
Structure–Activity Relationship and Molecular Docking of a Kunitz-Like Trypsin Inhibitor, Kunitzin-AH, from the Skin Secretion of Amolops hainanensis

 , , and
, , and
Abstract
:
1. Introduction
2. Results
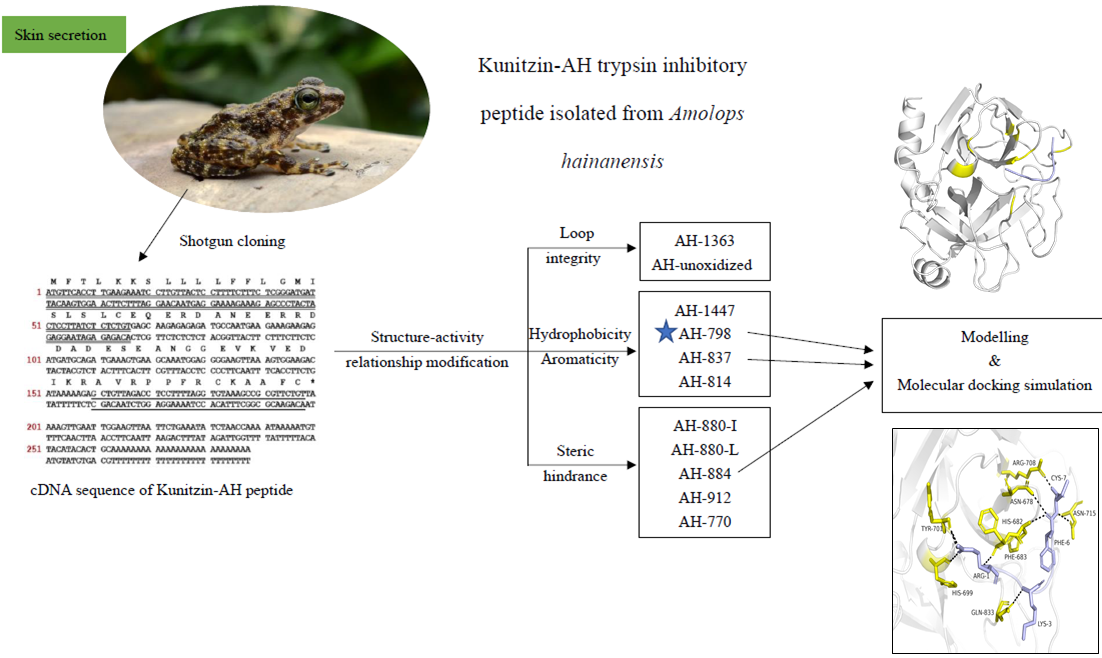
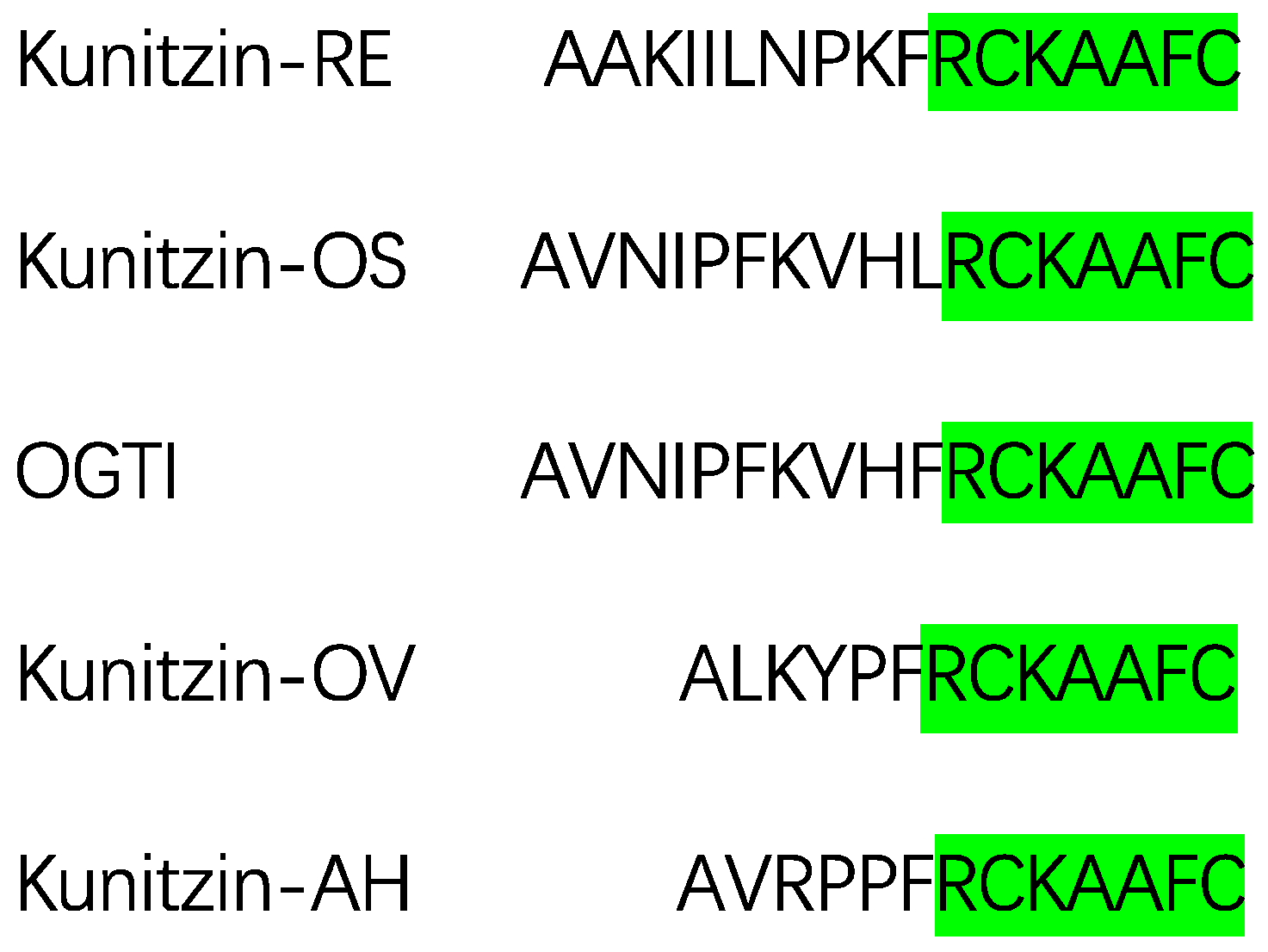
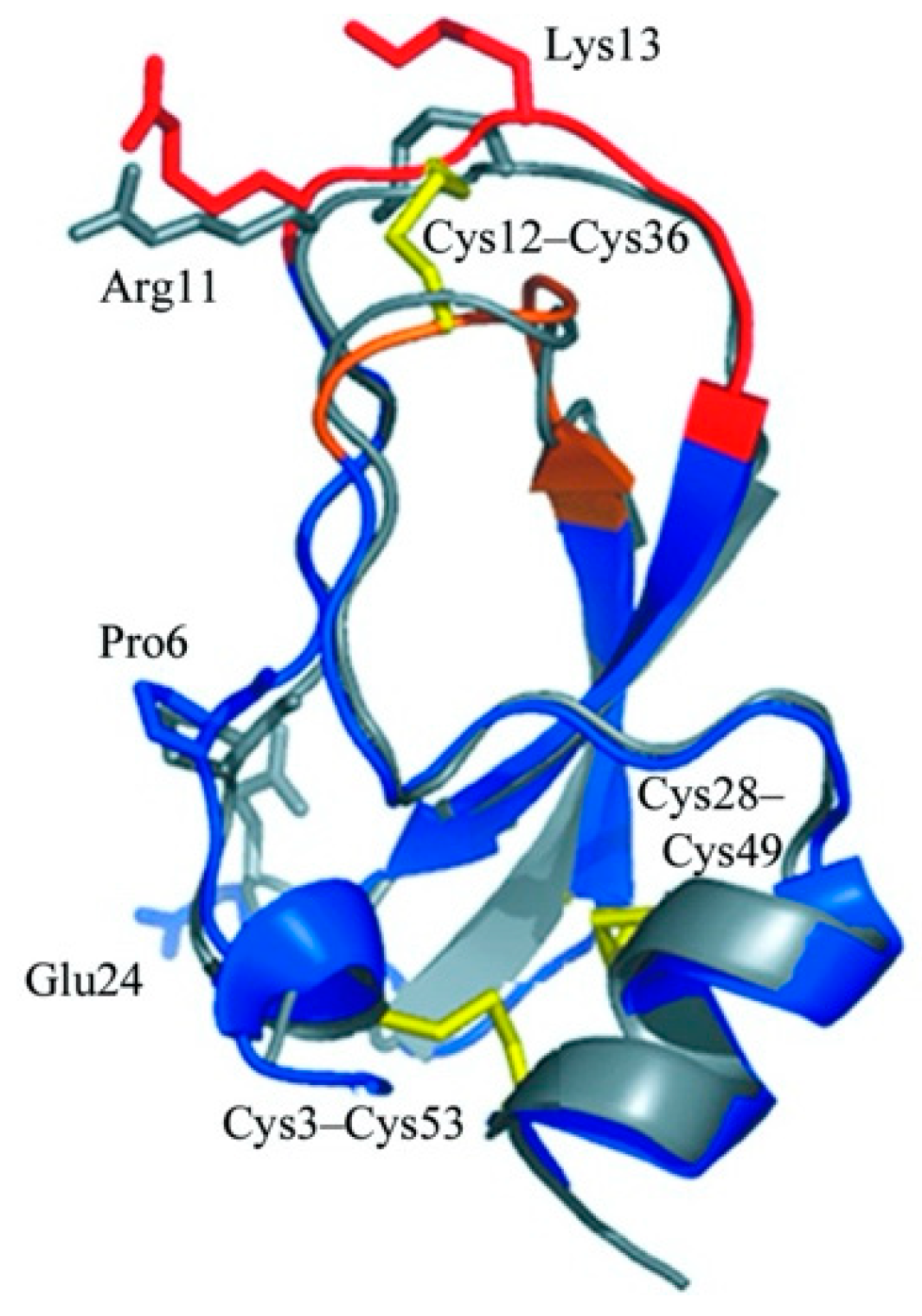
2.1. Identification and Characterization of Kunitzin-AH from the Frog Skin Secretion
2.2. Motif-Targeted Peptide Design
2.3. Trypsin Inhibition and Hemolysis Assays
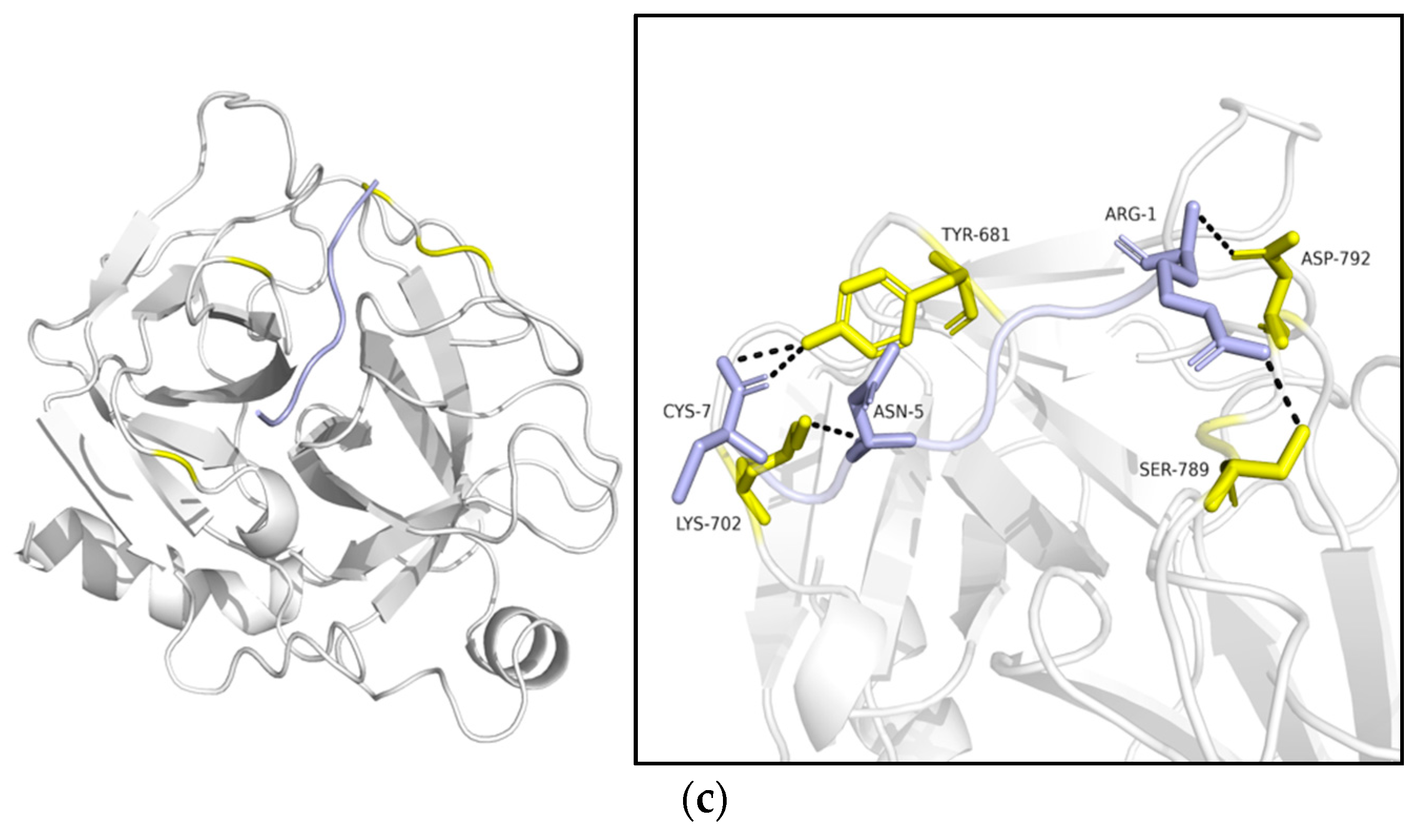
2.4. Modeling and Molecular Docking Simulation
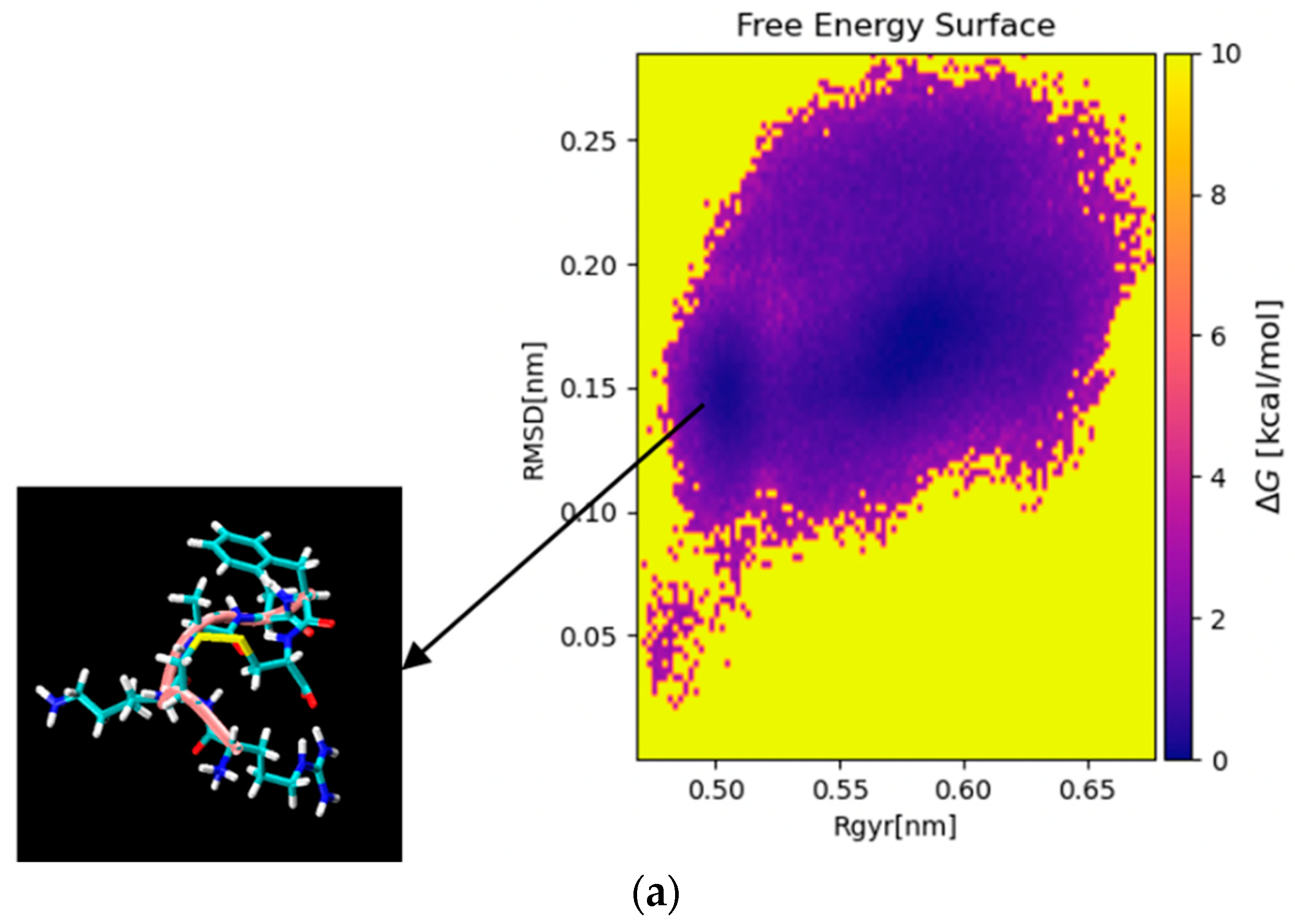
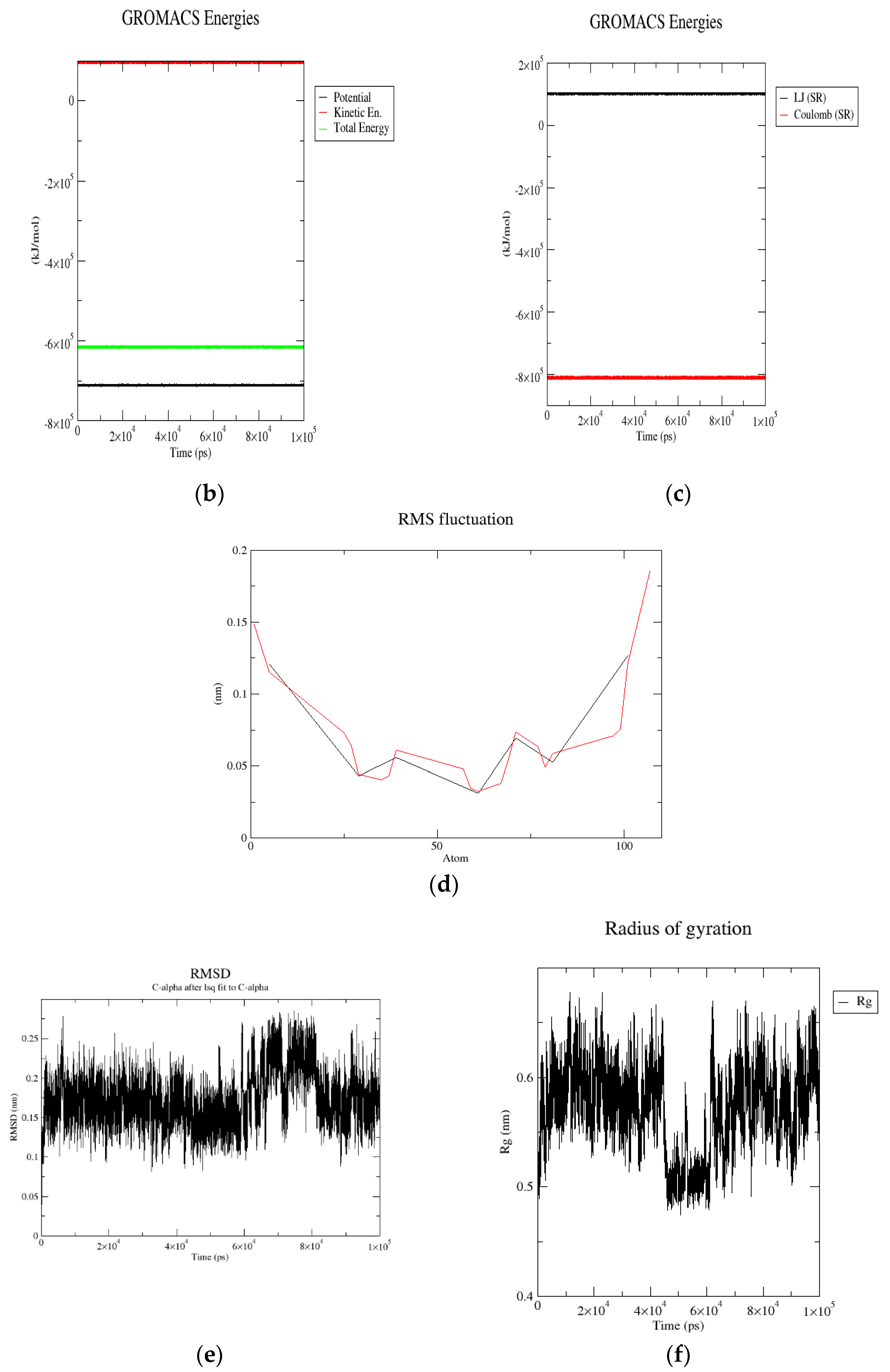
2.4.1. Molecular Dynamics (MD) Simulation
2.4.2. Ligand–Protein Docking Simulations with HDOCK
3. Discussion
4. Materials and Methods
4.1. The Acquisition of Skin Secretion from Amplops Hainanensis
4.2. Molecular Cloning of cDNA Library from Amolops Hainanensis Skin Secretion
4.3. Isolation and Identification of the Novel Peptide
4.4. Solid-Phase Peptide Synthesis
4.5. Trypsin Inhibition Assays
4.6. Hemolytic Activity Assay
4.7. Molecular Dynamics Simulation and Ligand–Protein Docking
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huma, H.; Khalid Majid, F. Plant protease inhibitors: A defense strategy in plants. Biotechnol. Mol. Biol. Rev. 2007, 2, 68–85. [Google Scholar]
