
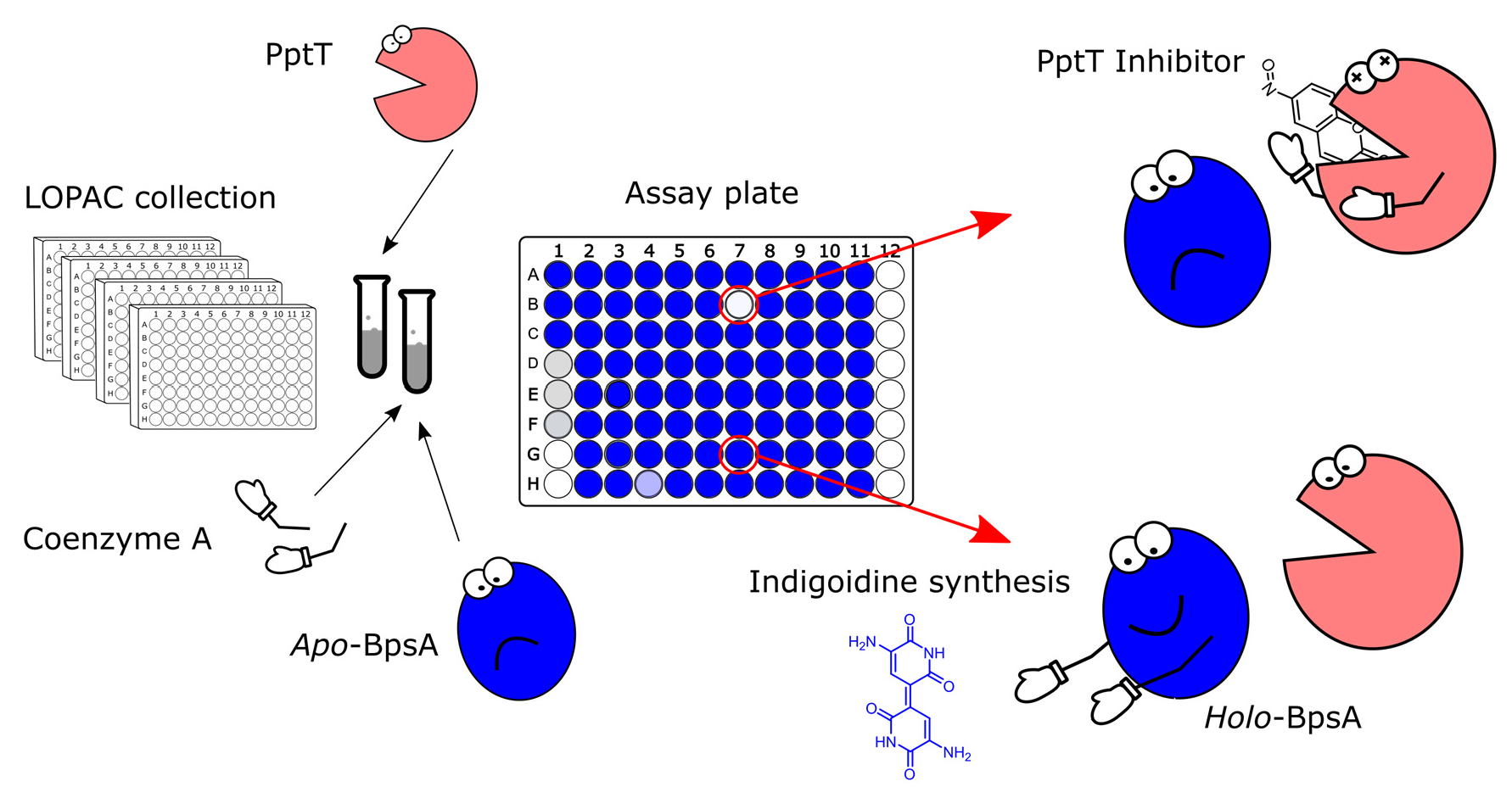
Inhibition of Indigoidine Synthesis as a High-Throughput Colourimetric Screen for Antibiotics Targeting the Essential Mycobacterium tuberculosis Phosphopantetheinyl Transferase PptT


