
The Role of PK/PD Analysis in the Development and Evaluation of Antimicrobials


Abstract
:1. Introduction
2. Pharmacokinetic/Pharmacodynamic Principles
3. Models to Study the PK/PD of Antimicrobials
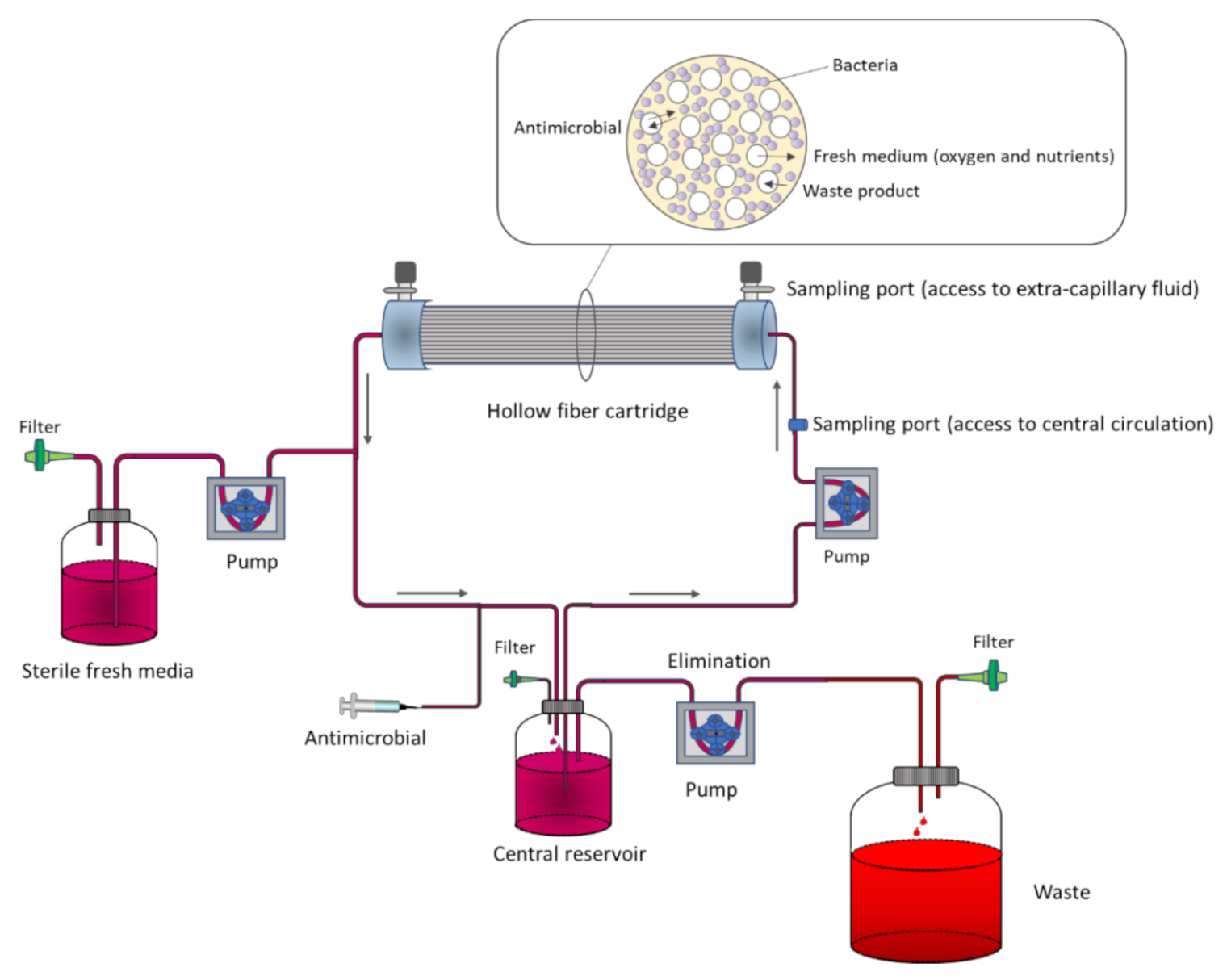
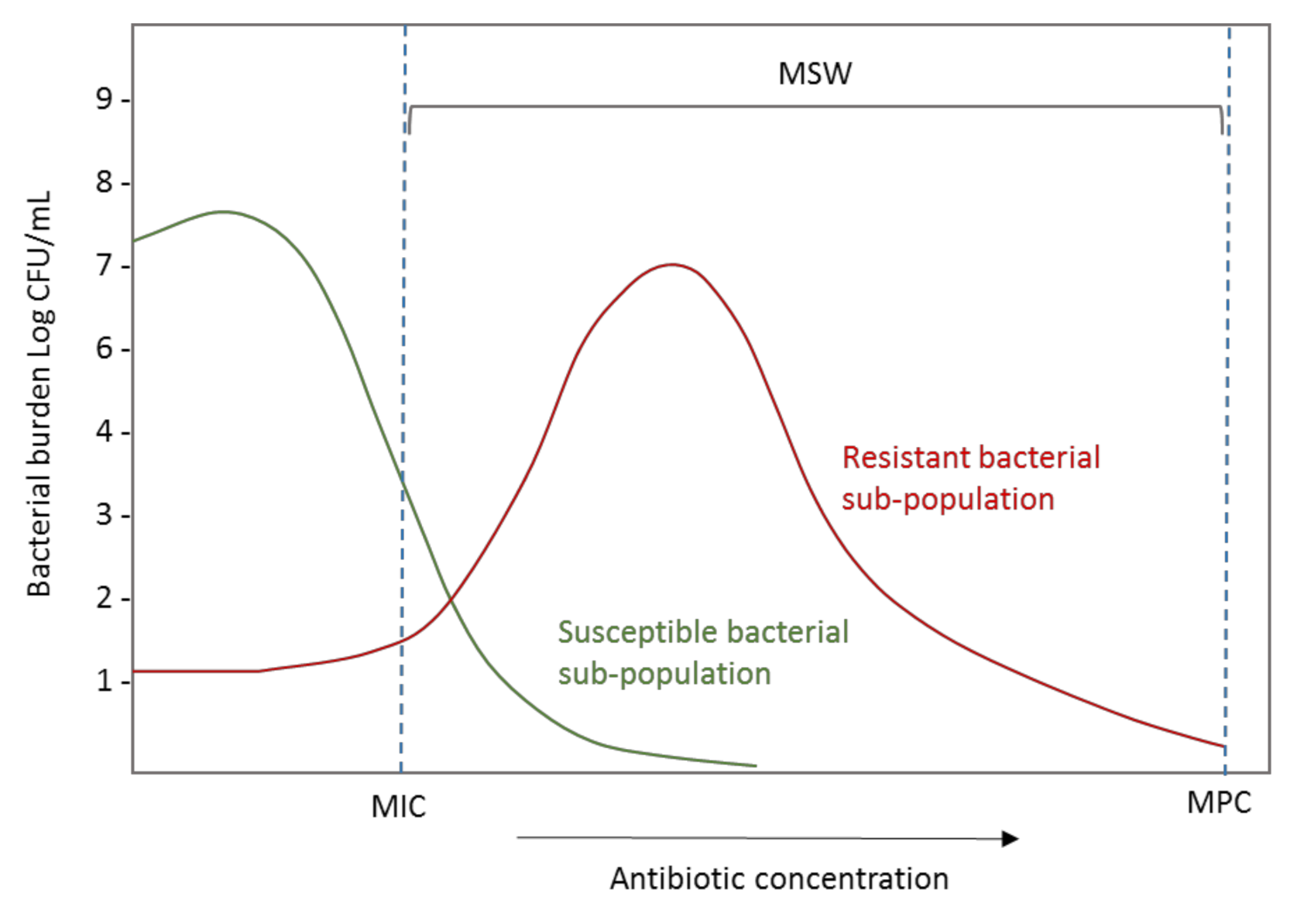
3.1. In Vitro Models
- -
- Describe the PK/PD relationships for representative organisms and a range of inocula;
- -
- Assess the effects of different PK profiles;
- -
- Study the relationships between rates of emergent resistance, drug exposure, and duration of therapy.