- Mehmood, S.; Imran, M.; Ali, A.; Munawar, A.; Khaliq, B.; Anwar, F.; Saeed, Q.; Buck, F.; Hussain, S.; Saeed, A. Model prediction of a Kunitz-type trypsin inhibitor protein from seeds of Acacia nilotica L. with strong antimicrobial and insecticidal activity. Turk. J. Biol. 2020, 44, 188–200. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, P.; Liu, X.; Wang, Z.; Li, S. Preparation and Irreversible Inhibition Mechanism Insight into a Recombinant Kunitz Trypsin Inhibitor from Glycine max L. Seeds. Appl. Biochem. Biotechnol. 2020, 191, 1207–1222. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, J.; Chen, Z.; Wang, X.; Yan, S.; Xu, Y.; San, M.; Tang, W.; Yang, F.; Cao, Z.; et al. Functional characterization of a new non-Kunitz serine protease inhibitor from the scorpion Lychas mucronatus. Int. J. Biol. Macromol. 2015, 72, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Rogozhin, E.A.; Solovyev, M.M.; Frolova, T.V.; Izvekova, G.I. Isolation and partial structural characterization of new Kunitz-type trypsin inhibitors from the pike cestode Triaenophorus nodulosus. Mol. Biochem. Parasitol. 2019, 233, 111217. [Google Scholar] [CrossRef]
- Marco, T.R.G.; Maria, L.O.; Miriam, T.P.L.; Carlos, E.S. Plant Proteinases and Inhibitors: An Overview of Biological Function and Pharmacological Activity. Curr. Protein Pept. Sci. 2011, 12, 417–436. [Google Scholar] [CrossRef]
- Oliva, M.L.V.; Silva, M.C.C.; Sallai, R.C.; Brito, M.V.; Sampaio, M.U. A novel subclassification for Kunitz proteinase inhibitors from leguminous seeds. Biochimie 2010, 92, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.L.; Mick, R.; Ware, J.; Metz, J.; Lustig, R.; Vapiwala, N.; Rengan, R.; Kennedy, A.R. Phase