
Formulation of Chewable Tablets Containing Carbamazepine-β-cyclodextrin Inclusion Complex and F-Melt Disintegration Excipient. The Mathematical Modeling of the Release Kinetics of Carbamazepine

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physical Mixture and Inclusion Complex Synthesis
2.3. Characterization
2.4. Quantitative Analysis of CBZ
2.5. In Vitro Release Kinetics Studies
3. Results and Discussion
3.1. Organoleptic Evaluation of the Compounds
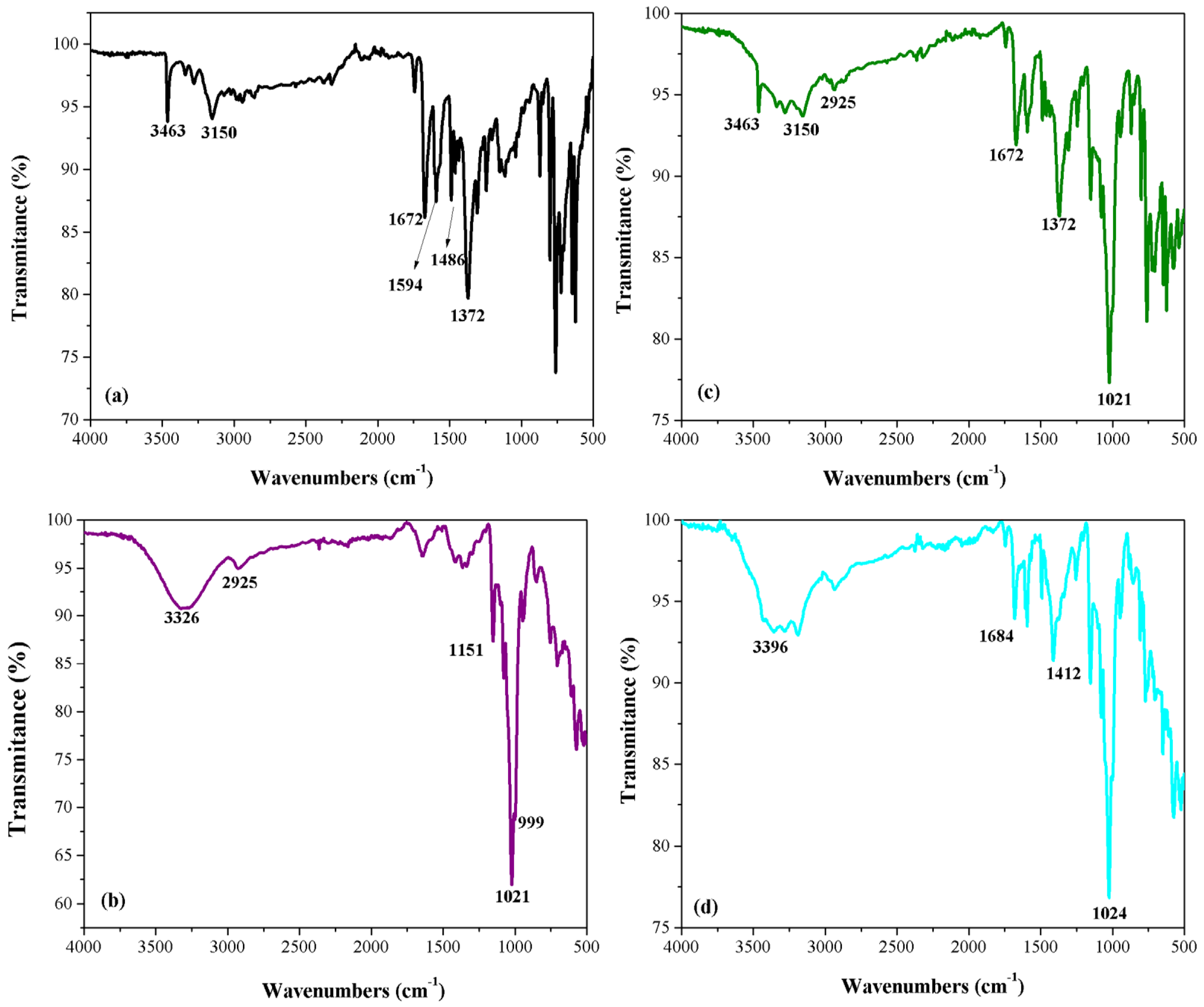
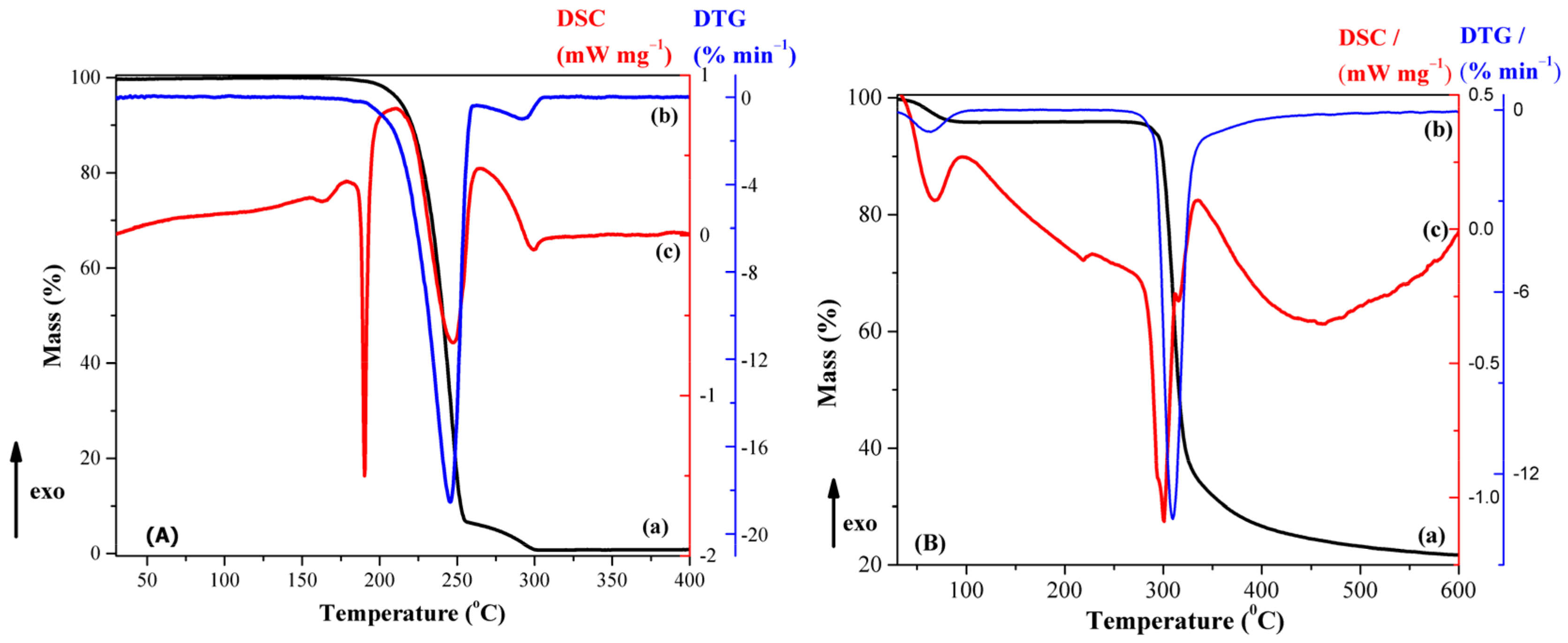
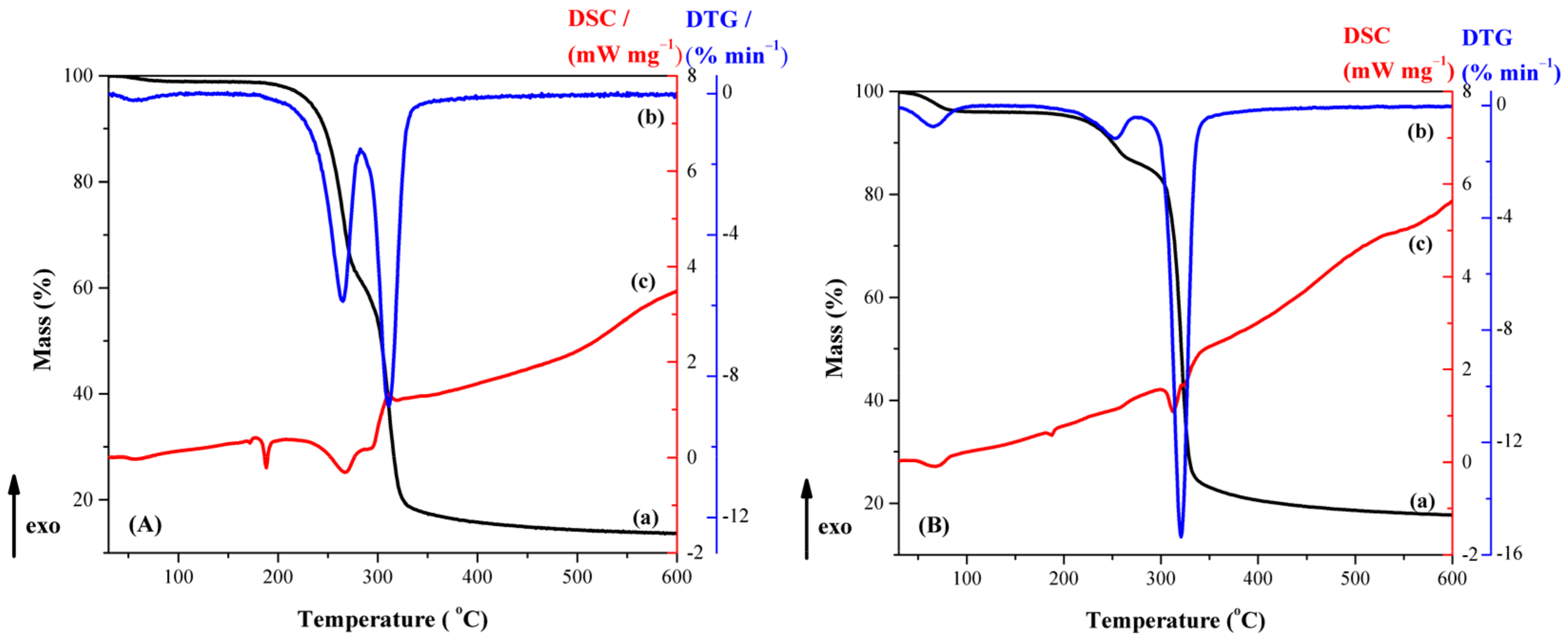
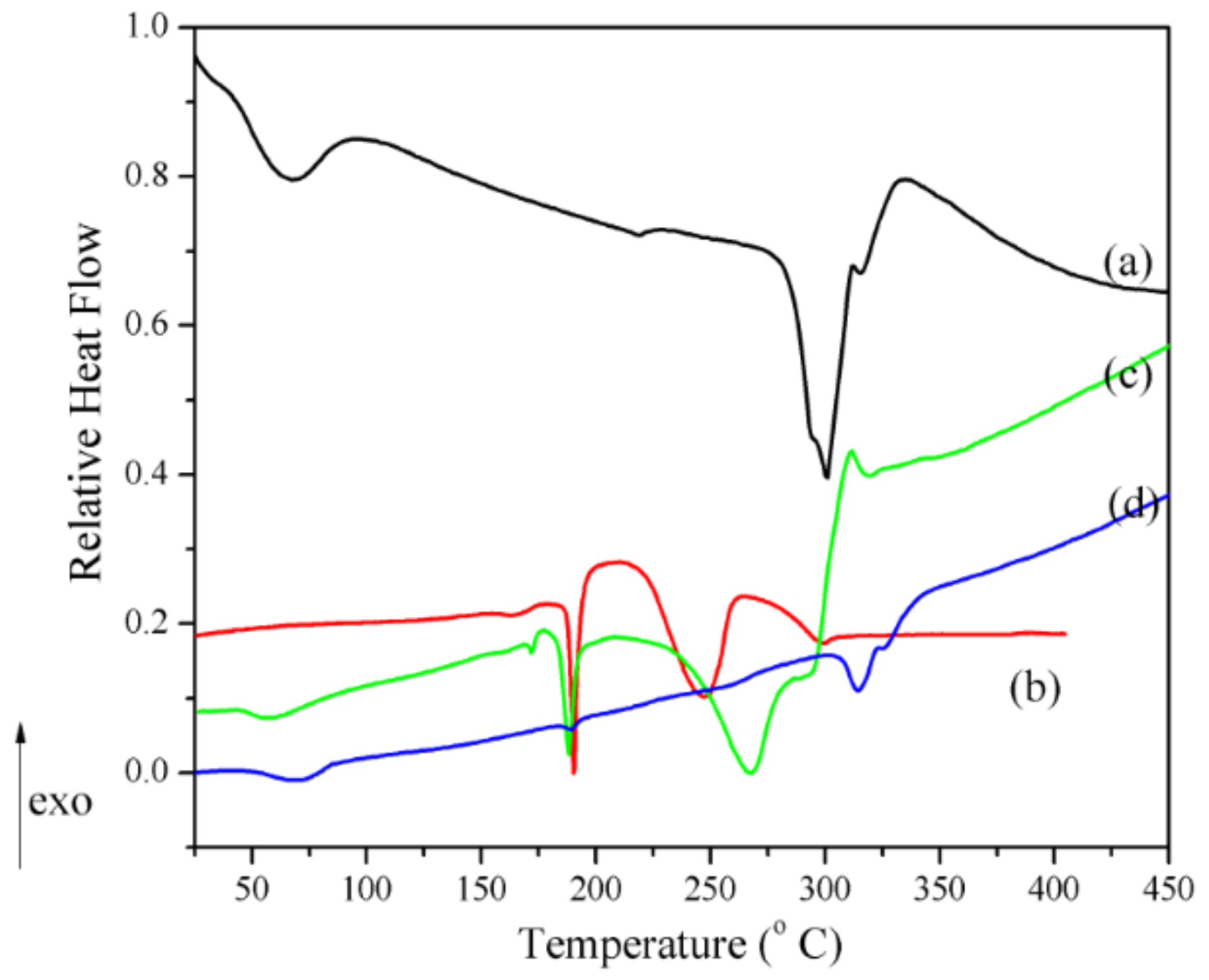
3.2. Physical–Chemical Characterization of Compounds
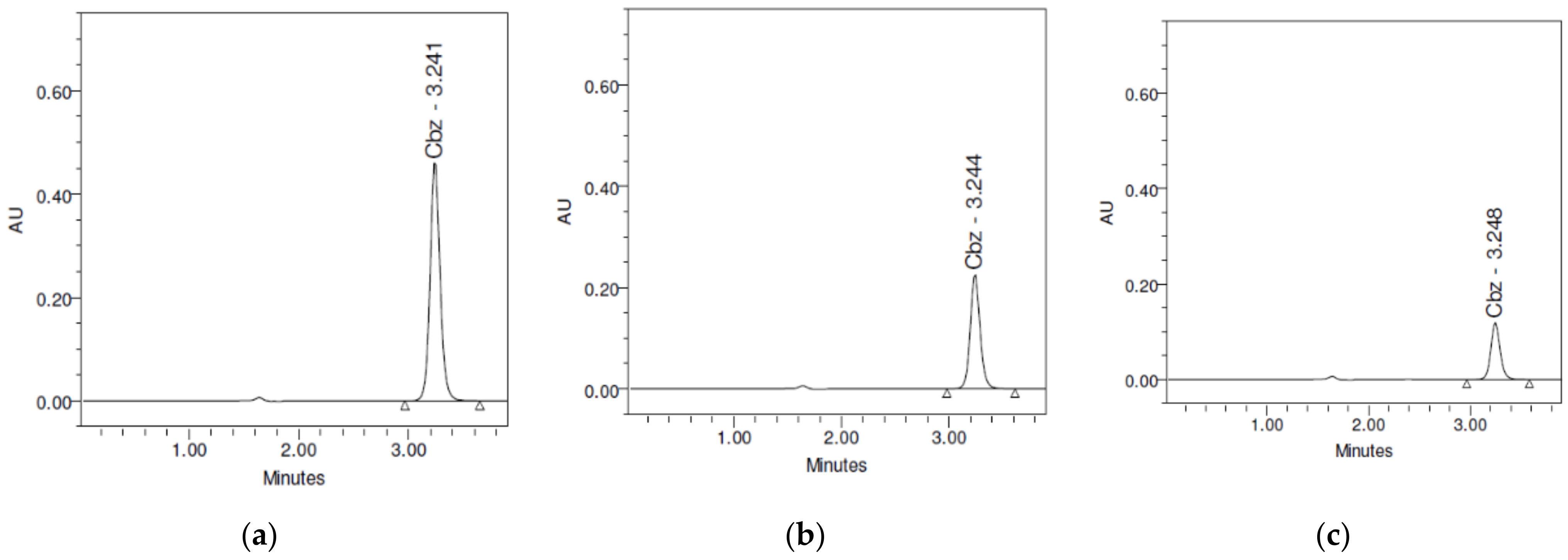
3.3. Quantitative Analysis of CBZ
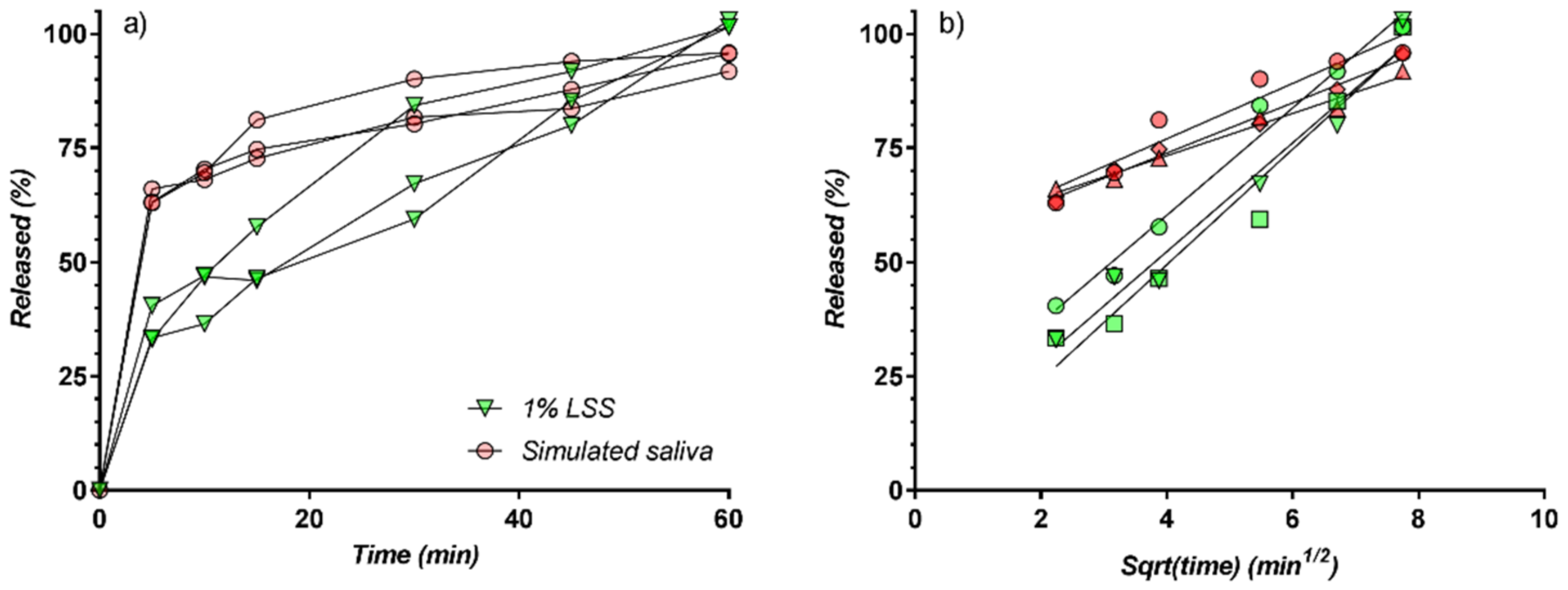
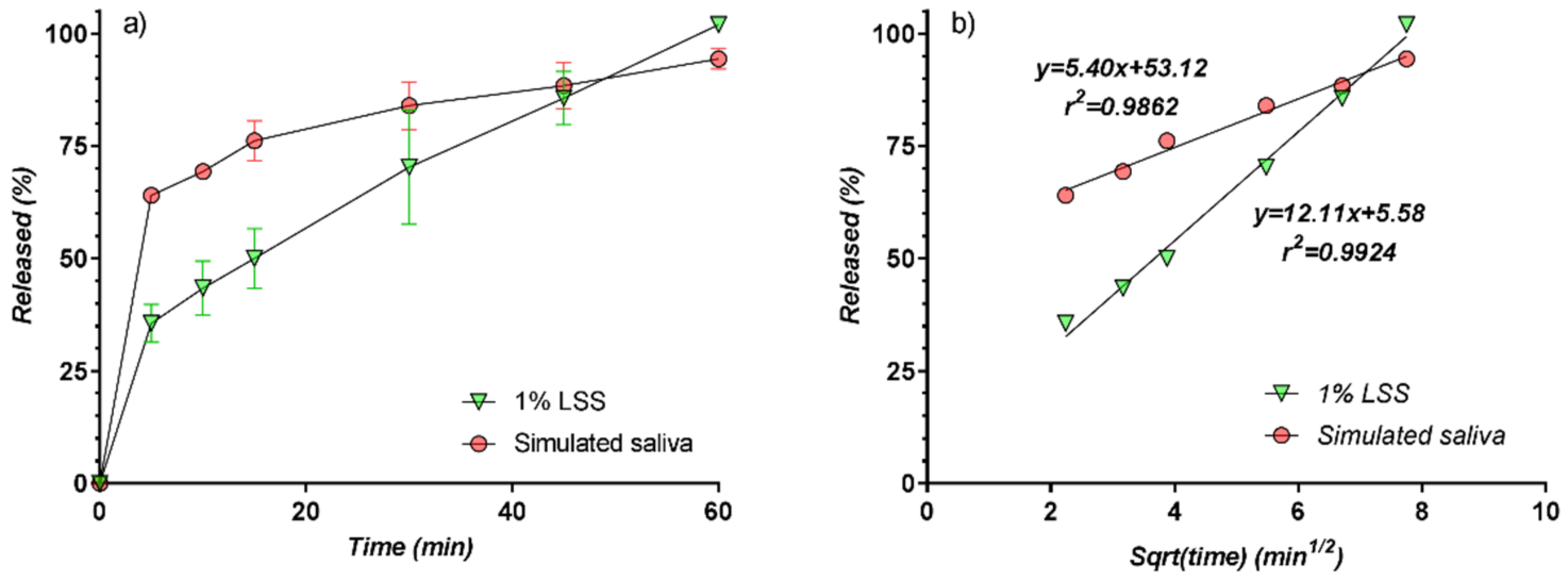
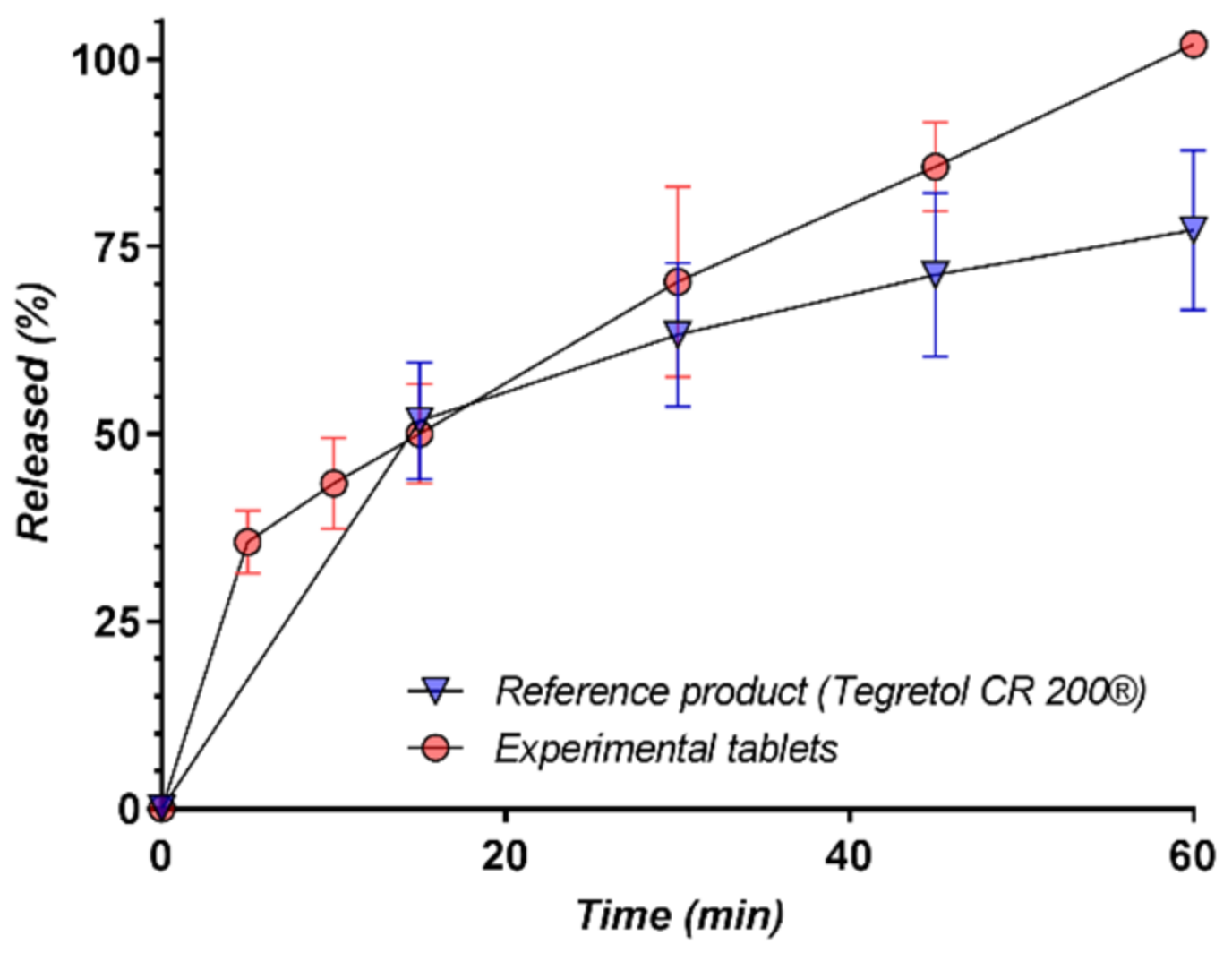
3.4. In Vitro Release Kinetics
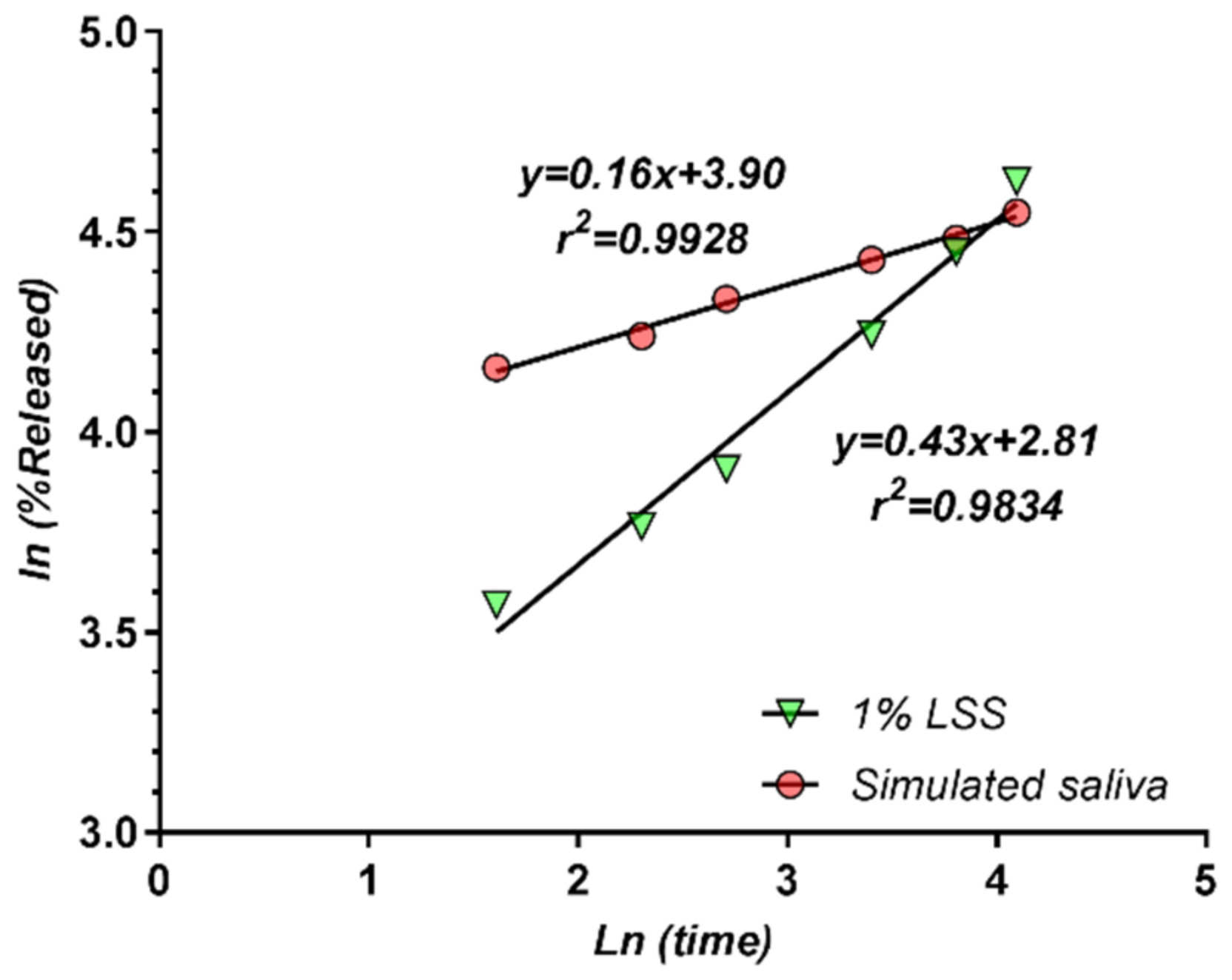
3.5. Modelling of Release Kinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. WHO Model List of Essential Medicines (19th List, PDF); World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Mircioiu, C.; Mircioiu, I.; Voicu, V.; Miron, D. Dissolution—Bioequivalence non-correlations. Basic Clin. Pharmacol. Toxicol. 2005, 96, 262–264. [Google Scholar] [CrossRef]