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Plasmid Construction
2.3. Protein Expression
2.4. Protein Purification
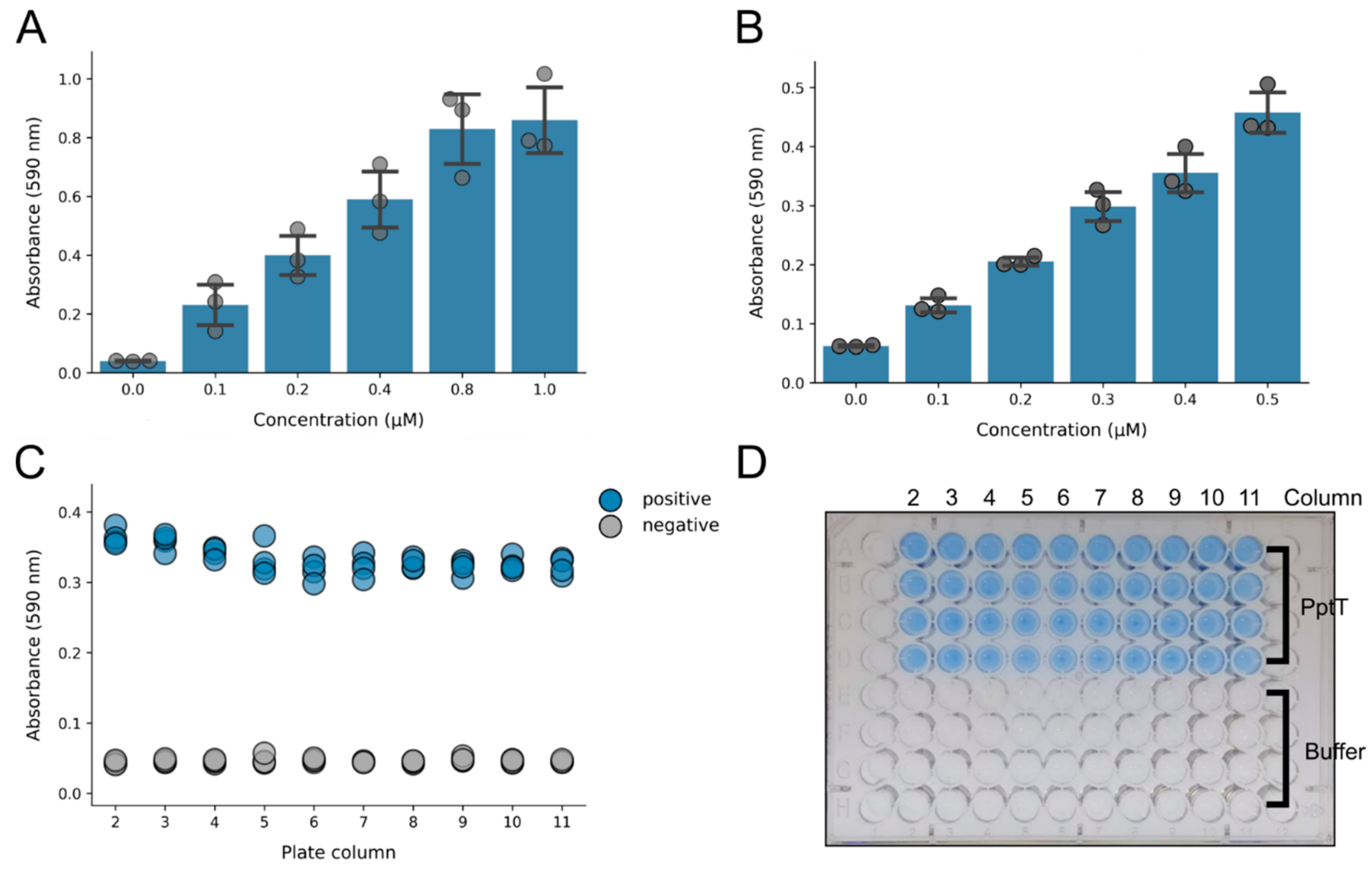
2.5. Optimisation of Enzyme Concentrations
