
Cell-Penetrating Peptides: Applications in Tumor Diagnosis and Therapeutics

Abstract
:1. Introduction
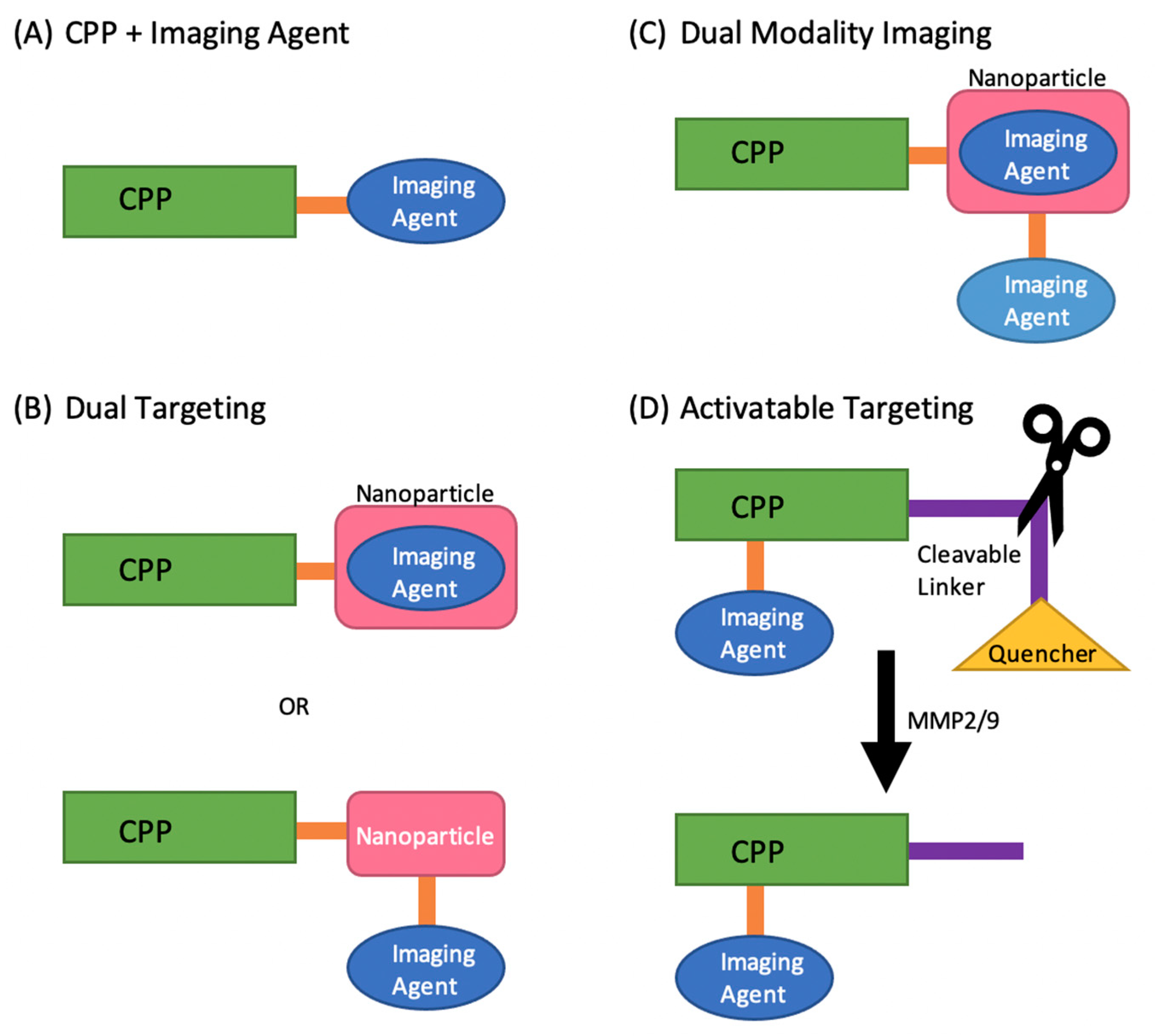
2. Cell-Penetrating Peptides as Tumor Imaging Agents
3. Cell-Penetrating Peptides as Vectors for Targeted Drug Delivery to Tumors
4. Clinical Trials Using CPPs as Cancer Therapeutics
5. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Disclosures
References
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.; Ishino, M.; Loewenstein, P.M. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell 1989, 58, 215–223. [Google Scholar] [CrossRef]
- DeRossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Smith, G.P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2019, 58, 14428–14437. [Google Scholar] [CrossRef] [Green Version]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, S.; White, S.J.; Watkins, S.J.; Hawkins, R.E.; Baker, A.H. Selective Targeting of Gene Transfer to Vascular Endothelial Cells by Use of Peptides Isolated by Phage Display. Circulation 2000, 102, 231–237. [Google Scholar] [CrossRef]
- Mi, Z.; Lu, X.; Mai, J.C.; Ng, B.G.; Wang, G.; Lechman, E.R.; Watkins, S.C.; Rabinowich, H.; Robbins, P.D. Identification of a synovial fibroblast-specific protein transduction domain for delivery of apoptotic agents to hyperplastic synovium. Mol. Ther. 2003, 8, 295–305. [Google Scholar] [CrossRef]
- Chamarthy, S.P.; Jia, L.; Kovacs, J.R.; Anderson, K.R.; Shen, H.; Firestine, S.M.; Meng, W.S. Gene delivery to dendritic cells facilitated by a tumor necrosis factor alpha-competing peptide. Mol. Immunol. 2004, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.K.; Bertera, S.; Bottino, R.; Balamurugan, A.N.; Mai, J.C.; Mi, Z.; Trucco, M.; Robbins, P.D. Protection of islets by in situ peptide-mediated transduction of the Ikappa B kinase inhibitor Nemo-binding domain peptide. J. Biol. Chem. 2003, 278, 9862–9868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.; Phillips, B.E.; Albers, S.M.; Giannoukakis, N.; Watkins, S.C.; Robbins, P.D. Identification of a Cardiac Specific Protein Transduction Domain by In Vivo Biopanning Using a M13 Phage Peptide Display Library in Mice. PLoS ONE 2010, 5, e12252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.; Robbins, P.D. Cell-type specific penetrating peptides: Therapeutic promises and challenges. Molecules 2015, 20, 13055–13070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, I.D.; Bechara, C.; Walrant, A.; Zaltsman, Y.; Jiao, C.-Y.; Sagan, S. Relationships between Membrane Binding, Affinity and Cell Internalization Efficacy of a Cell-Penetrating Peptide: Penetratin as a Case Study. PLoS ONE 2011, 6, e24096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, B.; Seal, B.L.; Brophy, C.M.; Panitch, A. Design of a bioactive cell-penetrating peptide: When a transduction domain does more than transduce. J. Pept. Sci. 2009, 15, 668–674. [Google Scholar] [CrossRef] [Green Version]
- Rennert, R.; Wespe, C.; Beck-Sickinger, A.G.; Neundorf, I. Developing novel hCT derived cell-penetrating peptides with improved metabolic stability. Biochim. Biophys. Acta Biomembr. 2006, 1758, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Neundorf, I.; Rennert, R.; Franke, J.; Kozle, I.; Bergmann, R. Detailed Analysis Concerning the Biodistribution and Metabolism of Human Calcitonin-Derived Cell-Penetrating Peptides. Bioconjugate Chem. 2008, 19, 1596–1603. [Google Scholar] [CrossRef]