3.1.1. Static Assays
3.1.2. Dynamic Assays
- One-compartment model
- Two-compartment model
3.1.3. In Vitro PK/PD Bladder Infection Models
3.2. Ex Vivo Models
3.3. In Vivo Animal Models
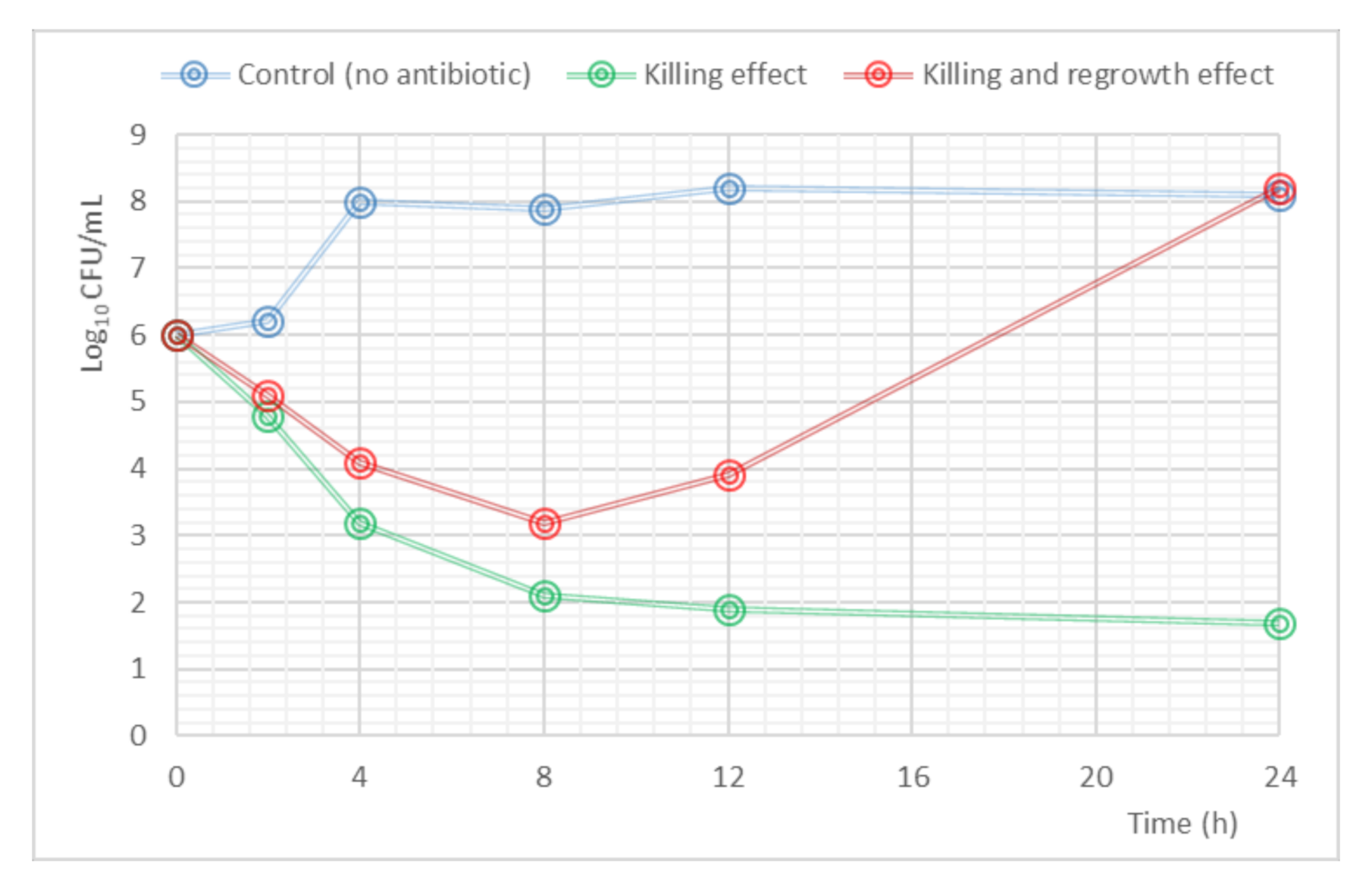
4. PK/PD Modeling of Microbial Kill-Curves
4.1. MIC-Based Approach
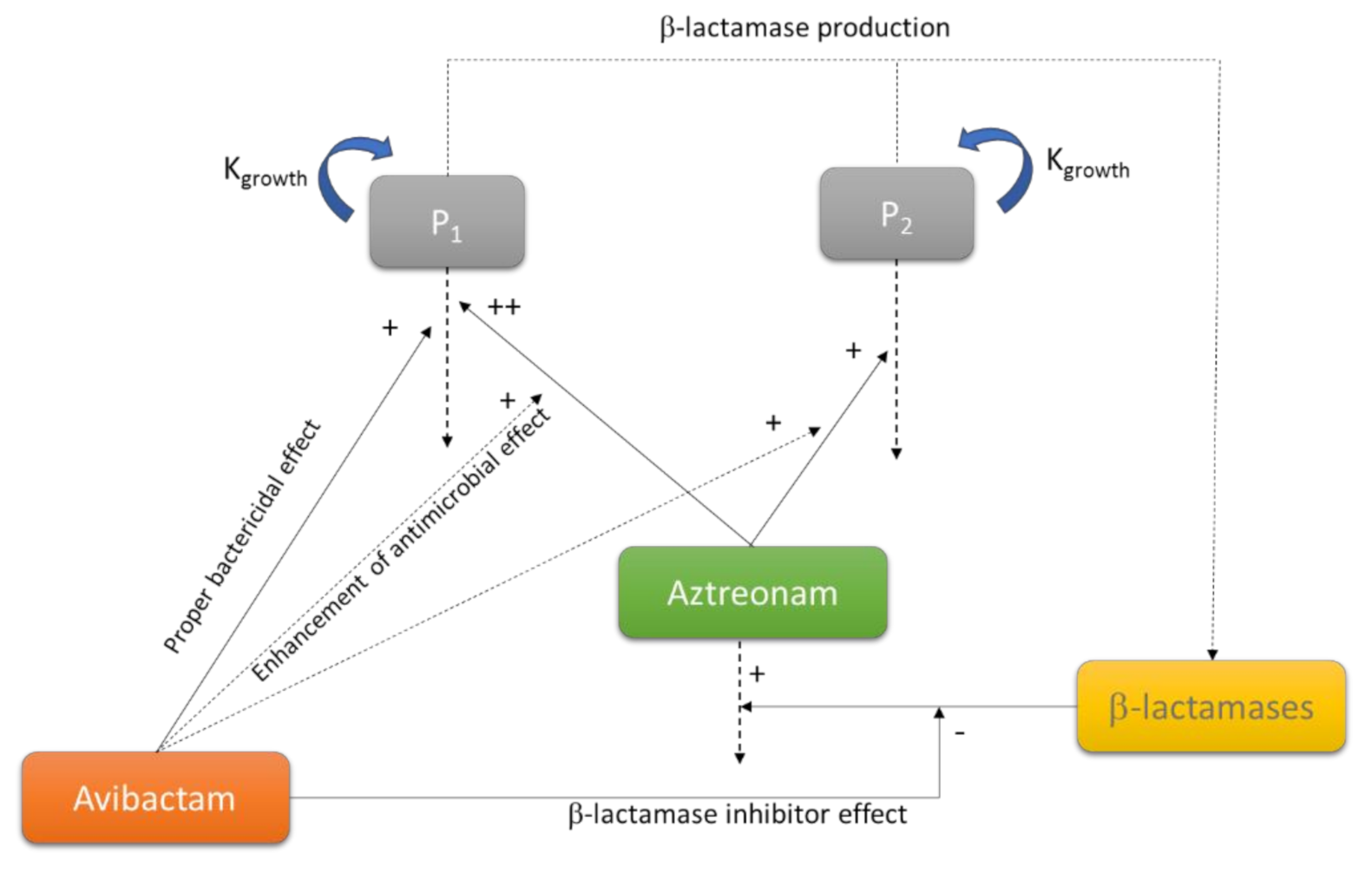
4.2. Mechanism-Based Models
5. Application of PK/PD Analysis and Population Pharmacokinetics for the Development and Optimization of Antimicrobial Treatments
5.1. PK/PD Analysis in Drug Development
5.2. PK/PD Analysis in Setting Susceptibility Breakpoints
5.3. PK/PD Analysis as A Tool For Surveillance of Antibacterial Activity
5.4. Population PK and PK/PD to Optimize Dosing Regimens. Therapeutic Drug Monitoring (TDM)
5.5. Application of PK/PD Modeling to Drug Resistance Prediction
5.6. Application of PK/PD Modeling in Veterinary Medicine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. World Health Organization Model List of Essential Medicines, 21st List, 2019; World Health Organization: Geneva, Switzerland, 2019; Licence: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- WHO. Antimicrobial Resistance. Available online: https://www.who.int/health-topics/antimicrobial-resistance (accessed on 11 May 2021).
- The 2019 Who Aware Classification of Antibiotics for Evaluation and Monitoring of Use; World Health Organization: Geneva, Switzerland, 2019; (WHO/EMP/IAU/2019.11). Licence: CC BY-NC-SA 3.0 IGO.