2.6. Z-Factor Calculation
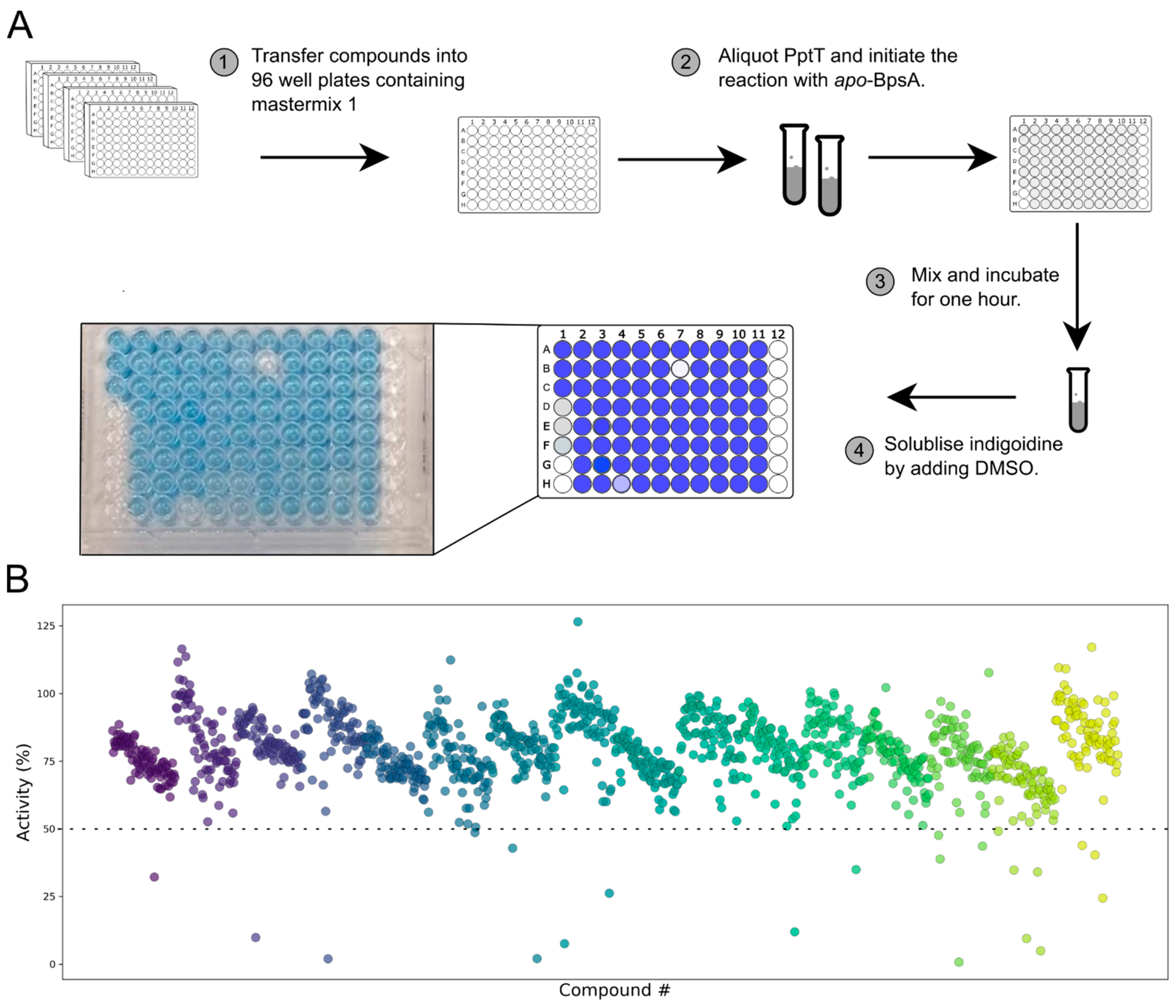
2.7. Screening of the LOPAC1280 Collection and EC50 Calculations
2.8. Kinetic Analysis of PptT Inhibition