- Pahomi, G.; Corlan, G.; Anuta, V.; Sandulovici, R.; Mircioiu, I. Study of the influence of bile salts and lecithin on distribution of ketoconazole between plasma and methylene chloride. Farmacia 2012, 60, 809–821. [Google Scholar]
- Mircioiu, C.; Voicu, V.A.; Ionescu, M.; Miron, D.S.; Radulescu, F.S.; Nicolescu, A.C. Evaluation of in vitro absorption, decontamination and desorption of organophosphorous compounds from skin and synthetic membranes. Toxicol. Lett. 2013, 219, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Betlach, C.A.; Gonzalez, M.A.; McKiernan, B.C.; Neff-Davis, C.; Bodor, N. Oral pharmacokinetics of carbamazepine in dogs from commercial tablets and a cyclodextrin complex. J. Pharm. Sci. 1993, 82, 256–258. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D.; Richter, A.; Schulz, H.-U.; Schürer, M.; Düsing, R.; Brewster, M.E. New injectable aqueous carbamazepine solution through complexing with 2-hydroxypropyl-β-cyclodextrin: Tolerability and pharmacokinetics after intravenous injection in comparison to a glycofurol-based formulation. Epilepsia 1995, 36, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Nelson, K.F. Improvement of oral bioavailability of carbamazepine by inclusion in 2-hydroxypropyl-β-cyclodextrin. J. Pharm. Sci. 1992, 85, 175–180. [Google Scholar] [CrossRef]