- Lindgren, M.E.; Hällbrink, M.M.; Elmquist, A.M.; Langel, Ü. Passage of cell-penetrating peptides across a human epithelial cell layer in vitro. Biochem. J. 2004, 377, 69–76. [Google Scholar] [CrossRef]
- Santra, S.; Yang, H.; Stanley, J.T.; Holloway, P.H.; Moudgil, B.M.; Walter, G.; Mericle, R.A. Rapid and effective labeling of brain tissue using TAT-conjugated CdS:Mn/ZnS quantum dots. Chem. Commun. 2005, 3144–3146. [Google Scholar] [CrossRef]
- Medintz, I.L.; Pons, T.; Delehanty, J.B.; Susumu, K.; Brunel, F.M.; Dawson, P.E.; Mattoussi, H. Intracellular Delivery of Quantum Dot−Protein Cargos Mediated by Cell Penetrating Peptides. Bioconjugate Chem. 2008, 19, 1785–1795. [Google Scholar] [CrossRef]
- Zhang, K.; Fang, H.; Chen, Z.; Taylor, J.-S.A.; Wooley, K.L. Shape Effects of Nanoparticles Conjugated with Cell-Penetrating Peptides (HIV Tat PTD) on CHO Cell Uptake. Bioconjugate Chem. 2008, 19, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Böhmová, E.; Pola, R.; Pechar, M.; Parnica, J.; Machová, D.; Janoušková, O.; Etrych, T. Polymer Cancerostatics Containing Cell-Penetrating Peptides: Internalization Efficacy Depends on Peptide Type and Spacer Length. Pharmaceutics 2020, 12, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonyarattanakalin, S.; Athavankar, S.; Sun, Q.; Peterson, B.R. Synthesis of an artificial cell surface receptor that enables oligohistidine affinity tags to function as metal-dependent cell-penetrating peptides. J. Am. Chem. Soc. 2006, 128, 386–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, C.-Y.; Delaroche, D.; Burlina, F.; Alves, I.; Chassaing, G.; Sagan, S. Translocation and Endocytosis for Cell-penetrating Peptide Internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hällbrink, M.; Florén, A.; Elmquist, A.; Pooga, M.; Bartfai, T.; Langel, Ü. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta Biomembr. 2001, 1515, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjugate Chem. 2007, 18, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Mäger, I.; Eiríksdóttir, E.; Langel, K.; El Andaloussi, S.; Langel, Ü. Assessing the uptake kinetics and internalization mechanisms of cell-penetrating peptides using a quenched fluorescence assay. Biochim. Biophys. Acta Biomembr. 2010, 1798, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Suhorutsenko, J.; Oskolkov, N.; Arukuusk, P.; Kurrikoff, K.; Eriste, E.; Copolovici, D.-M.; Lange, U. Cell-penetrating peptides, PepFects, show no evidence of toxicity and immunogenicity in vitro and in vivo. Bioconjugate Chem. 2011, 22, 2255–2262. [Google Scholar] [CrossRef]
- Richard, J.-P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Sauter, M.; Strieker, M.; Kleist, C.; Wischnjow, A.; Daniel, V.; Altmann, A.; Haberkorn, U.; Mier, W. Improving antibody-based therapies by chemical engineering of antibodies with multimeric cell-penetrating peptides for elevated intracellular delivery. J. Control Release 2020, 322, 200–208. [Google Scholar] [CrossRef]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.-R.; Shi, N.-Q.; Gao, W.; Xiang, B. Enhancing cellular uptake of activable cell-penetrating peptide–doxorubicin conjugate by enzymatic cleavage. Int. J. Nanomed. 