I randomized double-blind placebo-controlled single-dose safety studies of Bowman-Birk inhibitor concentrate. Oncol. Lett. 2014, 7, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Suzuki, M.; Kanayama, N.; Terao, T. A soybean Kunitz trypsin inhibitor suppresses ovarian cancer cell invasion by blocking urokinase upregulation. Clin. Exp. Metastasis 2004, 21, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Cristina Oliveira de Lima, V.; Piuvezam, G.; Leal Lima Maciel, B.; Heloneida de Araújo Morais, A. Trypsin inhibitors: Promising candidate satietogenic proteins as complementary treatment for obesity and metabolic disorders? J. Enzym. Inhib. Med. Chem. 2019, 34, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Tikhonova, I.G.; Jewhurst, H.L.; Drysdale, O.C.; Dvorak, J.; Robinson, M.W.; Cwiklinski, K.; Dalton, J.P. Unexpected Activity of a Novel Kunitz-type Inhibitor: Inhibition of cysteine proteases but not serine proteases. J. Biol. Chem. 2016, 291, 19220–19234. [Google Scholar] [CrossRef] [Green Version]
- Bendre, A.D.; Ramasamy, S.; Suresh, C.G. Analysis of Kunitz inhibitors from plants for comprehensive structural and functional insights. Int. J. Biol. Macromol. 2018, 113, 933–943. [Google Scholar] [CrossRef]
- Evette, S.R.; Daniel, E.K. A Clogged Gutter Mechanism for Protease Inhibitors. Cover 2002, 99, 10316–10321. [Google Scholar] [CrossRef] [Green Version]