- WHO. Global Action Plan on Antimicrobial Resistance. Available online: https://www.who.int/antimicrobial-resistance/global-action-plan/en/ (accessed on 11 May 2021).
- Jorda, A.; Zeitlinger, M. Preclinical Pharmacokinetic/Pharmacodynamic Studies and Clinical Trials in the Drug Development Process of EMA-Approved Antibacterial Agents: A Review. Clin. Pharmacokinet. 2020, 59, 1071–1084. [Google Scholar] [CrossRef]
- One Health Commission. Available online: https://www.onehealthcommission.org/ (accessed on 11 May 2021).
- Rhouma, M.; Tessier, M.; Aenishaenslin, C.; Sanders, P.; Carabin, H. Should the Increased Awareness of the One Health Approach Brought by the COVID-19 Pandemic Be Used to Further Tackle the Challenge of Antimicrobial Resistance? Antibiotics 2021, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- McEwen, S.A.; Collignon, P.J. Antimicrobial Resistance: A One Health Perspective. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Asín-Prieto, E.; Rodríguez-Gascón, A.; Isla, A. Applications of the pharmacokinetic/pharmacodynamic (PK/PD) analysis of antimicrobial agents. J. Infect. Chemother. 2015, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Balbas-Martinez, V.; Jergens, A.E.; Troconiz, I.F.; Allenspach, K.; Mochel, J.P. Model-Based Reverse Translation Between Veterinary and Human Medicine: The One Health Initiative. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabarinath, S.N.; Singh, R.P.; Derendorf, H. Pharmacokinetics I: PK-PD approaches—Antibiotic drug development. In Clinical Pharmacology: Current Topics and Case Studies; Müller, M., Ed.; Springer: Vienna, Austria, 2010; pp. 143–155. [Google Scholar]
- Heffernan, A.J.; Sime, F.B.; Lipman, J.; Roberts, J.A. Individualising Therapy to Minimize Bacterial Multidrug Resistance. Drugs 2018, 78, 621–641. [Google Scholar] [CrossRef]
- Cotta, M.O.; Roberts, J.A.; Lipman, J. Antibiotic dose optimization in critically ill patients. Med. Intensiva 2015, 39, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Sumi, C.D.; Heffernan, A.J.; Lipman, J.; Roberts, J.A.; Sime, F.B. What Antibiotic Exposures Are Required to Suppress the Emergence of Resistance for Gram-Negative Bacteria? A Systematic Review. Clin. Pharmacokinet. 2019, 58, 1407–1443. [Google Scholar] [CrossRef]
- EMA-CHMP. Guideline on the Use of Pharmacokinetics and Pharmacodynamics in the Development of Antimicrobial Medicinal Products (EMA/CHMP/594085/2015); European Medicines Agency: London, UK, 2016. [Google Scholar]
- Mouton, J.W.; Dudley, M.N.; Cars, O.; Derendorf, H.; Drusano, G.L. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: An update. J. Antimicrob. Chemother. 2005, 55, 601–607. [Google Scholar] [CrossRef] [Green Version]
- EMA-CHMP. Guideline on the Evaluation of Medicinal Products Indicated for the Treatment of Bacterial Infections: Revision 3 (EMA/844951/2018); European Medicines Agency: London, UK, 2018. [Google Scholar]
- EMA-CHMP. Addendum to the Guideline on the Evaluation of Medicinal Products Indicated for Treatment of Bacterial Infections (EMA/CHMP/351889/2013)); European Medicines Agency: London, UK, 2013. [Google Scholar]
- McAleenan, A.; Ambrose, P.G.; Bhavnani, S.M.; Drusano, G.L.; Hope, W.W.; Mouton, J.W.; Higgins, J.P.T.; MacGowan, A.P. Methodological features of clinical pharmacokinetic-pharmacodynamic studies of antibacterials and antifungals: A systematic review. J. Antimicrob. Chemother. 2020, 75, 1374–1389. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Hope, W.W.; Eakin, A.E.; Guina, T.; Tam, V.H.; Louie, A.; Drusano, G.L.; Hoover, J.L. Generating Robust and Informative Nonclinical In Vitro and In Vivo Bacterial Infection Model Efficacy Data to Support Translation to Humans. Antimicrob. Agents. Chemother. 2019, 63, e02307-18. [Google Scholar] [CrossRef] [Green Version]
- Toutain, P.L.; Pelligand, L.; Lees, P.; Bousquet-Mélou, A.; Ferran, A.A.; Turnidge, J.D. The pharmacokinetic/pharmacodynamic paradigm for antimicrobial drugs in veterinary medicine: Recent advances and critical appraisal. J. Vet. Pharmacol. Ther. 2021, 44, 172–200. [Google Scholar] [CrossRef]
- Tängdén, T.; Lundberg, C.V.; Friberg, L.E.; Huttner, A. How preclinical infection models help define antibiotic doses in the clinic. Int. J. Antimicrob. Agents 2020, 56, 106008. [Google Scholar] [CrossRef]
- Luo, W.; Chen, D.; Wu, M.; Li, Z.; Tao, Y.; Liu, Q.; Pan, Y.; Qu, W.; Yuan, Z.; Xie, S. Pharmacokinetics/Pharmacodynamics models of veterinary antimicrobial agents. J. Vet. Sci. 2019, 20, e40. [Google Scholar] [CrossRef]
- Velkov, T.; Bergen, P.J.; Lora-Tamayo, J.; Landersdorfer, C.B.; Li, J. PK/PD models in antibacterial development. Curr. Opin. Microbiol. 2013, 16, 573–579. [Google Scholar] [CrossRef] [Green Version]
- Montero, M.; Domene Ochoa, S.; López-Causapé, C.; VanScoy, B.; Luque, S.; Sorlí, L.; Campillo, N.; Angulo-Brunet, A.; Padilla, E.; Prim, N.; et al. Efficacy of ceftolozane-tazobactam in combination with colistin against extensively drug-resistant Pseudomonas aeruginosa, including high-risk clones, in an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 2020, 64, e02542-19. [Google Scholar] [CrossRef]
- Drusano, G.L. Pre-clinical in vitro infection models. Curr. Opin. Pharmacol. 2017, 36, 100–106. [Google Scholar] [CrossRef]
- Boorgula, G.D.; Jakkula, L.; Gumbo, T.; Jung, B.; Srivastava, S. Comparison of Rifamycins for Efficacy Against Mycobacterium avium Complex and Resistance Emergence in the Hollow Fiber Model System. Front. Pharmacol. 2021, 12, 645264. [Google Scholar] [CrossRef]
- Abbott, I.J.; Roberts, J.A.; Meletiadis, J.; Peleg, A.Y. Antimicrobial pharmacokinetics and preclinical in vitro models to support optimized treatment approaches for uncomplicated lower urinary tract infections. Expert. Rev. AntiInfect. Ther. 2021, 19, 271–295. [Google Scholar] [CrossRef]
- Abbott, I.J.; van Gorp, E.; van der Meijden, A.; Wijma, R.A.; Meletiadis, J.; Roberts, J.A.; Mouton, J.W.; Peleg, A.Y. Oral Fosfomycin Treatment for Enterococcal Urinary Tract Infections in a Dynamic In Vitro Model. Antimicrob. Agents Chemother. 2020, 64, e00342-20. [Google Scholar] [CrossRef]
- Zalewska-Piątek, B.; Olszewski, M.; Lipniacki, T.; Błoński, S.; Wieczór, M.; Bruździak, P.; Skwarska, A.; Nowicki, B.; Nowicki, S.; Piątek, R. A shear stress micromodel of urinary tract infection by the Escherichia coli producing Dr adhesin. PLoS Pathog. 2020, 16, e1008247. [Google Scholar] [CrossRef] [PubMed]
- Aliabadi, F.S.; Lees, P. Pharmacokinetics and pharmacokinetic/pharmacodynamic integration of marbofloxacin in calf serum, exudate and transudate. J. Vet. Pharmacol. Ther. 2002, 25, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, D.; Guan, J.; Xi, W.; Zhou, D.; Zhao, L.; Cui, J.; Xu, Y.; Gao, Z.; Liu, Y. In vitro and in vivo synergistic effects of tigecycline combined with aminoglycosides on carbapenem-resistant Klebsiella pneumoniae. J. Antimicrob. Chemother. 2021, 16, dkab122. [Google Scholar] [CrossRef] [PubMed]