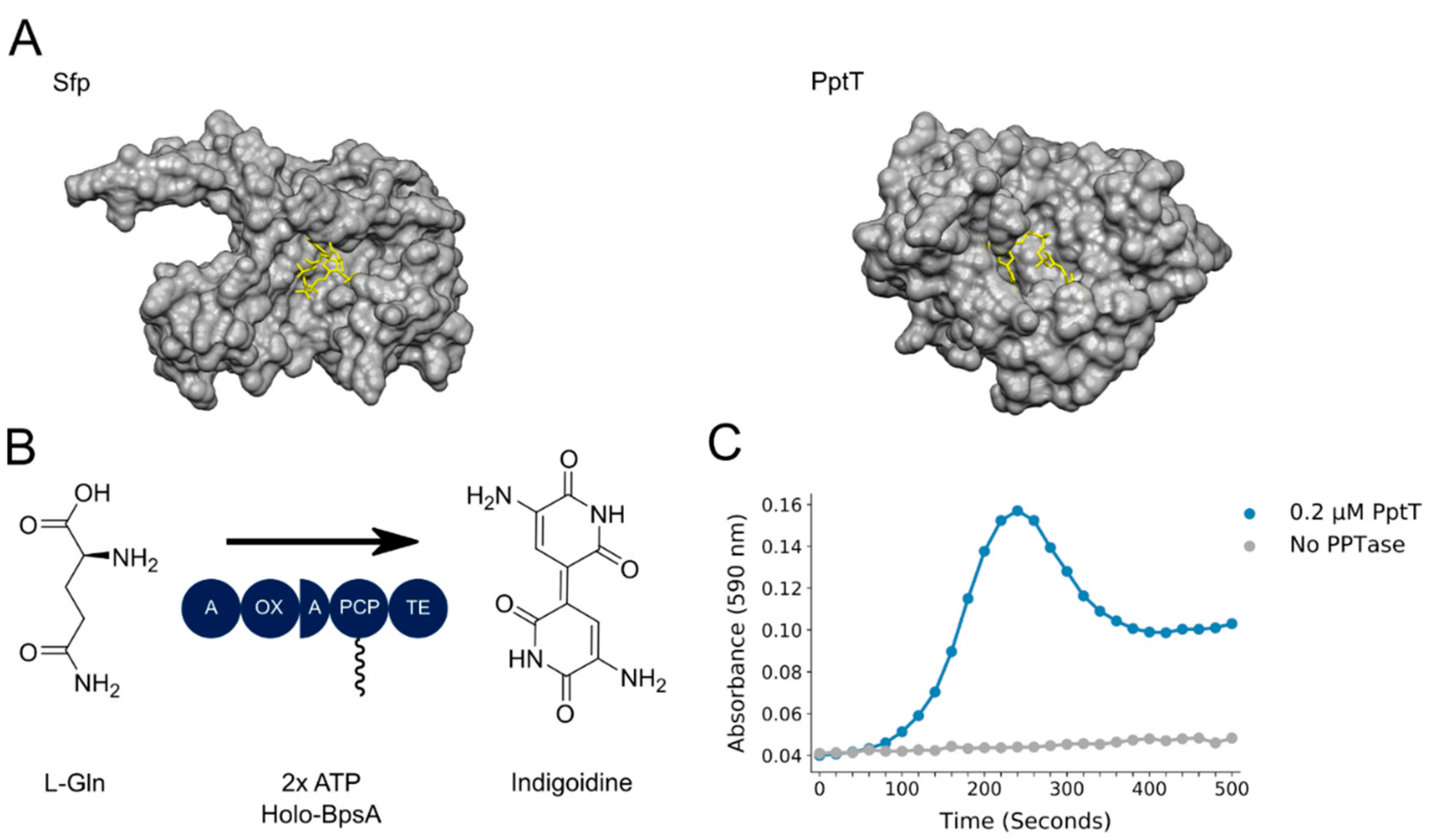
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Tuberculosis Report. 2020. Available online: https://www.who.int/publications-detail-redirect/9789240013131 (accessed on 5 May 2021).