- Bendre, A.D.; Suresh, C.; Shanmugam, D.; Ramasamy, S. Structural insights into the unique inhibitory mechanism of Kunitz type trypsin inhibitor from Cicer arietinum L. J. Biomol. Struct. Dyn. 2019, 37, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Young Moo, C.; Kwang Sik, L.; Hyung Joo, Y.; Yuling, Q.; Hu, W.; Mi Ri, S.; Hung Dae, S.; Byung Rae, J. Antifibrinolytic Role of a Bee Venom Serine Protease Inhibitor That Acts as a Plasmin Inhibitor. PLoS ONE 2012, 7, e32269. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, H.; Kimoto, H.; Yamauchi, Y.; Toriba, M.; Kubo, T. Functional characterization of Kunitz-type protease inhibitor Pr-mulgins identified from New Guinean Pseudechis australis. Toxicon 2012, 59, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, H.; Shen, Y.; Wang, L.; Zhou, M.; Chen, T.; Shaw, C. Kunitzins: Prototypes of a new class of protease inhibitor from the skin secretions of European and Asian frogs. Biochem. Biophys. Res. Commun. 2016, 477, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wu, J.; Wang, Y.; Xu, X.; Liu, T.; Lai, R.; Zhu, H. A small trypsin inhibitor from the frog of Odorrana grahami. Biochimie 2008, 90, 1356–1361. [Google Scholar] [CrossRef]
- Dong, Y.; Shi, D.; Ying, Y.; Xi, X.; Chen, X.; Wang, L.; Zhou, M.; Wu, Q.; Ma, C.; Chen, T. A Novel Kunitzin-Like Trypsin Inhibitor Isolated from Defensive Skin Secretion of Odorrana versabilis. Biomolecules 2019, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvin, A.; Anand, S.; Asha, R.; Reshmy, V.; Sanil, G.; Kumar, K.S. Structure-Activity