2012, 7, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Kolesinska, B.; Eyer, K.; Robinson, T. Interaction of beta(3) /beta(2) -peptides, consisting of Val-Ala-Leu segments, with POPC giant unilamellar vesicles (GUVs) and white blood cancer cells (U937)—A new type of cell-penetrating peptides, and a surprising chain-length dependence of their vesicle- and cell-lysing activity. Chem. Biodivers. 2015, 12, 697–732. [Google Scholar]
- Qiu, W.-X.; Liu, L.-H.; Li, S.-Y.; Lei, Q.; Luo, G.-F.; Zhang, X.-Z. ACPI Conjugated Gold Nanorods as Nanoplatform for Dual Image Guided Activatable Photodynamic and Photothermal Combined Therapy In Vivo. Small 2017, 13. [Google Scholar] [CrossRef]
- Polyakov, V.; Sharma, V.; Dahlheimer, J.L.; Pica, C.M.; Luker, G.D.; Piwnica-Worms, D. Novel Tat-peptide chelates for direct transduction of technetium-99m and rhenium into human cells for imaging and radiotherapy. Bioconjugate Chem. 2000, 11, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Konishi, Y.; Ueda, M.; Saji, H.; Futaki, S. Accumulation of arginine-rich cell-penetrating peptides in tumors and the potential for anticancer drug delivery in vivo. J. Control Release 2012, 159, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, J.; Kebebe, D.; Wu, Y.; Zhang, B.; Liu, Z. Cell penetrating peptides functionalized gambogic acid-nanostructured lipid carrier for cancer treatment. Drug Deliv. 2018, 25, 757–765. [Google Scholar] [CrossRef]
- Li, Y.; Hao, L.; Liu, F.; Yin, L.; Yan, S.; Zhao, H.; Ding, X.; Guo, Y.; Cao, Y.; Li, P.; et al. Cell penetrating peptide-modified nanoparticles for tumor targeted imaging and synergistic effect of sonodynamic/HIFU therapy. Int. J. Nanomed. 2019, 14, 5875–5894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Chen, J.; Luo, S.; Dong, J.; Hu, H.; Yang, Z.; Feng, X.; Liu, Y.; Liu, B.; Pan, G.; et al. Targeting and imaging colorectal cancer by activatable cell-penetrating peptides. Am. J. Transl. Res. 2020, 12, 1754–1766. [Google Scholar] [PubMed]
- Olson, E.S.; Jiang, T.; Aguilera, T.A.; Nguyen, Q.T.; Ellies, L.G.; Scadeng, M.; Tsien, R.Y. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc. Natl. Acad. Sci. USA 2010, 107, 4311–4316. [Google Scholar] [CrossRef] [Green Version]
- Collaborators GBDCoD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef] [Green Version]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.-M.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed]
- Primeau, A.J.; Rendon, A.; Hedley, D.; Lilge, L.; Tannock, I.F.; Koido, S.; Hara, E.; Homma, S.; Torii, A.; Toyama, Y.; et al. The Distribution of the Anticancer Drug Doxorubicin in Relation to Blood Vessels in Solid Tumors. Clin. Cancer Res. 2005, 11, 8782–8788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: Significance of elevated interstitial pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar] [PubMed]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Zhang, L. Therapeutic nanoparticles to combat cancer drug resistance. Curr. Drug Metab. 2009, 10, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S.; Sahoo, S.K. Polymeric nanoparticles for cancer therapy. J. Drug Target. 