- Greko, C.; Bengtsson, B.; Franklin, A.; Jacobsson, S.O.; Wiese, B.; Luthman, J. Efficacy of trimethoprim-sulfadoxine against Escherichia coli in a tissue cage model in calves. J. Vet. Pharmacol. Ther. 2002, 25, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Lepak, A.J.; Andes, D.R. Animal models in the pharmacokinetic/pharmacodynamic evaluation of antimicrobial agents. Bioorg. Med. Chem. 2016, 24, 6390–6400. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, N.; Cassat, J.E.; Monteith, A.; Miller, A.; Drury, S.; Skaar, E.; Serezani, C.H. Murine Models for Staphylococcal Infection. Curr. Protoc. 2021, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Mizgerd, J.P.; Skerrett, S.J. Animal models of human pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L387–L398. [Google Scholar] [CrossRef]
- Lepak, A.J.; Zhao, M.; Marchillo, K.; VanHecker, J.; Andes, D.R. In Vivo Pharmacodynamic Evaluation of Omadacycline against Staphylococcus aureus in the Neutropenic Mouse Pneumonia Model. Antimicrob. Agents Chemother. 2020, 64, e02058-19. [Google Scholar] [CrossRef]
- Coenye, T.; Nelis, H.J. In vitro and in vivo model systems to study microbial biofilm formation. J. Microbiol. Methods 2010, 83, 89–105. [Google Scholar] [CrossRef]
- Dalton, T.; Dowd, S.E.; Wolcott, R.D.; Sun, Y.; Watters, C.; Griswold, J.A.; Rumbaugh, K.P. An in vivo polymicrobial biofilm wound infection model to study interspecies interactions. PLoS ONE 2011, 6, e27317. [Google Scholar]
- Zhou, Y.F.; Tao, M.T.; He, Y.Z.; Sun, J.; Liu, Y.H.; Liao, X.P. In Vivo Bioluminescent Monitoring of Therapeutic Efficacy and Pharmacodynamic Target Assessment of Antofloxacin against Escherichia coli in a Neutropenic Murine Thigh Infection Model. Antimicrob. Agents Chemother. 2017, 62, e01281-17. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Sy, S.K.; Derendorf, H. Principles of Applied Pharmacokinetic-Pharmacodynamic Modeling. In Fundamentals of Antimicrobial Pharmacokinetics and Pharmadodynamics; Winks, A.A., Derendorf, H., Mouton, J.W., Eds.; Springer: New York, NY, USA, 2014; pp. 63–79. [Google Scholar]
- Nielsen, E.I.; Friberg, L.E. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol. Rev. 2013, 65, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Brill, M.J.E.; Kristoffersson, A.N.; Zhao, C.; Nielsen, E.I.; Friberg, L.E. Semi-mechanistic pharmacokinetic-pharmacodynamic modelling of antibiotic drug combinations. Clin. Microbiol. Infect. 2018, 24, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Rayner, C.R.; Smith, P.F.; Andes, D.; Andrews, K.; Derendorf, H.; Friberg, L.E.; Hanna, D.; Lepak, A.; Mills, E.; Polasek, T.M.; et al. Model-Informed Drug Development for Anti-Infectives: State of the Art and Future. Clin. Pharmacol. Ther. 2021, 109, 867–891. [Google Scholar] [CrossRef]
- Müller, M.; dela Peña, A.; Derendorf, H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: Distribution in tissue. Antimicrob. Agents Chemother. 2004, 48, 1441–1453. [Google Scholar] [CrossRef] [Green Version]
- Sy, S.K.; Zhuang, L.; Derendorf, H. Pharmacokinetics and pharmacodynamics in antibiotic dose optimization. Expert Opin. Drug Metab. Toxicol. 2016, 12, 93–114. [Google Scholar] [CrossRef]
- Chauzy, A.; Gaelzer Silva Torres, B.; Buyck, J.; de Jonge, B.; Adier, C.; Marchand, S.; Couet, W.; Grégoire, N. Semimechanistic Pharmacodynamic Modeling of Aztreonam-Avibactam Combination to Understand Its Antimicrobial Activity Against Multidrug-Resistant Gram-Negative Bacteria. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Lister, P.D. The role of pharmacodynamic research in the assessment and development of new antibacterial drugs. Biochem. Pharmacol. 2006, 71, 1057–1065. [Google Scholar] [CrossRef]
- Rizk, M.L.; Bhavnani, S.M.; Drusano, G.; Dane, A.; Eakin, A.E.; Guina, T.; Jang, S.H.; Tomayko, J.F.; Wang, J.; Zhuang, L.; et al. Considerations for Dose Selection and Clinical Pharmacokinetics/Pharmacodynamics for the Development of Antibacterial Agents. Antimicrob. Agents Chemother. 2019, 63, e02309-18. [Google Scholar] [CrossRef] [Green Version]
- FDA. Guidance for Industry Antibacterial Therapies for Patients with an Unmet Medical Need for the Treatment of Serious Bacterial Diseases; FDA: Silver Spring, MD, USA, 2017. [Google Scholar]
- Dalbavancin. Summary of product characteristics. European Agency Medicine. Available online: https://www.ema.europa.eu/en/documents/product-information/xydalba-epar-product-information_en.pdf (accessed on 25 May 2021).
- Dingemanse, J.; Appel-Dingemanse, S. Integrated pharmacokinetics and pharmacodynamics in drug development. Clin. Pharmacokinet. 2007, 46, 713–737. [Google Scholar] [CrossRef]
- Sherwin, C.M.; Kiang, T.K.; Spigarelli, M.G.; Ensom, M.H. Fundamentals of population pharmacokinetic modelling: Validation methods. Clin. Pharmacokinet. 2012, 51, 573–590. [Google Scholar] [CrossRef]
- De Velde, F.; Mouton, J.W.; de Winter, B.; van Gelder, T.; Koch, B. Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives. Pharmacol. Res. 2018, 134, 280–288. [Google Scholar] [CrossRef]
- EFPIA MID3 Workgroup; Marshall, S.F.; Burghaus, R.; Cosson, V.; Cheung, S.Y.; Chenel, M.; DellaPasqua, O.; Frey, N.; Hamrén, B.; Harnisch, L.; et al. Good Practices in Model-Informed Drug Discovery and Development: Practice, Application, and Documentation. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 93–122. [Google Scholar]
- Odenholt, I.; Cars, O.; Löwdin, E. Pharmacodynamic studies of amoxicillin against Streptococcus pneumoniae: Comparison of a new pharmacokinetically enhanced formulation (2000 mg twice daily) with standard dosage regimens. J. Antimicrob. Chemother. 2004, 54, 1062–1066. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chai, H.; Li, Y.; Chai, X.; Zhao, Y.; Zhao, Y.; Tao, T.; Xiang, X. A Three-Pulse Release Tablet for Amoxicillin: Preparation, Pharmacokinetic Study and Physiologically Based Pharmacokinetic Modeling. PLoS ONE 2016, 11, e0160260. [Google Scholar]
- Horwitz, E.; Kagan, L.; Chamisha, Y.; Gati, I.; Hoffman, A.; Friedman, M.; Lavy, E. Novel gastroretentive controlled-release drug delivery system for amoxicillin therapy in veterinary medicine. J. Vet. Pharmacol. Therap. 2011, 34, 487–493. [Google Scholar] [CrossRef]
- Sou, T.; Soukarieh, F.; Williams, P.; Stocks, M.J.; Cámara, M.; Bergström, C. Model-Informed Drug Discovery and Development in Pulmonary Delivery: Biopharmaceutical Pharmacometric Modeling for Formulation Evaluation of Pulmonary Suspensions. ACS Omega 2020, 5, 25733–25746. [Google Scholar] [CrossRef] [PubMed]
- Mouton, J.W.; Brown, D.F.; Apfalter, P.; Cantón, R.; Giske, C.G.; Ivanova, M.; MacGowan, A.P.; Rodloff, A.; Soussy, C.J.; Steinbakk, M.; et al. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: The EUCAST approach. Clin. Microbiol. Infect. 2012, 18, E37–E45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asín, E.; Isla, A.; Canut, A.; Rodríguez Gascón, A. Comparison of antimicrobial pharmacokinetic/pharmacodynamic breakpoints with EUCAST and CLSI clinical breakpoints for Gram-positive bacteria. Int. J. Antimicrob. Agents 2012, 40, 313–322. [Google Scholar] [CrossRef]
- Ambrose, P.G. Antimicrobial susceptibility breakpoints: PK-PD and susceptibility breakpoints. Treat. Respir. Med. 2005, 4, 5–11. [Google Scholar] [CrossRef]
- Mouton, J.W. Setting Clinical MIC Breakpoints from a PK/PD Point of View: It Is the Dose That Matters. In Fundamentals of Antimicrobial Pharmacokinetics and Pharmacodynamics; Vinks, A., Derendorf, H., Mouton, J., Eds.; Springer: New York, NY, USA, 2014; pp. 45–61. [Google Scholar]