- Brewster, M.E.; Anderson, W.R.; Meinsma, D.; Moreno, D.; Webb, A.I.; Pablo, L.; Estes, K.S.; Derendorf, H.; Bodor, N.; Sawchuk, R.; et al. Intravenous and oral pharmacokinetics evaluation of a 2-hydroxypropyl-β-cyclodextrin-based formulation of carbamazepine in the dog: Comparison with commercially available tablets and suspensions. J. Pharm. Sci. 1997, 86, 335–339. [Google Scholar] [CrossRef] [PubMed]
- El-Nahhas, A.S. Physico-chemical characteristics of carbamazepine-β-cyclodextrin inclusion compounds and carbamazepine–PEG solid dispersions. Pharmazie 1996, 51, 960–963. [Google Scholar]
- El-Zein, H.; Riad, L.; El-Bary, A.A. Enhancement of carbamazepine dissolution: In vitro and in vivo evaluation. Int. J. Pharm. 1998, 168, 209–220. [Google Scholar] [CrossRef]
- El-Gindy, G.A.; Mohammed, F.A.; Salem, S.Y. Preparation, pharmacokinetic and pharmacodynamic evaluation of carbamazepine inclusion complexes with cyclodextrins. STP Pharma Sci. 2002, 12, 369–378. [Google Scholar]
- Tasic, L.Z.; Milic-Askabic, J.; Rajic, D. Effect of Cyclodextrins on the Physical Properties and Dissolution Rate of Carbamazepine Tablets. In Proceedings of the Eighth International Symposium on Cyclodextrins, Budapest, Hungary, 31 March–2 April 1996; Szejtli, J., Szente, L., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; pp. 449–452. [Google Scholar] [CrossRef]