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Neyrolles, O.; Guilhot, C. Recent advances in deciphering the contribution of Mycobacterium tuberculosis lipids to pathogenesis. Tuberculosis 2011, 91, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Daffé, M. The cell envelope of tubercle bacilli. Tuberculosis 2015, 95, S155–S158. [Google Scholar] [CrossRef] [PubMed]
- Karakousis, P.C.; Bishai, W.R.; Dorman, S.E. Mycobacterium tuberculosiscell envelope lipids and the host immune response. Cell. Microbiol. 2004, 6, 105–116. [Google Scholar] [CrossRef]
- Gokhale, R.S.; Saxena, P.; Chopra, T.; Mohanty, D. Versatile polyketide enzymatic machinery for the biosynthesis of complex mycobacterial lipids. Nat. Prod. Rep. 2007, 24, 267–277. [Google Scholar] [CrossRef]
- McMahon, M.D.; Rush, J.S.; Thomas, M.G. Analyses of MbtB, MbtE, and MbtF Suggest Revisions to the Mycobactin Biosynthesis Pathway in Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 2809–2818. [Google Scholar] [CrossRef] [Green Version]
- Beld, J.; Sonnenschein, E.; Vickery, C.R.; Noel, J.P.; Burkart, M.D. The phosphopantetheinyl transferases: Catalysis of a post-translational modification crucial for life. Nat. Prod. Rep. 2014, 31, 61–108. [Google Scholar] [CrossRef] [Green Version]
- Foley, T.L.; Young, B.S.; Burkart, M.D. Phosphopantetheinyl transferase inhibition and secondary metabolism. FEBS J. 2009, 276, 7134–7145. [Google Scholar] [CrossRef]
- Chalut, C.; Botella, L.; de Sousa-D’Auria, C.; Houssin, C.; Guilhot, C. The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of Mycobacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 8511–8516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimhony, O.; Schwarz, A.; Raitses-Gurevich, M.; Peleg, Y.; Dym, O.; Albeck, S.; Burstein, Y.; Shakked, Z. AcpM, the Meromycolate Extension Acyl Carrier Protein of Mycobacterium tuberculosis, Is Activated by the 4′-Phosphopantetheinyl Transferase PptT, a Potential Target of the Multistep Mycolic Acid Biosynthesis. Biochemistry 2015, 54, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. USA 2001, 98, 12712–12717. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, C.; Prudhomme, T.; Tabouret, G.; Ray, A.; Burbaud, S.; Cabantous, S.; Mourey, L.; Guilhot, C.; Chalut, C. 4′-Phosphopantetheinyl Transferase PptT, a New Drug Target Required for Mycobacterium tuberculosis Growth and Persistence In Vivo. PLOS Pathog. 2012, 8, e1003097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadri, L.E.; Sello, J.; Keating, T.A.; Weinreb, P.; Walsh, C.T. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 1998, 5, 631–645. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, E.; Mosior, J.; Hartman, T.; Burns-Huang, K.; Gold, B.; Morris, R.; Goullieux, L.; Blanc, I.; Vaubourgeix, J.; Lagrange, S.; et al. Opposing reactions in coenzyme A metabolism sensitize Mycobacterium tuberculosis to enzyme inhibition. Science 2019, 363, eaau8959. [Google Scholar] [CrossRef] [PubMed]
- Vickery, C.R.; Kosa, N.M.; Casavant, E.P.; Duan, S.; Noel, J.P.; Burkart, M.D. Structure, Biochemistry, and Inhibition of Essential 4′-Phosphopantetheinyl Transferases from Two Species of Mycobacteria. ACS Chem. Biol. 2014, 9, 1939–1944. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Bashiri, G.; Johnston, J.M.; Brown, A.S.; Ackerley, D.F.; Baker, E.N. Crystal structure of the essential Mycobacterium tuberculosis phosphopantetheinyl transferase PptT, solved as a fusion protein with maltose binding protein. J. Struct. Biol. 2014, 188, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Reuter, K.; Mofid, M.R.; Marahiel, M.A.; Ficner, R. Crystal structure of the surfactin synthetase-activating enzyme Sfp: A prototype of the 4′-phosphopantetheinyl transferase superfamily. EMBO J. 1999, 18, 6823–6831. [Google Scholar] [CrossRef]