- DeRyke, C.A.; Kuti, J.L.; Nicolau, D.P. Reevaluation of current susceptibility breakpoints for Gram-negative rods based on pharmacodynamic assessment. Diagn. Microbiol. Infect. Dis. 2007, 58, 337–344. [Google Scholar] [CrossRef]
- Frei, C.R.; Wiederhold, N.P.; Burgess, D.S. Antimicrobial breakpoints for gram-negative aerobic bacteria based on pharmacokinetic-pharmacodynamic models with Monte Carlo simulation. J. Antimicrob. Chemother. 2008, 61, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Burgess, D.S.; Frei, C.R.; Lewis Ii, J.S.; Fiebelkorn, K.R.; Jorgensen, J.H. The contribution of pharmacokinetic-pharmacodynamic modelling with Monte Carlo simulation to the development of susceptibility breakpoints for Neisseria meningitidis. Clin. Microbiol. Infect. 2007, 13, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, D.; Pasipanodya, J.G.; Mpagama, S.G.; Bendet, P.; Srivastava, S.; Koeuth, T.; Lee, P.S.; Bhavnani, S.M.; Ambrose, P.G.; Thwaites, G.; et al. Levofloxacin Pharmacokinetics/Pharmacodynamics, Dosing, Susceptibility Breakpoints, and Artificial Intelligence in the Treatment of Multidrug-resistant Tuberculosis. Clin. Infect. Dis. 2018, 67, S293–S302. [Google Scholar] [CrossRef]
- Zuur, M.A.; Pasipanodya, J.G.; van Soolingen, D.; van der Werf, T.S.; Gumbo, T.; Alffenaar, J.C. Intermediate Susceptibility Dose-Dependent Breakpoints for High-Dose Rifampin, Isoniazid, and Pyrazinamide Treatment in Multidrug-Resistant Tuberculosis Programs. Clin. Infect. Dis. 2018, 67, 1743–1749. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 30th ed.; CLSI Supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020. [Google Scholar]
- Rempel, O.R.; Laupland, K.B. Surveillance for antimicrobial resistant organisms: Potential sources and magnitude of bias. Epidemiol. Infect. 2009, 137, 1665–1673. [Google Scholar] [CrossRef]
- Valero, A.; Isla, A.; Rodríguez-Gascón, A.; Calvo, B.; Canut, A.; Solinís, M.A. Pharmacokinetic/pharmacodynamic analysis as a tool for surveillance of the activity of antimicrobials against Pseudomonas aeruginosa strains isolated in critically ill patients. Enferm. Infecc. Microbiol. Clin. 2019, 37, 380–386. [Google Scholar] [CrossRef]
- Valero, A.; Isla, A.; Rodríguez-Gascón, A.; Canut, A.; Solinís, M.A. Susceptibility of Pseudomonas aeruginosa and antimicrobial activity using PK/PD analysis: An 18-year surveillance study. Enferm. Infecc. Microbiol. Clin. 2019, 37, 626–633. [Google Scholar] [CrossRef]
- Drew, R.H. Antimicrobial stewardship programs: How to start and steer a successful program. J. Manag. Care Pharm. 2009, 15, S18–S23. [Google Scholar] [CrossRef]
- Lee, C.R.; Cho, I.H.; Jeong, B.C.; Lee, S.H. Strategies to Minimize Antibiotic Resistance. Int. J. Environ. Res. Public. Health 2013, 10, 4274–4305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenitsky, S.A.; Rubinstein, E.; Ariano, R.E.; Zhanel, G.G. Canadian Antimicrobial Resistance Alliance Integrating pharmacokinetics, pharmacodynamic sand MIC distributions to assess changing antimicrobial activity against clinical isolates of Pseudomonas aeruginosa causing infections in Canadian hospitals (CANWARD). J. Antimicrob. Chemother. 2013, 68, i67–i72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torumkuney, D.; Chaiwarith, R.; Reechaipichitkul, W.; Malatham, K.; Chareonphaibul, V.; Rodrigues, C.; Chitins, D.S.; Dias, M.; Anandan, S.; Kanakapura, S.; et al. Results from the Survey of Antibiotic Resistance (SOAR) 2012–14 in Thailand, India, South Korea and Singapore. J. Antimicrob. Chemother. 2016, 71, i3–i19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kacou-Ndouba, A.; Revathi, G.; Mwathi, P.; Seck, A.; Diop, A.; Kabedi-Bajani, M.J.; Mwiti, W.; Anguibi-Pokou, M.J.; Morrissey, I.; Torumkuney, D. Results from the Survey of Antibiotic Resistance (SOAR) 2011–14 in the Democratic Republic of Congo, Ivory Coast, Republic of Senegal and Kenya. J. Antimicrob. Chemother. 2016, 71, i21–i31. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Zhu, D.; Wang, F.; Morrissey, I.; Wang, J.; Torumkuney, D. Results from the Survey of Antibiotic Resistance (SOAR) 2009–11 and 2013–14 in China. J. Antimicrob. Chemother. 2016, 71, i33–i43. [Google Scholar] [CrossRef]
- Jamsheer, A.; Rafay, A.M.; Daoud, Z.; Morrissey, I.; Torumkuney, D. Results from the Survey of Antibiotic Resistance (SOAR) 2011–13 in the Gulf States. J. Antimicrob. Chemother. 2016, 71, i45–i61. [Google Scholar] [CrossRef] [Green Version]
- Feshchenko, Y.; Dzyublik, A.; Pertseva, T.; Bratus, E.; Dzyublik, Y.; Gladka, G.; Morrissey, I.; Torumkuney, D. Results from the Survey of Antibiotic Resistance (SOAR) 2011–13 in Ukraine. J. Antimicrob. Chemother. 2016, 71, i63–i69. [Google Scholar] [CrossRef] [Green Version]
- Soyletir, G.; Altinkanat, G.; Gur, D.; Altun, B.; Tunger, A.; Aydemir, S.; Kayacan, C.; Aktas, Z.; Gunaydin, M.; Karadag, A.; et al. Results from the Survey of Antibiotic Resistance (SOAR) 2011–13 in Turkey. J. Antimicrob. Chemother. 2016, 71, i71–i83. [Google Scholar] [CrossRef] [Green Version]
- Torumkuney, D.; Gur, D.; Soyletir, G.; Gurler, N.; Aktas, Z.; Sener, B.; Tunger, A.; Bayramoglu, G.; Koksal, I.; Yalcin, A.N.; et al. Results from the Survey of Antibiotic Resistance (SOAR) 2002–09 in Turkey. J. Antimicrob. Chemother. 2016, 71, i85–i91. [Google Scholar] [CrossRef] [Green Version]
- Van, P.H.; Binh, P.T.; Minh, N.H.; Morrissey, I.; Torumkuney, D. Results from the Survey of Antibiotic Resistance (SOAR) 2009–11 in Vietnam. J. Antimicrob. Chemother. 2016, 71, i93–i102. [Google Scholar] [CrossRef] [Green Version]
- Zafar, A.; Hasan, R.; Nizamuddin, S.; Mahmood, N.; Mukhtar, S.; Ali, F.; Morrissey, I.; Barker, K.; Torumkuney, D. Antibiotic susceptibility in Streptococcus pneumoniae, Haemophilus influenzae and Streptococcus pyogenes in Pakistan: A review of results from the Survey of Antibiotic Resistance (SOAR) 2002–15. J. Antimicrob. Chemother. 2016, 71, i103–i109. [Google Scholar] [CrossRef]
- Torumkuney, D.; Nica, M.; Nistor, I.; Vatcheva-Dobrevska, R.; Petrovic, V.; Stoica, A.; Hanicar, B.; Antic, D.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2014–16 in Bulgaria, Romania, Serbia and Croatia. J. Antimicrob. Chemother. 2018, 73, v2–v13. [Google Scholar] [CrossRef]
- Torumkuney, D.; Mayanskiy, N.; Edelstein, M.; Sidorenko, S.; Kozhevin, R.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2014–16 in Russia. J. Antimicrob. Chemother. 2018, 73, v14–v21. [Google Scholar] [CrossRef]
- Torumkuney, D.; Zemlickova, H.; Maruscak, M.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2014–16 in the Czech Republic. J. Antimicrob. Chemother. 2018, 73, v22–v27. [Google Scholar] [CrossRef]
- Torumkuney, D.; Pertseva, T.; Bratus, E.; Dziublik, A.; Yachnyk, V.; Liskova, A.; Sopko, O.; Malynovska, K.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2014–16 in Ukraine and the Slovak Republic. J. Antimicrob. Chemother. 2018, 73, v28–v35. [Google Scholar] [CrossRef]
- Torumkuney, D.; Papaparaskevas, J.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2014–16 in Greece. J. Antimicrob. Chemother. 2018, 73, v36–v42. [Google Scholar] [CrossRef]