- Koester, L.S.; Xavier, C.R.; Mayorga, P.; Bassani, V.L. Influence of beta-cyclodextrin complexation on carbamazepine release from hydroxypropyl methylcellulose matrix tablets. Eur. J. Pharm. Biopharm. 2003, 55, 85–91. [Google Scholar] [CrossRef]
- Koester, L.S.; Bertuol, J.B.; Groch, K.R.; Xavier, C.R.; Moellerke, R.; Mayorga, P.; Dalla Costa, T.; Bassani, V.L. Bioavailability of carbamazepine: Beta-cyclodextrin complex in beagle dogs from hydroxypropylmethylcellulose matrix tablets. Eur. J. Pharm. Sci. 2004, 22, 201–207. [Google Scholar] [CrossRef]
- Sarasija, S.; Shivakumar, H.N.; Gorisetty, K.K. Effect of β Cyclodextrin complexation on the solubility and dissolution rate of carbamazepine from tablets. Indian J. Pharm. Sci. 2006, 68, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Ali, W.; Badawi, A.A.; Mahdy, M.A.; El-Nahas, H.M. Formulation ţand Evaluation of Carbamazepine 200 mg immediate release tablets using polyethylene glycol 6000. Int. J. Pharm. Pharm. Sci. 2013, 5, 114–119. [Google Scholar]
- Ali, W.; Badawi, A.A.; Mahdy, M.A.; El-Nahas, H.M. Formulation and Evaluation of Carbamazepine 200 mg Controlled Release Tablets Using Different HPMC Grades. J. Pharm. Res. 2013, 3, 632–647. [Google Scholar] [CrossRef]