2008, 16, 108–123. [Google Scholar] [CrossRef]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.; Shin, D.M. Therapeutic Nanoparticles for Drug Delivery in Cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Khawli, L.A.; Hu, P.; Epstein, A.L. NHS76/PEP2, a Fully Human Vasopermeability-Enhancing Agent to Increase the Uptake and Efficacy of Cancer Chemotherapy. Clin. Cancer Res. 2005, 11, 3084–3093. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, R.; Ishima, Y.; Chuang, V.T.G.; Nakamura, H.; Fang, J.; Watanabe, H.; Shimizu, T.; Okuhira, K.; Ishida, T.; Maeda, H.; et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials 2017, 140, 162–169. [Google Scholar] [CrossRef]
- Huwyler, J.; Cerletti, A.; Fricker, G.; Eberle, A.N.; Drewe, J. By-passing of P-glycoprotein Using Immunoliposomes. J. Drug Target. 2002, 10, 73–79. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J. Drug Target. 2007, 15, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Colagiuri, B.; Dhillon, H.; Butow, P.N.; Jansen, J.; Cox, K.; Jacquet, J. Does Assessing Patients’ Expectancies About Chemotherapy Side Effects Influence Their Occurrence? J. Pain Symptom Manag. 2013, 46, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, V.J.; D’Angelo, S.; Butler, K.S.; Theron, C.; Smith, T.L.; Marchiò, S.; Gelovani, J.G.; Sidman, R.L.; Dobroff, A.S.; Brinker, C.J.; et al. Ligand-targeted theranostic nanomedicines against cancer. J. Control Release 2016, 240, 267–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruoslahti, E. Peptides as Targeting Elements and Tissue Penetration Devices for Nanoparticles. Adv. Mater. 2012, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Akashi, Y.; Oda, T.; Ohara, Y.; Miyamoto, R.; Kurokawa, T.; Hashimoto, S.; Enomoto, T.; Yamada, K.M.; Satake, M.; Ohkohchi, N. Anticancer effects of gemcitabine are enhanced by co-administered iRGD peptide in murine pancreatic cancer models that overexpressed neuropilin-1. Br. J. Cancer 2014, 110, 1481–1487. [Google Scholar] [CrossRef]
- Sha, H.; Zou, Z.; Xin, K.; Bian, X.; Cai, X.; Lu, W.; Chen, J.; Chen, G.; Huang, L.; Blair, A.M.; et al. Tumor-penetrating peptide fused EGFR single-domain antibody enhances cancer drug penetration into 3D multicellular spheroids and facilitates effective gastric cancer therapy. J. Control Release 2015, 200, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Schmithals, C.; Köberle, V.; Korkusuz, H.; Pleli, T.; Kakoschky, B.; Augusto, E.A.; Ibrahim, A.A.; Arencibia, J.M.; Vafaizadeh, V.; Groner, B.; et al. Improving Drug Penetrability with iRGD Leverages the Therapeutic Response to Sorafenib and Doxorubicin in Hepatocellular Carcinoma. Cancer Res. 2015, 75, 3147–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipman, S.D.; Oldham, F.B.; Pezzoni, G.; Singer, J.W. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer–drug conjugate. Int. J. Nanomed. 2006, 1, 375–383. [Google Scholar] [CrossRef]