- Yasgar, A.; Foley, T.L.; Jadhav, A.; Inglese, J.; Burkart, M.D.; Simeonov, A. A strategy to discover inhibitors of Bacillus subtilis surfactin-type phosphopantetheinyl transferase. Mol. BioSyst. 2009, 6, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, B.P.; Aldrich, C.C. Development of a high-throughput fluorescence polarization assay for the discovery of phosphopantetheinyl transferase inhibitors. Anal. Biochem. 2010, 403, 13–19. [Google Scholar] [CrossRef]
- Kosa, N.M.; Foley, T.L.; Burkart, M.D. Fluorescent techniques for discovery and characterization of phosphopantetheinyl transferase inhibitors. J. Antibiot. 2013, 67, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.G.; Copp, J.N.; Ackerley, D.F. Rapid and flexible biochemical assays for evaluating 4′-phosphopantetheinyl transferase activity. Biochem. J. 2011, 436, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Kumagai, T.; Kitani, K.; Mori, M.; Matoba, Y.; Sugiyama, M. Cloning and Characterization of a Streptomyces Single Module Type Non-ribosomal Peptide Synthetase Catalyzing a Blue Pigment Synthesis. J. Biol. Chem. 2007, 282, 9073–9081. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S.; Robins, K.; Ackerley, D.F. A sensitive single-enzyme assay system using the non-ribosomal peptide synthetase BpsA for measurement of L-glutamine in biological samples. Sci. Rep. 2017, 7, srep41745. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.G.; Robins, K.; Parachin, N.; Ackerley, D.F. A functional screen for recovery of 4′-phosphopantetheinyl transferase and associated natural product biosynthesis genes from metagenome libraries. Environ. Microbiol. 2012, 14, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- The Pandas Development Team. Pandas-Dev/Pandas: Pandas 1.2.4. Zenodo 2021. [Google Scholar] [CrossRef]
- Waskom, M.L. seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Rottier, K.; Faille, A.; Prudhomme, T.; Leblanc, C.; Chalut, C.; Cabantous, S.; Guilhot, C.; Mourey, L.; Pedelacq, J.-D. Detection of soluble co-factor dependent protein expression in vivo: Application to the 4′-phosphopantetheinyl transferase PptT from Mycobacterium tuberculosis. J. Struct. Biol. 2013, 183, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Calcott, M.J.; Collins, V.M.; Owen, J.G.; Ackerley, D.F. The indigoidine synthetase BpsA provides a colorimetric ATP assay that can be adapted to quantify the substrate preferences of other NRPS enzymes. Biotechnol. Lett. 2020, 42, 2665–2671. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.S.; Ackerley, D.F.; Calcott, M.J. High-Throughput Screening for Inhibitors of the SARS-CoV-2 Protease Using a FRET-Biosensor. Molecules 2020, 25, 4666. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Sissons, J.A.; Owen, J.G.; Ackerley, D.F. Directed Evolution of the Nonribosomal Peptide Synthetase BpsA to Enable Recognition by the Human Phosphopantetheinyl Transferases for Counter-Screening Antibiotic Candidates. ACS Infect. Dis. 2020, 6, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Johns, A.; Scharf, D.; Gsaller, F.; Schmidt, H.; Heinekamp, T.; Straßburger, M.; Oliver, J.D.; Birch, M.; Beckmann, N.; Dobb, K.S.; et al. A Nonredundant Phosphopantetheinyl Transferase, PptA, Is a Novel Antifungal Target That Directs Secondary Metabolite, Siderophore, and Lysine Biosynthesis in Aspergillus fumigatus and Is Critical for Pathogenicity. mBio 2017, 8, e01504-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, A.M.; Kirisits, M.; Toy, P.; Nunn, D.S.; Failli, A.; Dushin, E.G.; Novikova, E.; Petersen, P.J.; Joseph-McCarthy, D.; McFadyen, I.; et al. Anthranilate 4H-oxazol-5-ones: Novel small molecule antibacterial acyl carrier protein synthase (AcpS) inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 37–41. [Google Scholar] [CrossRef]