- Rao, N.G.R.; Patel, T.; Gandhi, S. Development and evaluation of carbamazepine fast dissolving tablets tablets prepared with a complex by direct compression technique. Asian J. Pharm. 2009, 3, 97–103. [Google Scholar]
- Rao, N.G.R.; Kulkarni, U. Development of carbamazepine fast dissolving tablets: Effect of functionality of hydrophilic carriers on solid dispersion technique. Asian J. Pharm. Clin. Res. 2010, 3, 114–117. [Google Scholar]
- Szejtli, J. Cyclodextrin Complexed Generic Drugs are Generally not Bio-equivalent with the Reference Products: Therefore the Increase in Number of Marketed Drug/Cyclodextrin Formulations is so Slow. J. Incl. Phenom. Macrocycl. Chem. 2005, 52, 1–11. [Google Scholar] [CrossRef]
- Manne, A.S.N.; Hegde, A.R.; Raut, S.Y.; Rao, R.R.; Kulkarni, V.I.; Mutalik, S. Hot liquid extrusion assisted drug-cyclodextrin complexation: A novel continuous manufacturing method for solubility and bioavailability enhancement of drugs. Drug Deliv. Transl. Res. 2021, 11, 1273–1287. [Google Scholar] [CrossRef]
- Conceição, J.; Adeoye, O.; Cabral-Marques, H.; Concheiro, A.; Alvarez-Lorenzo, C.; Sousa Lobo, J.M. Orodispersible Carbamazepine/Hydroxypropyl-β-Cyclodextrin Tablets Obtained by Direct Compression with Five-in-One Co-processed Excipients. AAPS Pharm. Sci. Tech. 