Relationship and Mode of Action of a Frog Secreted Antibacterial Peptide B1CTcu5 Using Synthetically and Modularly Modified or Deleted (SMMD) Peptides. PLoS ONE 2015, 10, e0124210. [Google Scholar] [CrossRef]
- Chen, X.; Chen, D.; Huang, L.; Chen, X.; Zhou, M.; Xi, X.; Ma, C.; Chen, T.; Wang, L. Identification and Target-Modification of SL-BBI: A Novel Bowman–Birk Type Trypsin Inhibitor from Sylvirana latouchii. Biomolecules 2020, 10, 1254. [Google Scholar] [CrossRef]
- Gitlin-Domagalska, A.; Maciejewska, A.; Dębowski, D. Bowman-Birk Inhibitors: Insights into Family of Multifunctional Proteins and Peptides with Potential Therapeutical Applications. Pharmaceuticals 2020, 13, 421. [Google Scholar] [CrossRef]
- Proaño-Bolaños, C.; Li, R.; Zhou, M.; Wang, L.; Xi, X.; Tapia, E.E.; Coloma, L.A.; Chen, T.; Shaw, C. Novel Kazal-type proteinase inhibitors from the skin secretion of the Splendid leaf frog, Cruziohyla calcarifer. EuPA Open Proteom. 2017, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef]
- BogdanowichKnipp, S.J.; Chakrabarti, S.; Siahaan, T.J.; Williams, T.D.; Dillman, R.K. Solution stability of linear vs. cyclic RGD peptides. J. Pept. Res. 1999, 53, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Long, Q.; Xu, Y.; Guo, S.; Chen, T.; Wang, L.; Zhou, M.; Zhang, Y.; Shaw, C.; Walker, B. A structural and functional analogue of a Bowman–Birk-type protease inhibitor from Odorrana schmackeri. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, R.; Pons, T.; Meyer, A.; Perbandt, M.; Gonzalez-Gonzalez, Y.; Gil, D.; de los Angeles Chávez, M.; Betzel, C.; Redecke, L. Structure of the recombinant BPTI/Kunitz-type inhibitor rShPI-1A from the marine invertebrate Stichodactyla helianthus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1289–1293. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; De Veer, S.J.; White, A.M.; Chen, X.; Harris, J.M.; Swedberg, J.E.; Craik, D.J. Amino Acid Scanning at P5′ within the Bowman–Birk Inhibitory Loop Reveals Specificity Trends for Diverse Serine Proteases. J. Med. Chem. 2019, 62, 3696–3706. [Google Scholar] [CrossRef]
- de Veer, S.J.; White, A.M.; Craik, D.J. Sunflower trypsin Inhibitor-1 (SFTI-1): Sowing seeds in the fields of chemistry and biology. Angew. Chem. Int. Ed. 2021, 60, 8050–8071. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, H.; Jiang, Y.; Yu, Y.; Wang, L.; Zhou, M.; Zhang, Y.; Chen, T.; Shaw, C. A novel Kazal-type trypsin inhibitor from the skin secretion of the Central American red-eyed leaf frog, Agalychnis callidryas. Biochimie 2012, 94, 1376–1381. [Google Scholar] [CrossRef]
- Yuan, Y.; Zai, Y.; Xi, X.; Ma, C.; Wang, L.; Zhou, M.; Shaw, C.; Chen, T. A novel membrane-disruptive antimicrobial peptide from frog skin secretion against cystic fibrosis isolates and evaluation of anti-MRSA effect using Galleria mellonella model. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 849–856. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Van Aalst, E.; Yekefallah, M.; Mehta, A.K.; Eason, I.; Wylie, B. Codon Harmonization of a Kir3. 1-KirBac1. 3 Chimera for Structural Study Optimization. Biomolecules 2020, 10, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Hsieh, T.-C.; Wu, J.M.; Wang, X.; Christopher, J.S.; Pham, A.H.; Swaby, J.D.-L.; Lou, L.; Xie, Z.-R. Elucidating the Inhibitory Effect of Resveratrol and Its Structural Analogs on Selected Nucleotide-Related Enzymes. Biomolecules 2020, 10, 1223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, T.; Zhou, S.; Cheng, J.; Yuan, S.; Lo, G.V.; Dou, Y. Molecular Dynamics Simulation of Transmembrane Transport of Chloride Ions in Mutants of Channelrhodopsin. Biomolecules 2019, 9, 852. [Google Scholar] [CrossRef] [Green Version]
- Asoodeh, A.; Haghighi, L.; Chamani, J.; Ansari-Ogholbeyk, M.A.; Mojallal-Tabatabaei, Z.; Lagzian, M. Potential angiotensin I converting enzyme inhibitory peptides from gluten hydrolysate: Biochemical characterization and molecular docking study. J. Cereal Sci. 2014, 60, 92–98. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 39, 2607–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Ki Value/µM | Hemolysis/µM |
|---|---|---|---|
| Kunitzin-AH | AVRPPFRCKAAFC | 1.18 ± 0.08 | >512 |
| AH-1363 | AVRPPFRCKAAF | 96.94 ± 1.20 ** | >512 |
| AH-unoxidized | AVRPPFRCKAAFC | 59.85 ± 2.82 ** | >512 |
| AH-1447 | AVRPPFRCKAAKC | 349.20 ± 0.93 *** | >512 |
| AH-798 | RCKAAFC | 1.76 ± 0.13 * | >512 |
| AH-837 | RCKAAWC | 14.74 ± 5.23 ** | >512 |