- Torumkuney, D.; Hammami, A.; Mezghani Maalej, S.; Ayed, N.B.; Revathi, G.; Zerouali, K.; Elmdaghri, N.; Gachii, A.K.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2015–18 in Tunisia, Kenya and Morocco: Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i2–i18. [Google Scholar] [CrossRef] [PubMed]
- Torumkuney, D.; Van, P.H.; Thinh, L.Q.; Koo, S.H.; Tan, S.H.; Lim, P.Q.; Sivhour, C.; Lamleav, L.; Somary, N.; Sosorphea, S.; et al. Results from the Survey of Antibiotic Resistance (SOAR) 2016–18 in Vietnam, Cambodia, Singapore and the Philippines: Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i19–i42. [Google Scholar] [CrossRef]
- Torumkuney, D.; Smayevsky, J.; Relloso, M.S.; Sucari, A.; Pennini, M.; Brilla, E.; Vilches, V.; De la Cruz, Y.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2015–17 in Latin America (Argentina, Chile and Costa Rica): Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i43–i59. [Google Scholar] [CrossRef] [PubMed]
- Torumkuney, D.; Mokaddas, E.; Jiman-Fatani, A.; Ageel, A.; Daoud, Z.; Bouferraa, Y.; Zerdan, M.B.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2015–17 in the Middle East (Kuwait, Lebanon and Saudi Arabia): Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i60–i75. [Google Scholar] [CrossRef]
- Torumkuney, D.; Anwar, S.; Nizamuddin, S.; Malik, N.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2015–17 in Pakistan: Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i76–i87. [Google Scholar] [CrossRef]
- Torumkuney, D.; Tunger, A.; Sancak, B.; Bıçakçıgil, A.; Altun, B.; Aktas, Z.; Kayacan, C.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2015–17 in Turkey: Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i88–i99. [Google Scholar] [CrossRef] [PubMed]
- Torumkuney, D.; Bratus, E.; Yuvko, O.; Pertseva, T.; Morrissey, I. Results from the Survey of Antibiotic Resistance (SOAR) 2016–17 in Ukraine: Data based on CLSI, EUCAST (dose-specific) and pharmacokinetic/pharmacodynamic (PK/PD) breakpoints. J. Antimicrob. Chemother. 2020, 75, i100–i111. [Google Scholar] [CrossRef] [PubMed]
- Seña, A.C.; Bachmann, L.; Johnston, C.; Wi, T.; Workowski, K.; Hook, E.W., 3rd; Hocking, J.S.; Drusano, G.; Unemo, M. Optimising treatments for sexually transmitted infections: Surveillance, pharmacokinetics and pharmacodynamics, therapeutic strategies, and molecular resistance prediction. Lancet Infect. Dis. 2020, 20, e181–e191. [Google Scholar] [CrossRef]
- Ibar-Bariain, M.; Rodríguez-Gascón, A.; Isla, A.; Solinís, M.A.; Canut-Blasco, A. Evaluation of the adequacy of the antimicrobial therapy of invasive Haemophilus influenzae infections: A pharmacokinetic/pharmacodynamic perspective. Enferm. Infecc. Microbiol. Clin. 2021, 39, 65–71. [Google Scholar] [CrossRef]
- Ibar-Bariain, M.; Rodríguez-Gascón, A.; Isla, A.; Solinís, M.A.; Canut-Blasco, A. Application of pharmacokinetic/pharmacodynamic analysis to evaluate the adequacy of antimicrobial therapy for pediatric acute otitis media in Spain before and after the introduction of the PCV7 vaccine. Rev. Esp. Quimioter. 2019, 32, 121–129. [Google Scholar]
- Owens, R.C., Jr.; Bulik, C.C.; Andes, D.R. Pharmacokinetics-pharmacodynamics, computer decision support technologies, and antimicrobial stewardship: The compass and rudder. Diagn. Microbiol. Infect. Dis. 2018, 91, 371–382. [Google Scholar] [CrossRef]
- Dellit, T.H.; Owens, R.C.; McGowan, J.E., Jr.; Gerding, D.N.; Weinstein, R.A.; Burke, J.P.; Huskins, W.C.; Paterson, D.L.; Fishman, N.O.; Carpenter, C.F.; et al. Infectious Diseases Society of America; Society for Healthcare Epidemiology of America. Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship. Clin. Infect. Dis. 2007, 44, 159–177. [Google Scholar]
- Pea, F.; Viale, P. Pharmacodynamics of antibiotics to treat multidrug-resistant Gram-positive hospital infections. Expert Rev. Anti. Infect. Ther. 2007, 5, 255–270. [Google Scholar] [CrossRef]
- Balbas-Martinez, V.; Michelet, R.; Edginton, A.N.; Meesters, K.; Trocóniz, I.F.; Vermeulen, A. Physiologically-Based Pharmacokinetic model for Ciprofloxacin in children with complicated Urinary Tract Infection. Eur. J. Pharm. Sci. 2019, 128, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Schlender, J.F.; Teutonico, D.; Coboeken, K.; Schnizler, K.; Eissing, T.; Willmann, S.; Jaehde, U.; Stass, H. A Physiologically-Based Pharmacokinetic Model to Describe Ciprofloxacin Pharmacokinetics over the Entire Span of Life. Clin. Pharmacokinet. 2018, 57, 1613–1634. [Google Scholar] [CrossRef] [Green Version]
- Montanha, M.C.; Diniz, A.; Silva, N.; Kimura, E.; Paixão, P. Physiologically-Based Pharmacokinetic Model on the Oral Drug Absorption in Roux-en-Y Gastric Bypass Bariatric Patients: Amoxicillin Tablet and Suspension. Mol. Pharm. 2019, 16, 5025–5034. [Google Scholar] [CrossRef]
- Thémans, P.; Marquet, P.; Winkin, J.J.; Musuamba, F.T. Towards a Generic Tool for Prediction of Meropenem Systemic and Infection-Site Exposure: A Physiologically Based Pharmacokinetic Model for Adult Patients with Pneumonia. Drugs R D. 2019, 19, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Cordes, H.; Thiel, C.; Aschmann, H.E.; Baier, V.; Blank, L.M.; Kuepfer, L. A Physiologically Based Pharmacokinetic Model of Isoniazid and Its Application in Individualizing Tuberculosis Chemotherapy. Antimicrob. Agents Chemother. 2016, 60, 6134–6145. [Google Scholar] [CrossRef] [Green Version]
- Hornik, C.P.; Wu, H.; Edginton, A.N.; Watt, K.; Cohen-Wolkowiez, M.; Gonzalez, D. Development of a Pediatric Physiologically-Based Pharmacokinetic Model of Clindamycin Using Opportunistic Pharmacokinetic Data. Clin. Pharmacokinet. 2017, 56, 1343–1353. [Google Scholar] [CrossRef]
- Rimmler, C.; Lanckohr, C.; Akamp, C.; Horn, D.; Fobker, M.; Wiebe, K.; Redwan, B.; Ellger, B.; Koeck, R.; Hempel, G. Physiologically based pharmacokinetic evaluation of cefuroxime in perioperative antibiotic prophylaxis. Br. J. Clin. Pharmacol. 2019, 85, 2864–2877. [Google Scholar] [CrossRef]
- Joyner, M.L.; Manning, C.C.; Forbes, W.; Bobola, V.; Frazier, W. Modeling Ertapenem: The impact of body mass index on distribution of the antibiotic in the body. Math. Biosci. Eng. 2019, 16, 713–726. [Google Scholar] [CrossRef]
- Tod, M.; Lagneau, F.; Jullien, V.; Mimoz, O. A physiological model to evaluate drug kinetics in patients with hemorrhagic shock followed by fluid resuscitation. Application to amoxicillin-clavulanate. Pharm. Res. 2008, 25, 1431–1439. [Google Scholar] [CrossRef]
- Chant, C.; Leung, A.; Friedrich, J.O. Optimal dosing of antibiotics in critically ill patients by using continuous/extended infusions: A systematic review and meta-analysis. Crit. Care 2013, 17, R279. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, E.; Spapen, H.; Pierard, D. Prolonged vs intermittent infusion of piperacillin/tazobactam in critically ill patients: A narrative and systematic review. J. Crit Care 2014, 29, 1089e95. [Google Scholar] [CrossRef]
- Sakka, S.G.; Glauner, A.K.; Bulitta, J.B.; Kinzig-Schippers, M.; Pfister, W.; Drusano, G.L.; Sörgel, F. Population pharmacokinetics and pharmacodynamics of continuous versus short-term infusion of imipenem-cilastatin in critically ill patients in a randomized, controlled trial. Antimicrob. Agents. Chemother. 2007, 51, 3304e10. [Google Scholar] [CrossRef] [Green Version]