- Gao, Y.; Kuang, Y.; Guo, Z.F.; Guo, Z.; Krauss, I.J.; Xu, B. Enzyme-instructed molecular self-assembly confers nanofibers and a supramolecular hydrogel of taxol derivative. J. Am. Chem. Soc. 2009, 131, 13576–13577. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Cheng, J. Drug-Initiated, Controlled Ring-Opening Polymerization for the Synthesis of Polymer-Drug Conjugates. Macromolecules 2012, 45, 2225–2232. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, X.; Yang, Z.; Zhao, J.; Wang, X.; Zhang, Q.; Yuan, M.; Xie, L.; Liu, H.; He, Q. The potential efficacy of R8-modified paclitaxel-loaded liposomes on pulmonary arterial hypertension. Pharm. Res. 2013, 30, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Dubikovskaya, E.A.; Thorne, S.H.; Pillow, T.H.; Contag, C.H.; Wender, P.A. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc. Natl. Acad. Sci. USA 2008, 105, 12128–12133. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.; Khwaja, A.; Liapi, E.; Torbenson, M.S.; Georgiades, C.S.; Geschwind, J.-F.H. New Intra-arterial Drug Delivery System for the Treatment of Liver Cancer: Preclinical Assessment in a Rabbit Model of Liver Cancer. Clin. Cancer Res. 2006, 12, 2563–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M.; et al. Expression of a multidrug resistance gene in human cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cheetham, A.G.; Lock, L.L.; Cui, H. Cellular Uptake and Cytotoxicity of Drug–Peptide Conjugates Regulated by Conjugation Site. Bioconjugate Chem. 2013, 24, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Takenobu, T.; Tomizawa, K.; Matsushita, M.; Li, S.-T.; Moriwaki, A.; Lu, Y.-F.; Matsui, H. Development of p53 protein transduction therapy using membrane-permeable peptides and the application to oral cancer cells. Mol. Cancer Ther. 2002, 1, 1043–1049. [Google Scholar] [PubMed]
- Michiue, H.; Tomizawa, K.; Wei, F.-Y.; Matsushita, M.; Lu, Y.-F.; Ichikawa, T.; Tamiya, T.; Date, I.; Matsui, H. The NH2 Terminus of Influenza Virus Hemagglutinin-2 Subunit Peptides Enhances the Antitumor Potency of Polyarginine-mediated p53 Protein Transduction. J. Biol. Chem. 2005, 280, 8285–8289. [Google Scholar] [CrossRef] [Green Version]
- Jauset, T.; Beaulieu, M.E. Bioactive cell penetrating peptides and proteins in cancer: A bright future ahead. Curr. Opin. Pharmacol. 2019, 47, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Turchick, A.; Hegan, D.C.; Jensen, R.B.; Glazer, P.M. A cell-penetrating antibody inhibits human RAD51 via direct binding. Nucleic Acids Res. 2017, 45, 11782–11799. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-M.; Choi, D.-K.; Jung, K.; Bae, J.; Kim, J.-S.; Park, S.-W.; Song, K.-H.; Kim, Y.-S. Antibody targeting intracellular oncogenic ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090. [Google Scholar] [CrossRef] [Green Version]
- Van Impe, K.; Bethuyne, J.; Cool, S.; Impens, F.; Ruano-Gallego, D.; De Wever, O.; Vanloo, B.; Van Troys, W.; Lambein, K.; Boucherie, C.; et al. A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast Cancer Res. 2013, 15, R116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agemy, L.; Friedmann-Morvinski, D.; Kotamraju, V.R.; Roth, L.; Sugahara, K.N.; Girard, O.; Mattrey, R.F.; Verma, I.M.; Ruoslahti, E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, A.M.; Aidoudi-Ahmed, S.; Sharma, S.; Kotamraju, V.R.; Foster, P.J.; Sugahara, K.N.; Ruoslahti, E.; Rutt, B.K. Nanoparticles coated with the tumor-penetrating peptide iRGD reduce experimental breast cancer metastasis in the brain. J. Mol. Med. 2015, 93, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Braun, G.B.; De Mendoza, T.H.; Kotamraju, V.R.; French, R.P.; Lowy, A.M.; Teesalu, T.; Ruoslahti, E. Tumor-Penetrating iRGD Peptide Inhibits Metastasis. Mol. Cancer Ther. 2015, 14, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, M.; Ouyang, J.; Wei, Y.; Zhang, J.; Lan, Q.; Deng, C.; Zhong, Z. Selective Cell Penetrating Peptide-Functionalized Envelope-Type Chimeric Lipopepsomes Boost Systemic RNAi Therapy for Lung Tumors. Adv. Heal. Mater. 2019, 8, e1900500. [Google Scholar] [CrossRef]