2020, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Conceição, J.; Adeoye, O.; Cabral-Marques, H.; Concheiro, A.; Alvarez-Lorenzo, C.; Sousa Lobo, J.M. Carbamazepine bilayer tablets combining hydrophilic and hydrophobic cyclodextrins as a quick/slow biphasic release system. J. Drug Deliv. Sci. Technol. 2020, 57, 101611. [Google Scholar] [CrossRef]
- Abou-Taleb, H.A.; Fathalla, Z.; Abdelkader, H. Comparative studies of the effects of novel excipients amino acids with cyclodextrins on enhancement of dissolution and oral bioavailability of the non-ionizable drug carbamazepine. Eur. J. Pharm. Sci. 2020, 155, 105562. [Google Scholar] [CrossRef]
- Agenţia Naţională a Medicamentului. Farmacopeea Romana, Xth ed.; Editura Medicala: Bucharest, Romania, 2009; p. 805. [Google Scholar]
- Abdalrb, G.A.; Budura, E.A.; Toderescu, C.D.; Sarbu, I. Formulation and Manufacturing Process of Some Chewable Tablets Containing Carbamazepine-β-Cyclodextrin Inclusion Complex. Stud. Univ. VG SSV 2017, 27, 105–110. [Google Scholar]
- Miclea, L.-M.; Vlaia, L.; Vlaia, V. Preparation and Characterisation of Inclusion Complexes of Meloxicam and Alpha-Cyclodextrin and β-Cyclodextrin. Farmacia 2010, 58, 583–593. [Google Scholar]
- ICH Expert Working Group. Q2(R1) Guideline.pdf. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 5 May 2021).
