lipase | Cyclodextrin | Standard Assay | Assay in FeSSIF | ||
|---|---|---|---|---|---|
| c (%) | UF (U × g−1) | c (%) | UF (U × g−1) | ||
| Nanofibrous formula | |||||
| PS Lip | - | 74.9 ± 4.8 | 137 ± 9 | 38.7 ± 0.5 | 191 ± 3 |
| B-CD | 102.6 ± 4.7 | 188 ± 9 | 39.6 ± 0.5 | 196 ± 3 | |
| HPB-CD | 30.7 ± 0.5 | 56 ± 1 | 43.6 ± 3.5 | 216 ± 18 | |
| AO Lip | - | 99.1 ± 0.8 | 182 ± 10 | 22.7 ± 0.3 | 112 ± 1 |
| HPB-CD | 77.3 ± 11.1 | 142 ± 20 | 24.6 ± 4.1 | 122 ± 20 | |
| SBB-CD | 96.3 ± 7.0 | 177 ± 13 | 21.0 ± 0.4 | 104 ± 2 | |
| Marketed pharmaceutical product | |||||
| Kreon® 25000 | 6.7 ± 1.3 | 12 ± 2 | 10.9 ± 0.1 | 54 ± 1 | |
| Pangrol® 25000 | 3.4 ± 0.3 | 6 ± 1 | 10.8 ± 0.5 | 54 ± 3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, G.D.; Kazsoki, A.; Gyarmati, B.; Szilágyi, A.; Vasvári, G.; Katona, G.; Szente, L.; Zelkó, R.; Poppe, L.; Balogh-Weiser, D.; et al. Nanofibrous Formulation of Cyclodextrin Stabilized Lipases for Efficient Pancreatin Replacement Therapies. Pharmaceutics 2021, 13, 972. https://doi.org/10.3390/pharmaceutics13070972
Tóth GD, Kazsoki A, Gyarmati B, Szilágyi A, Vasvári G, Katona G, Szente L, Zelkó R, Poppe L, Balogh-Weiser D, et al. Nanofibrous Formulation of Cyclodextrin Stabilized Lipases for Efficient Pancreatin Replacement Therapies. Pharmaceutics. 2021; 13(7):972. https://doi.org/10.3390/pharmaceutics13070972
Chicago/Turabian StyleTóth, Gergő Dániel, Adrienn Kazsoki, Benjámin Gyarmati, András Szilágyi, Gábor Vasvári, Gábor Katona, Lajos Szente, Romána Zelkó, László Poppe, Diána Balogh-Weiser, and et al. 2021. "Nanofibrous Formulation of Cyclodextrin Stabilized Lipases for Efficient Pancreatin Replacement Therapies" Pharmaceutics 13, no. 7: 972. https://doi.org/10.3390/pharmaceutics13070972