- Ben Djemaa, S.; Herve-Aubert, K.; Lajoie, L.; Falanga, A.; Galdiero, S.; Nedellec, S.; Souce, M.; Munnier, E.; Chourpa, I.; David, S.; et al. gH625 Cell-Penetrating Peptide Promotes the Endosomal Escape of Nanovectorized siRNA in a Triple-Negative Breast Cancer Cell Line. Biomacromolecules 2019, 20, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gao, Y.; Gong, C.; Han, Z.; Qiang, L.; Tai, Z.; Tian, J.; Gao, S. Dual-Blockade Immune Checkpoint for Breast Cancer Treatment Based on a Tumor-Penetrating Peptide Assembling Nanoparticle. ACS Appl. Mater. Interfaces 2019, 11, 39513–39524. [Google Scholar] [CrossRef]
- Furukawa, K.; Tanaka, M.; Oba, M. siRNA delivery using amphipathic cell-penetrating peptides into human hepatoma cells. Bioorganic Med. Chem. 2020, 28, 115402. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, W.; Wu, Y.; Wan, X.; Gong, Y. Co-delivery of EGFR and BRD4 siRNA by cell-penetrating peptides-modified redox-responsive complex in triple negative breast cancer cells. Life Sci. 2021, 266, 118886. [Google Scholar] [CrossRef]
- Aileron Therapeutics. ALRN-6924 in Patients with Advanced Solid Tumors or Lymphomas; National Library of Medicine: Bethesda, MD, USA, 2014; NCT02264613 (ClinicalTrials.gov Identifier). [Google Scholar]
- Aileron Therapeutics. Safety Study of ALRN-6924 in Patients with Acute Myeloid Leukemia or Advanced Myelodysplastic Syndrome; National Library of Medicine: Bethesda, MD, USA, 2016; NCT02909972 (ClinicalTrials.gov Identifier). [Google Scholar]
- Aileron Therapeutics. ALRN-6924 and Paclitaxel in Treating Patients with Advanced, Metastatic, or Unresectable Solid Tumors; National Library of Medicine: Bethesda, MD, USA, 2018; NCT03725436 (ClinicalTrials.gov Identifier). [Google Scholar]
- Aileron Therapeutics. Phase 1 Study of the Dual MDM2/MDMX Inhibitor ALRN-6924 in Pediatric Cancer; National Library of Medicine: Bethesda, MD, USA, 2018; NCT03654716 (ClinicalTrials.gov Identifier). [Google Scholar]
- Aileron Therapeutics. A Study of ALRN-6924 for the Prevention of Topotecan-induced Myelosuppression During Treatment for Small Cell Lung Cancer; National Library of Medicine: Bethesda, MD, USA, 2019; NCT04022876 (ClinicalTrials.gov Identifier). [Google Scholar]
- Cancer Research, UK. BT1718 in Patients with Advanced Solid Tumours; National Library of Medicine: Bethesda, MD, USA, 2018; NCT03486730 (ClinicalTrials.gov Identifier). [Google Scholar]
- Das Gupta, T.K. Safety Study of a Cell Penetrating Peptide (p28) to Treat Solid Tumors That Resist Standard Methods of Treatmen; National Library of Medicine: Bethesda, MD, USA, 2009; NCT00914914 (ClinicalTrials.gov Identifier). [Google Scholar]
- Pediatric Brain Tumor Consortium. p28 in Treating Younger Patients with Recurrent or Progressive Central Nervous System Tumors; National Library of Medicine: Bethesda, MD, USA, 2013; NCT01975116 (ClinicalTrials.gov Identifier). [Google Scholar]