- Phe, K.; Heil, E.L.; Tam, V.H. Optimizing Pharmacokinetics-Pharmacodynamics of Antimicrobial Management in Patients with Sepsis: A Review. J. Infect. Dis. 2020, 222, S132–S141. [Google Scholar] [CrossRef]
- Dulhunty, J.M.; Roberts, J.A.; Davis, J.S.; Webb, S.; Bellomo, R.; Gomersall, C.; Shirwadkar, C.; Eastwood, G.M.; Myburgh, J.; Paterson, D.L.; et al. Continuous Infusion of Beta-Lactam Antibiotics in Severe Sepsis: A Multicenter Dou-ble-Blind, Randomized Controlled Trial. Clin. Infect. Dis. 2013, 56, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, M.H.; Sulaiman, H.; Mat-Nor, M.B.; Rai, V.; Wong, K.K.; Hasan, M.S.; Abd Rahman, A.N.; Jamal, J.A.; Wallis, S.C.; Lipman, J.; et al. Beta-Lactam Infusion in Severe Sepsis (BLISS): A prospective, two-centre, open-labelled randomised controlled trial of continuous versus intermittent beta-lactam infusion in critically ill patients with severe sepsis. Intensive Care Med. 2016, 42, 1535–1545. [Google Scholar] [CrossRef]
- Barrasa, H.; Soraluce, A.; Usón, E.; Sainz, J.; Martín, A.; Sánchez-Izquierdo, J.Á.; Maynar, J.; Rodríguez-Gascón, A.; Isla, A. Impact of augmented renal clearance on the pharmacokinetics of linezolid: Advantages of continuous infusion from a pharmacokinetic/pharmacodynamic perspective. Int. J. Infect. Dis. 2020, 93, 329–338. [Google Scholar] [CrossRef]
- Soraluce, A.; Barrasa, H.; Asín-Prieto, E.; Sánchez-Izquierdo, J.Á.; Maynar, J.; Isla, A.; Rodríguez-Gascón, A. Novel Population Pharmacokinetic Model for Linezolid in Critically Ill Patients and Evaluation of the Adequacy of the Current Dosing Recommendation. Pharmaceutics 2020, 12, 54. [Google Scholar] [CrossRef] [Green Version]
- Smyth, A.; Tan, K.H.; Hyman-Taylor, P.; Mulheran, M.; Lewis, S.; Stableforth, D.; Prof Knox, A.; TOPIC Study Group. Once versus three-times daily regimens of tobramycin treatment for pulmonary exacerbations of cystic fibrosis—The TOPIC study: A randomized controlled trial. Lancet 2005, 365, 573e8. [Google Scholar] [CrossRef]
- Nicolau, D.P.; Freeman, C.D.; Belliveau, P.P.; Nightingale, C.H.; Ross, J.W.; Quintiliani, R. Experience with a once-daily aminoglycoside program administered to 2,184 adult patients. Antimicrob. Agents Chemother. 1995, 39, 650e5. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Seike, K.; Yasuda, M.; Matsumoto, T. Evaluation by Monte Carlo simulation of levofloxacin dosing for complicated urinary tract infections caused by Escherichia coli or Pseudomonas aeruginosa. J. Infect. Chemother. 2011, 17, 726e30. [Google Scholar] [CrossRef]
- Pea, F.; Viale, P.; Furlanut, M. Antimicrobial therapy in critically ill patients: A review of pathophysiological conditions responsible for altered disposition and pharmacokinetic variability. Clin. Pharmacokinet. 2005, 44, 1009–1034. [Google Scholar] [CrossRef]
- Roberts, J.A.; Norris, R.; Paterson, D.L.; Martin, J.H. Therapeutic drug monitoring of antimicrobials. Br. J. Clin. Pharmacol. 2012, 73, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Septimus, E.J.; Owens, R.C., Jr. Need and potential of antimicrobial stewardship in community hospitals. Clin. Infect. Dis. 2011, 53, S8–S14. [Google Scholar] [CrossRef] [PubMed]
- Barlam, T.F.; Cosgrove, S.E.; Abbo, L.M.; MacDougall, C.; Schuetz, A.N.; Septimus, E.J.; Srinivasan, A.; Dellit, T.H.; Falck-Ytter, Y.T.; Fishman, N.O.; et al. Implementing an Antibiotic Stewardship Program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin. Infect. Dis. 2016, 15, e51–e77. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhao, X.; Kreiswirth, B.N.; Drlica, K. Mutant prevention concentration as a measure of antibiotic potency: Studies with clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2000, 44, 2581–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondeau, J.M.; Zhao, X.; Hansen, G.; Drlica, K. Mutant prevention concentrations of fluoroquinolones for clinical isolates of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.J.; Nichol, K.A.; Hoban, D.J.; Zhanel, G.G. Stretching the mutant prevention concentration (MPC) beyond its limits. J. Antimicrob. Chemother. 2003, 51, 1323–1325. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Hope, W.W.; Roberts, J.A. Applying pharmacokinetic/pharmacodynamic principles incritically ill patients: Optimizing efficacy and reducing resistance development. Semin. Respir. Crit. Care Med. 2015, 36, 136–153. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. The Evolving Threat of Antimicrobial Resistance: Options for Action. World Health Organization. 2012. Available online: https://apps.who.int/iris/handle/10665/44812 (accessed on 11 May 2021).
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Huang, L.; Hao, H.; Sanders, P.; Yuan, Z. Application of PK/PD Modeling in Veterinary Field: Dose Optimization and Drug Resistance Prediction; published correction appears in Biomed. Res. Int. 2017, 2017, 1408737. Biomed. Res. Int. 2016, 2016, 5465678. [Google Scholar]
- Koritz, G.D.; Bevill, R.F. Role of pharmacokinetics in the outcome of infections. Acta Vet. Scand. 1991, 87, 18–27. [Google Scholar]
- Renard, L.; Sanders, P.; Laurentie, M.; Delmas, J.M. Pharmacokinetic-pharmacodynamic model for spiramycin in staphylococcal mastitis. J. Vet. Pharmacol. Ther. 1996, 19, 95–103. [Google Scholar] [CrossRef]
- Burch, D.; Sperling, D. Amoxicillin-current use in swine medicine. J. Vet. Pharmacol. Ther. 2018, 41, 356–368. [Google Scholar] [CrossRef] [Green Version]
- El Badawy, S.A.; Amer, A.; Kamel, G.M.; Eldeib, K.M.; Constable, P.D. Pharmacokinetics and pharmacodynamics of intramammary cefquinome in lactating goats with and without experimentally induced Staphylococcus aureus mastitis. J. Vet. Pharmacol. Ther. 2019, 42, 452–460. [Google Scholar] [CrossRef]
- Lei, Z.; Liu, Q.; Zhu, Q.; Yang, B.; Khaliq, H.; Sun, A.; Qi, Y.; Moku, G.K.; Su, Y.; Wang, J.; et al. Comparative Pharmacokinetics and Preliminary Pharmacodynamics Evaluation of Piscidin 1 Against PRV and PEDV in Rats. Front. Chem. 2018, 6, 244. [Google Scholar] [CrossRef] [Green Version]
- Vercelli, C.; Łebkowska-Wieruszewska, B.; Barbero, R.; Lisowski, A.; Re, G.; Giorgi, M. Pharmacokinetics of levofloxacin in non-lactating goats and evaluation of drug effects on resistance in coliform rectal flora. Res. Vet. Sci. 2020, 133, 283–288. [Google Scholar] [CrossRef]
- Birhanu, B.T.; Lee, E.B.; Park, S.C. Evaluation of the pharmacokinetic-pharmacodynamic integration of marbofloxacin in combination with methyl gallate against Salmonella Typhimurium in rats. PLoS ONE 2020, 15, e0234211. [Google Scholar] [CrossRef]
- Li, Y.; Xie, M.; Zhou, J.; Lin, H.; Xiao, T.; Wu, L.; Ding, H.; Fang, B. Increased Antimicrobial Activity of Colistin in Combination with Gamithromycin Against Pasteurella multocida in a Neutropenic Murine Lung Infection Model. Front. Microbiol. 2020, 11, 511356. [Google Scholar] [CrossRef]
- Zeng, D.; Sun, M.; Lin, Z.; Li, M.; Gehring, R.; Zeng, Z. Pharmacokinetics and Pharmacodynamics of Tildipirosin Against Pasteurella multocida in a Murine Lung Infection Model. Front. Microbiol. 2018, 9, 1038. [Google Scholar] [CrossRef]