| AH-814 | RCKAAYC | 77.94 ± 7.84 ** | >512 |
| AH-880-I | RCKIIFC | 16.05 ± 1.20 ** | >512 |
| AH-880-L | RCKLLFC | 17.51 ± 1.20 * | >512 |
| AH-884 | RCKNNFC | 133.20 ± 4.10 ** | >512 |
| AH-912 | RCKQQFC | 105.00 ± 5.02 ** | >512 |
| AH-770 | RCKGGFC | 169.20 ± 0.86 *** | >512 |
| Name | Sequence | Docking Scores 1 |
|---|---|---|
| AH-798 | RCKAAFC | −184.79 |
| AH-837 | RCKAAWC | −181.58 |
| AH-884 | RCKNNFC | −171.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Xi, X.; Ma, C.; Zhou, M.; Chen, X.; Ye, Z.; Ge, L.; Wu, Q.; Chen, T.; Wang, L.; et al. Structure–Activity Relationship and Molecular Docking of a Kunitz-Like Trypsin Inhibitor, Kunitzin-AH, from the Skin Secretion of Amolops hainanensis. Pharmaceutics 2021, 13, 966. https://doi.org/10.3390/pharmaceutics13070966
Chen Y, Xi X, Ma C, Zhou M, Chen X, Ye Z, Ge L, Wu Q, Chen T, Wang L, et al. Structure–Activity Relationship and Molecular Docking of a Kunitz-Like Trypsin Inhibitor, Kunitzin-AH, from the Skin Secretion of Amolops hainanensis. Pharmaceutics. 2021; 13(7):966. https://doi.org/10.3390/pharmaceutics13070966
Chicago/Turabian StyleChen, Yuqing, Xinping Xi, Chengbang Ma, Mei Zhou, Xiaoling Chen, Zhuming Ye, Lilin Ge, Qinan Wu, Tianbao Chen, Lei Wang, and et al. 2021. "Structure–Activity Relationship and Molecular Docking of a Kunitz-Like Trypsin Inhibitor, Kunitzin-AH, from the Skin Secretion of Amolops hainanensis" Pharmaceutics 13, no. 7: 966. https://doi.org/10.3390/pharmaceutics13070966