- Institut Curie. First-in-human Phase I to Evaluate PEP-010 as Single Agent and in Combination with Paclitaxel (CleverPeptide); National Library of Medicine: Bethesda, MD, USA, 2021; NCT04733027 (ClinicalTrials.gov Identifier). [Google Scholar]
- Amal Therapeutics. Phase 1b Study to Evaluate ATP128, With or Without BI 754091, in Patients with Stage IV Colorectal Cancer (KISIMA-01); National Library of Medicine: Bethesda, MD, USA, 2019; NCT04046445 (ClinicalTrials.gov Identifier). [Google Scholar]


{kind=link}
| Sponsor | ClinicalTrials.govIdentifier | Study Stage | CPP Employed | Cancer Targeted | Drug Employed with CPP | Study Size |
|---|---|---|---|---|---|---|
| Aileron Therapeutics [90] | NCT02264613 | Phase 1—Completed Phase 2a—Completed | ALRN-6924 | Solid tumor, lymphoma, and peripheral T-cell lymphoma | ALRN-6924—alone and in combination withpalbociclib | 149 |
| Aileron Therapeutics [91] | NCT02909972 | Phase 1—Completed | ALRN-6924 | Acute myeloid leukemia, and advanced myelodysplastic syndrome | ALRN-6924—alone and in combination with cytarabine | 55 |
| Aileron Therapeutics [92] | NCT03725436 | Phase 1 | ALRN-6924 | Advanced, metastatic or unresectable solid tumors | ALRN-6924—in combination with paclitaxel | 45 |
| Aileron Therapeutics [93] | NCT03654716 | Phase 1 | ALRN-6924 | Pediatric leukemia, pediatric brain tumor, pediatric solid tumor, pediatric lymphoma | ALRN-6924—alone or in combination with cytarabine for patients with leukemia | 69 |
| Aileron Therapeutics [94] | NCT04022876 | Phase 1a—Completed Phase 1b Phase 2 | ALRN-6924 | Small cell lung cancer | Phase 1b—ALRN-6924 with topotecan Phase 2—topotecan alone and in combination with ALRN-6924 | 120 |
| Cancer Research UK [95] | NCT03486730 | Phase 1 Phase 2 | BT1718 | Advanced solid tumors, non-small cell lung cancer, non-small cell lung sarcoma, and esophageal cancer | BT1718—alone | 130 |
| CDG Therapeutics and Dr. Tapas K. Das Gupta [96] | NCT00914914 | Phase 1—Completed | P28 | Refractory solid tumors | P28—alone | 15 |
| Pediatric Brain Tumor Consortium/National Cancer Institute (NCI) [97] | NCT01975116 | Phase 1—Completed | P28 | Recurrent or progressive central nervous system tumors | P28—alone | 18 |
| Institut Curie [98] | NCT04733027 | Phase 1 | PEP-010 | Metastatic solid tumor cancer | PEP-010—alone PEP-010—in combination with paclitaxel | 56 |
| Amal Therapeutics [99] | NCT04046445 | Phase 1a—Completed Phase 1b | ATP128 | Stage IV colorectal cancer | ATP128—alone and in combination with BI 754091 | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiltner, J.; McCandless, K.; Zahid, M. Cell-Penetrating Peptides: Applications in Tumor Diagnosis and Therapeutics. Pharmaceutics 2021, 13, 890. https://doi.org/10.3390/pharmaceutics13060890
Stiltner J, McCandless K, Zahid M. Cell-Penetrating Peptides: Applications in Tumor Diagnosis and Therapeutics. Pharmaceutics. 2021; 13(6):890. https://doi.org/10.3390/pharmaceutics13060890
Chicago/Turabian StyleStiltner, Jeffrey, Kayla McCandless, and Maliha Zahid. 2021. "Cell-Penetrating Peptides: Applications in Tumor Diagnosis and Therapeutics" Pharmaceutics 13, no. 6: 890. https://doi.org/10.3390/pharmaceutics13060890