- USP 32. United States Pharmacopeia 32 National Formulary 27; United States Pharmacopoeial Convention Inc.: Rockville, MD, USA, 2009. [Google Scholar]
- Gohel, M.C.; Parikh, R.K.; Aghara, P.Y.; Nagori, S.A.; Delvadia, R.R.; Dabhi, M.R. Application of simplex lattice design and desirability function for the formulation development of mouth dissolving film of salbutamol sulphate. Curr. Drug Deliv. 2009, 6, 486–494. [Google Scholar] [CrossRef]
- Marques, M.R.; Loebenberg, R.; Almukainzi, M. Simulated biological fluids with possible application in dissolution testing. Dissolut. Technol. 2011, 18, 15–28. [Google Scholar] [CrossRef]
- Moore, J.W.; Flanner, H.H. Mathematical comparison of dissolution profiles. Pharm. Tech. 1996, 20, 64–75. [Google Scholar]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Langenbucher, F. Letters to the Editor: Linearization of dissolution rate curves by the Weibull distribution. J. Pharm. Pharm. 1972, 24, 979–981. [Google Scholar] [CrossRef]
- Korsmeyer, R.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N. Mechanisms of potassium chloride release from compressed, hydrophilic, polymeric matrices: Effect of entrapped air. J. Pharm. Sci. 1983, 72, 1189–1191. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Council of Europe. European Pharmacopoeia, 10th ed.; EDQM, Council of Europe: Strasbourg, France, 2019. [Google Scholar]
- Thilak Kumar, R.; Umamaheswari, S. FTIR, FTR and UV-Vis Analysis of Carbamazepine. Res. J. Pharm. Biol. Chem. Sci. 2011, 2, 685–693. [Google Scholar]
- Balaci, T.; Velescu, B.; Karampelas, O.; Musuc, A.M.; Nițulescu, G.M.; Ozon, E.A.; Nițulescu, G.; Gîrd, C.E.; Fița, C.; Lupuliasa, D. Physico-Chemical and Pharmaco-Technical Characterization of Inclusion Complexes Formed by Rutoside with β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin Used to Develop Solid Dosage Forms. Processes 2021, 9, 26. [Google Scholar] [CrossRef]
- Musuc, A.M.; Anuta, V.; Atkinson, I.; Popa, V.T.; Sarbu, I.; Mircioiu, C.; Abdalrb, G.A.; Mitu, M.A.; Ozon, E.A. Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex. Pharmaceutics 2020, 12, 744. [Google Scholar] [CrossRef]
- Lowes, M.M.J.; Caira, M.R.; Lötter, A.P.; Van der Watt, J.G. Physicochemical properties and X-ray structural studies of the trigonal polymorph of carbamazepine. J. Pharm. Sci. 1987, 76, 744–752. [Google Scholar] [CrossRef]
- de Azevedo, M.d.B.M.; Tasic, L.; Fattori, J.; Rodrigues, F.H.S.; Cantos, F.C.; Ribeiro, L.P.; de Paula, V.; Ianzer, D.; Santos, R.A.S. New formulation of an old drug in hypertension treatment: The sustained release of captopril from cyclodextrin nanoparticles. Int. J. Nanomed. 2011, 6, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Childs, S.L.; Rodríguez-Hornedo, N.; Reddy, L.S.; Jayasankar, A.; Maheshwari, C.; Mc Causland, L.; Shipplett, R.; Stahly, B.C. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. Cryst. Eng. Comm. 2008, 10, 856–864. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzeger, A.J. Comparison of the Four Anhydrous Polymorphs of Carbamazepine and the Crystal Structure of Form. Int. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef] [Green Version]
- Rustichelli, C.; Gamberini, G.; Ferioli, V.; Gamberini, M.C.; Ficarra, R.; Tommasini, S. Solid-state study of polymorphic drugs: Carbamazepine. J. Pharm. Biomed. Anal. 2000, 23, 41–54. [Google Scholar] [CrossRef]
- Sethia, S.; Squillante, E. Solid dispersion of carbamazepine in PVPK30 by conventional solvent evaporation and supercriticalmethods. Int. J. Pharm. 2004, 272, 1–10. [Google Scholar] [CrossRef]
- Martins, R.M.; Siqueira, S.; Tacon, L.; Freitas, L. Microstructured ternary solid dispersions to improve carbamazepine solubility. Powder Technol. 2012, 215–216, 156–165. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, S. Preparation and characterization of inclusion complexes of prazosin hydrochloride with β-cyclodextrin and hydroxypropyl- β–cyclodextrin. J. Pharm. Biomed. Anal. 2006, 40, 122–127. [Google Scholar] [CrossRef]
- Musuc, A.M.; Badea-Doni, M.; Jecu, L.; Rusu, A.; Popa, V.T. FTIR, XRD, and DSC analysis of the rosemary extract effect on polyethylene structure and biodegradability. J. Therm. Anal. Calorim. 2013, 114, 169–177. [Google Scholar] [CrossRef]
- Mircioiu, I.; Anuta, V.; Ibrahim, N.; Mircioiu, C. Dissolution of Tamoxifen in Biorelevant Media. A Two Phase Release Model. Farmacia 2012, 60, 315–324. [Google Scholar]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Oxford University Press: London, UK, 1975. [Google Scholar]
- Peppas, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Colombo, P.; Bettini, R.; Massimo, G.; Catellani, P.L.; Santi, P.; Peppas, N. Diffusion front movement is important indrug release from swellable matrix tablets. J. Pharm. Sci. 1995, 84, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Ferrefo, C.; Muñoz-Ruiz, A.; Jiménez-Castellanos, M.R. Front’s movement as a useful tool for hydrophilic matrixrelease mechanism elucidation. Int. J. Pharm. 2000, 39, 231–237. [Google Scholar] [CrossRef]
- Bettini, R.; Catellani, P.L.; Santi, P.; Massimo, G.; Peppas, N.A.; Colombo, P. Translocation of drug particles in HPMC matrix gel layer:effect of drug solubility and influence on release rate. J. Control. Release 2001, 70, 383–391. [Google Scholar] [CrossRef]