- Maan, M.K.; Sattar, A.; Mi, K.; Bakr Shabbir, M.A.; Xie, S.; Xin. L.; Ahmed, S.; Algharib, S.A.; Huang, L.; Yuan, Z. Integration of PK/PD for dose optimization of aditoprim against Trueperella pyogenes causing endometritis in bovines. Microb. Pathog. 2020, 142, 104097. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, Z.; Xia, X.; Gu, X.; Cai, Q.; Shen, X.; Yang, H.; Ding, H. Pharmacokinetic and Pharmacodynamic Integration and Resistance Analysis of Tilmicosin Against Mycoplasma gallisepticum in an In Vitro Dynamic Model. Front. Pharmacol. 2019, 10, 670. [Google Scholar] [CrossRef]
- Fernández-Varón, E.; Cárceles-García, C.; Serrano-Rodríguez, J.M.; Cárceles-Rodríguez, C.M. Pharmacokinetics (PK), pharmacodynamics (PD), and PK-PD integration of ceftiofur after a single intravenous, subcutaneous and subcutaneous-LA administration in lactating goats. BMC Vet. Res. 2016, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Cheng, P.; Xiao, T.; Ulziikhutag, J.; Yu, H.; Li, J.; Liu, R.; Muhammad, I.; Zhang, X. Pharmacokinetics and pharmacodynamics integration of danofloxacin against Escherichia coli in piglet ileum ultrafiltration probe model. Sci. Rep. 2021, 11, 681. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, Y.F.; Sun, J.; Shi, W.; Liao, X.P.; Liu, Y.H. Pharmacokinetic and pharmacodynamic modeling of sarafloxacin against avian pathogenic Escherichia coli in Muscovy ducks. BMC Vet. Res. 2017, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Cazer, C.L.; Ducrot, L.; Volkova, V.V.; Gröhn, Y.T. Monte Carlo Simulations Suggest Current Chlortetracycline Drug-Residue Based Withdrawal Periods Would Not Control Antimicrobial Resistance Dissemination from Feedlot to Slaughterhouse. Front. Microbiol. 2017, 8, 1753. [Google Scholar] [CrossRef] [Green Version]
- EMA-CVMP. Guideline for the Demonstration of Efficacy for Veterinary Medicinal Products Containing Antimicrobial Substances; European Medicines Agency: London, UK, 2002; EMEA/CVMP/627/2001. [Google Scholar]
- Lees, P.; AliAbadi, F.S.; Toutain, P.L. PK-PD modelling: An alternative to dose titration studies for antimicrobial drug dosage selection. Regul. Aff. J. Pharma 2004, 15, 175–180. [Google Scholar]
- EMA-CVMP. Guideline for the Demonstration of Efficacy for Veterinary Medicinal Products Containing Antimicrobial Substances; European Medicines Agency: London, UK, 2016; EMA/CVMP/627/2001-Rev.1. [Google Scholar]
- EMA-CVMP. Reflection Paper on Dose Optimisation of Established Veterinary Antibiotics in the Context of SPC Harmonization; European Medicines Agency: London, UK, 2021; EMA/CVMP/849775/2017. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Activity | PK/PD Index | ||
|---|---|---|---|
| Concentration-dependent activity | |||
| Aminoglycosides | fCmax/MIC | ||
| Quinolones | fAUC24/MIC | ||
| Time-dependent activity | |||
| β-lactams | fT>MIC | ||
| Penicillins | |||
| Cephalosporins | |||
| Carbapenems | |||
| Concentration-dependent activity with time-dependence | |||
| Vancomycin | Fosfomycin | fAUC24/MIC | |
| Linezolid | Fluoroquinolones | ||
| Daptomycin | Colistin | ||
| Reference | Bacteria | Antimicrobials | |
|---|---|---|---|
| Betalactams | Others | ||
| DeRyke et al. [64] | P. aeruginosa A. baumanii E. coli Klebsiella spp. | Cefepime Ceftazidime Ceftriaxona Imipenem Meropenem Piperacillin/tazabactam | Ciprofloxacin Levofloxacin |
| Frei et al. [65] |
Enterobateriaceae P. aeruginosa A. baumannii | Aztreonam Cefepime Ceftizoxime Cetazidime Ertapenem Imipenem Meropenem Piperacillin/tazobactam | Ciprofloxacin Gentamicin Levofloxacin Tobramycin |
| Asín et al. [61] | Enterococcus Staphylococcus β-Haemolytic streptococci Other streptococci S. pneumoniae | Amoxicillin Cefepime Cefotaxime Cloxacillin Ertapenem Imipenem Meropenem Piperacillin/tazobactam | Levofloxacin Vancomycin Daptomycin Tigecycline Linezolid |
| Burgess et al. [66] | Neisseria meningitidis | Ampicillin Cefotaxime Ceftriaxone Ciprofloxacin Meropenem Penicillin G | Azithromycin Chloramphenicol Doxycycline Levofloxacin Minocycline Rifampicin Sulphafurazole Tetracycline Co-Trimoxazole |
| Zuur et al. [68] | Mycobacterium tuberculosis | Isoniazid Pyrazinamide Rifampin | |
| Deshpande et al. [67] | Mycobacterium tuberculosis | Levofloxacin | |
| Reference | Patient Population | Antimicrobial | Route of Administration |
|---|---|---|---|
| Balbas-Martinez et al. [103] | Children with complicated urinary tract infection | Ciprofloxacin | Oral/intravenous |
| Schlender et al. [104] | Pediatric/adult/geriatric | Ciprofloxacin | Oral/intravenous |
| Montanha et al. [105] | Bariatric patients | Amoxicillin | Oral tablet/suspension |
| Thémans et al. [106] | Critical/non critical/obese | Meropenem | Intravenous |
| Cordes et al. [107] | Patients with tuberculosis | Isoniazid | Oral |
| Hornik et al. [108] | Pediatric | Clindamycin | Intravenous |
| Rimmler et al. [109] | Perioperative patients | Cefuroxime | Intravenous |
| Joyner et al. [110] | Patients with different body mass indexes | Ertapenem | Intravenous |
| Tod et al. [111] | Patients with hemorrhagic shock followed by fluid resuscitation | Amoxicillin-clavulanate | Intravenous |
| Reference | Year | Antimicrobial | Study Description |
|---|---|---|---|
| Burch et al. [136] | 2018 | Amoxicillin | Review the PK and PD in pigs |
| El Badawy et al. [137] | 2019 | Cefquinome | PK and PD in lactating goats |
| Lei et al. [138] | 2018 | Piscidin | Evaluation of an antimicrobial peptide in a rat animal model for a future use in veterinary medicine |
| Vercelli et al. [139] | 2020 | Levofloxacin | PK/PD of levofloxacin in non-lactating goats |
| Birhanu et al. [140] | 2020 | Marbofloxacin in combination with methyl gallate | PK/PD analysis of marbofloxacin in combination with methyl gallate in a rat animal model |
| Li et al. [141] | 2020 | Colistin in Combination With Gamithromycin | In vitro susceptibility and time-kill tests and in vivo PK and PD assays using a neutropenic murine lung infection model |
| Zeng et al. [142] | 2018 | Tildipirosin | PK/PD modeling in a murine lung infection model |
| Maan et al. [143] | 2020 | Aditoprim | In vivo intrauterine PK in cattles and in vitro and ex vivo PD |
| Huang et al. [144] | 2019 | Tilmicosin | PK/PD analysis in an in vitro dynamic model |
| Fernández-Varón et al. [145] | 2016 | Ceftiofur | PK in lactating goats, in vitro and ex vivo activity, and determination of PK/PD index |
| Yang et al. [146] | 2021 | Danofloxacin | PK in piglet and PK/PD analysis in vivo and ex vivo |
| Yu et al. [147] | 2017 | Sarafloxacin | PK/PD in Muscovy ducks |
| Cazer et al. [148] | 2017 | Chlortetracycline | PK/PD and the enteric bacterial population dynamics in beef cattle |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Gascón, A.; Solinís, M.Á.; Isla, A. The Role of PK/PD Analysis in the Development and Evaluation of Antimicrobials. Pharmaceutics 2021, 13, 833. https://doi.org/10.3390/pharmaceutics13060833
Rodríguez-Gascón A, Solinís MÁ, Isla A. The Role of PK/PD Analysis in the Development and Evaluation of Antimicrobials. Pharmaceutics. 2021; 13(6):833. https://doi.org/10.3390/pharmaceutics13060833
Chicago/Turabian StyleRodríguez-Gascón, Alicia, María Ángeles Solinís, and Arantxa Isla. 2021. "The Role of PK/PD Analysis in the Development and Evaluation of Antimicrobials" Pharmaceutics 13, no. 6: 833. https://doi.org/10.3390/pharmaceutics13060833