- Foley, T.L.; Rai, G.; Yasgar, A.; Daniel, T.; Baker, H.L.; Attene-Ramos, M.; Kosa, N.M.; Leister, W.; Burkart, M.D.; Jadhav, A.; et al. 4-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-methoxypyridin-2-yl)piperazine-1-carbothioamide (ML267), a Potent Inhibitor of Bacterial Phosphopantetheinyl Transferase That Attenuates Secondary Metabolism and Thwarts Bacterial Growth. J. Med. Chem. 2014, 57, 1063–1078. [Google Scholar] [CrossRef]
- Matano, L.M.; Morris, H.G.; Wood, B.M.; Meredith, T.C.; Walker, S. Accelerating the discovery of antibacterial compounds using pathway-directed whole cell screening. Bioorg. Med. Chem. 2016, 24, 6307–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.H.; Nisa, S.; Dempsey, S.; Jack, C.; O’Toole, R. Modifying Culture Conditions in Chemical Library Screening Identifies Alternative Inhibitors of Mycobacteria. Antimicrob. Agents Chemother. 2009, 53, 5279–5283. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, K.A.; Besra, G.S. Mycobacterial drug discovery. RSC Med. Chem. 2020, 11, 1354–1365. [Google Scholar] [CrossRef]
- Kana, B.D.; Karakousis, P.C.; Parish, T.; Dick, T. Future target-based drug discovery for tuberculosis? Tuberculosis 2014, 94, 551–556. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Known Drug Activities | % Activity in PptT Screen | EC50 (µM) |
|---|---|---|---|
| Aurintricarboxylic acid | DNA topoisomerase II inhibitor | 32 | 4.8 ± 0.9 |
| SCH-202676 hydrobromide | Allosteric agonist and antagonist of GPCRs | 0.7 | 5.9 ± 0.1 |
| Tyrphostin AG 537 | EGFR protein tyrosine kinase inhibitor | 35 | 5.9 ± 0.5 |
| Disulfiram | Alcohol dehydrogenase inhibitor | 9 | 6.0 ± 1.5 |
| Tyrphostin AG 538 | (IGF-1) receptor protein tyrosine kinase inhibitor | 24 | 6.4 ± 0.7 |
| Bay 11-7085 | Inhibitor of NFκB | 10 | 7.4 ± 0.4 |
| U-73122 | Phospholipase C and A2 inhibitor | 44 | 8.1 ± 0.5 |
| 6-Nitroso-1,2-benzopyrone | Poly(ADP-ribose) ligand | 12 | 8.3 ± 1.7 |
| 6-Hydroxy-DL-DOPA | Precursor of the catecholaminergic neurotoxin | 26 | N.D. 1 |
| Tyrphostin AG 808 | Protein tyrosine kinase inhibitor | 34 | N.D. 1 |
| I-OMe-Tyrphostin AG 538 | (IGF-1) receptor protein tyrosine kinase inhibitor | 40 | N.D. 1 |
| Reactive Blue 2 | P2Y receptor antagonist | 38 | N.D. 1 |
| GW5074 | cRaf1 kinase inhibitor | 43 | N.D. 1 |
| PD 404, 182 | KDO-8-P synthase inhibitor | 35 | N.D. 1 |
| Sanguinarine chloride | Inhibitor of ATPase | 44 | N.D. 1 |
| 7,7-Dimethyl-(5z,8z)-eicosadienoic acid | Phospholipase A2 and lipoxygenase inhibitor | 49 | N.D. 1 |
| Rottlerin | PKC and CaM kinase III inhibitor | 48 | N.D. 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, A.S.; Owen, J.G.; Jung, J.; Baker, E.N.; Ackerley, D.F. Inhibition of Indigoidine Synthesis as a High-Throughput Colourimetric Screen for Antibiotics Targeting the Essential Mycobacterium tuberculosis Phosphopantetheinyl Transferase PptT. Pharmaceutics 2021, 13, 1066. https://doi.org/10.3390/pharmaceutics13071066
Brown AS, Owen JG, Jung J, Baker EN, Ackerley DF. Inhibition of Indigoidine Synthesis as a High-Throughput Colourimetric Screen for Antibiotics Targeting the Essential Mycobacterium tuberculosis Phosphopantetheinyl Transferase PptT. Pharmaceutics. 2021; 13(7):1066. https://doi.org/10.3390/pharmaceutics13071066
Chicago/Turabian StyleBrown, Alistair S., Jeremy G. Owen, James Jung, Edward N. Baker, and David F. Ackerley. 2021. "Inhibition of Indigoidine Synthesis as a High-Throughput Colourimetric Screen for Antibiotics Targeting the Essential Mycobacterium tuberculosis Phosphopantetheinyl Transferase PptT" Pharmaceutics 13, no. 7: 1066. https://doi.org/10.3390/pharmaceutics13071066