- Rinaki, E.; Dokoumetzidis, A.; Macheras, P. The mean dissolution time depends on the dose/solubility ratio. Pharm. Res. 2003, 20, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Prasacu, I.; Mircioiu, C.; Sandulovici, R.; Enache, F. Release of metoprolol from solid dosage forms. Choice and validation of theoretical model. Farmacia 2009, 1, 89–98. [Google Scholar]
- Sandulovici, R.; Prasacu, I.; Mircioiu, C.; Voicu, V.; Medvedovici, A.; Anuta, V. Mathematical and phenomenological criteria in selection of pharmacokinetic model for M1 metabolite of pentoxyphylline. Farmacia 2009, 57, 235–246. [Google Scholar]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Jourghanian, P.; Ghaffari, S.; Ardjmand, M.; Haghighat, S.; Mohammadnejad, M. Sustained release curcumin loaded solid lipid nanoparticles. Adv. Pharm Bull. 2016, 6, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Udeni Gunathilake, T.M.S.; Ching, Y.C.; Chuah, C.H. Enhancement of Curcumin Bioavailability Using Nanocellulose Reinforced Chitosan Hydrogel. Polymers 2017, 9, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurniawan, K.; Posangi, J.; Rampengan, N. Association between public knowledge regarding antibiotics and self-medication with antibiotics in TelingAtas Community Health Center, East Indonesia. Med. J. Indones. 2017, 26, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Zidan, A.S.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A.; Hussain, M.D.; Khan, M.A.; Habib, M.J. Formulation of anastrozolemicroparticles as biodegradable anticancer drug carriers. AAPS Pharm. Sci. Tech. 2006, 7, E38–E46. [Google Scholar] [CrossRef] [Green Version]
- Sonaje, K.; Italia, J.L.; Sharma, G.; Bhardwaj, V.; Tikoo, K.; Kumar, M.N. Development of biodegradable nanoparticles for oral delivery of ellagic acid and evaluation of their antioxidant efficacy against cyclosporine A-induced nephrotoxicity in rats. Pharm. Res. 2007, 24, 899–908. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Dashevsky, A.; Bodmeier, R. Drug release from and sterilization of in situ cubic phase forming monoglyceride drug delivery systems. Eur. J. Pharm. Biopharm. 2010, 75, 375–380. [Google Scholar] [CrossRef]
- Brniak, W.; Jachowicz, R.; Krupa, A.; Skorka, T.; Niwinski, K. Evaluation of co-processed excipients used for direct compression of orally disintegrating tablets (ODT) using novel disintegration apparatus. Pharm. Dev. Technol. 2013, 18, 464–474. [Google Scholar] [CrossRef]
- Bergese, P.; Alessandri, I.; Colombo, I.; Coceani, N.; Depero, L.E. Microstructure and morphology of nimesulide/crospovidone nanocomposites by Raman and electron microscopies. Compos. Part A Appl. Sci. Manuf. 2005, 36, 443–448. [Google Scholar] [CrossRef]
- Mircioiu, C. Release of drug from an infinite reservoir. An alternative method to derive the square-root (Higuchi) law. In Proceedings of the Perspectives in Percutaneous Penetration Sixth International Conference, Leiden, The Netherlands, 22–26 September 1998. [Google Scholar]
- Mircioiu, I.; Anuta, V.; Purcaru, S.-O.; Radulescu, F.; Miron, D.; Dumitrescu, I.B.; Ibrahim, N.; Mircioiu, C. In Vitro Dissolution of Poorly Soluble Drugs in The Presence of Surface Active Agents—In Vivo Pharmacokinetics Correlations. II. Nimesulide. Farmacia 2013, 61, 88–102. [Google Scholar]
- Koester, L.S.; Ortega, G.G.; Mayorga, P.; Bassani, V.L. Mathematical evaluation of in vitro release profiles of hydroxypropylmethylcellulose matrix tablets containing carbamazepine associated to beta-cyclodextrin. Eur. J. Pharm. Biopharm. 2004, 58, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Anuta, V.; Nitulescu, G.M.; Dinu-Pîrvu, C.E.; Olaru, O.T. Biopharmaceutical Profiling of New Antitumor Pyrazole Derivatives. Molecules 2014, 19, 16381–16401. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | LSS 1% | Simulated Saliva | ||||
|---|---|---|---|---|---|---|
| Mean | SD | RSD (%) | Mean | SD | RSD (%) | |
| 5 | 35.60 | 4.19 | 11.78 | 64.04 | 1.72 | 2.69 |
| 10 | 43.42 | 6.02 | 13.86 | 69.34 | 1.15 | 1.65 |
| 15 | 50.01 | 6.63 | 13.26 | 76.15 | 4.43 | 5.82 |
| 30 | 70.29 | 12.73 | 18.12 | 83.99 | 5.31 | 6.32 |
| 45 | 85.67 | 5.90 | 6.89 | 88.44 | 5.19 | 5.86 |
| 60 | 101.99 | 0.84 | 0.82 | 94.42 | 2.29 | 2.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musuc, A.M.; Anuta, V.; Atkinson, I.; Sarbu, I.; Popa, V.T.; Munteanu, C.; Mircioiu, C.; Ozon, E.A.; Nitulescu, G.M.; Mitu, M.A. Formulation of Chewable Tablets Containing Carbamazepine-β-cyclodextrin Inclusion Complex and F-Melt Disintegration Excipient. The Mathematical Modeling of the Release Kinetics of Carbamazepine. Pharmaceutics 2021, 13, 915. https://doi.org/10.3390/pharmaceutics13060915
Musuc AM, Anuta V, Atkinson I, Sarbu I, Popa VT, Munteanu C, Mircioiu C, Ozon EA, Nitulescu GM, Mitu MA. Formulation of Chewable Tablets Containing Carbamazepine-β-cyclodextrin Inclusion Complex and F-Melt Disintegration Excipient. The Mathematical Modeling of the Release Kinetics of Carbamazepine. Pharmaceutics. 2021; 13(6):915. https://doi.org/10.3390/pharmaceutics13060915
Chicago/Turabian StyleMusuc, Adina Magdalena, Valentina Anuta, Irina Atkinson, Iulian Sarbu, Vlad Tudor Popa, Cornel Munteanu, Constantin Mircioiu, Emma Adriana Ozon, George Mihai Nitulescu, and Mirela Adriana Mitu. 2021. "Formulation of Chewable Tablets Containing Carbamazepine-β-cyclodextrin Inclusion Complex and F-Melt Disintegration Excipient. The Mathematical Modeling of the Release Kinetics of Carbamazepine" Pharmaceutics 13, no. 6: 915. https://doi.org/10.3390/pharmaceutics13060915