
Reprogramming Extracellular Vesicles for Protein Therapeutics Delivery

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
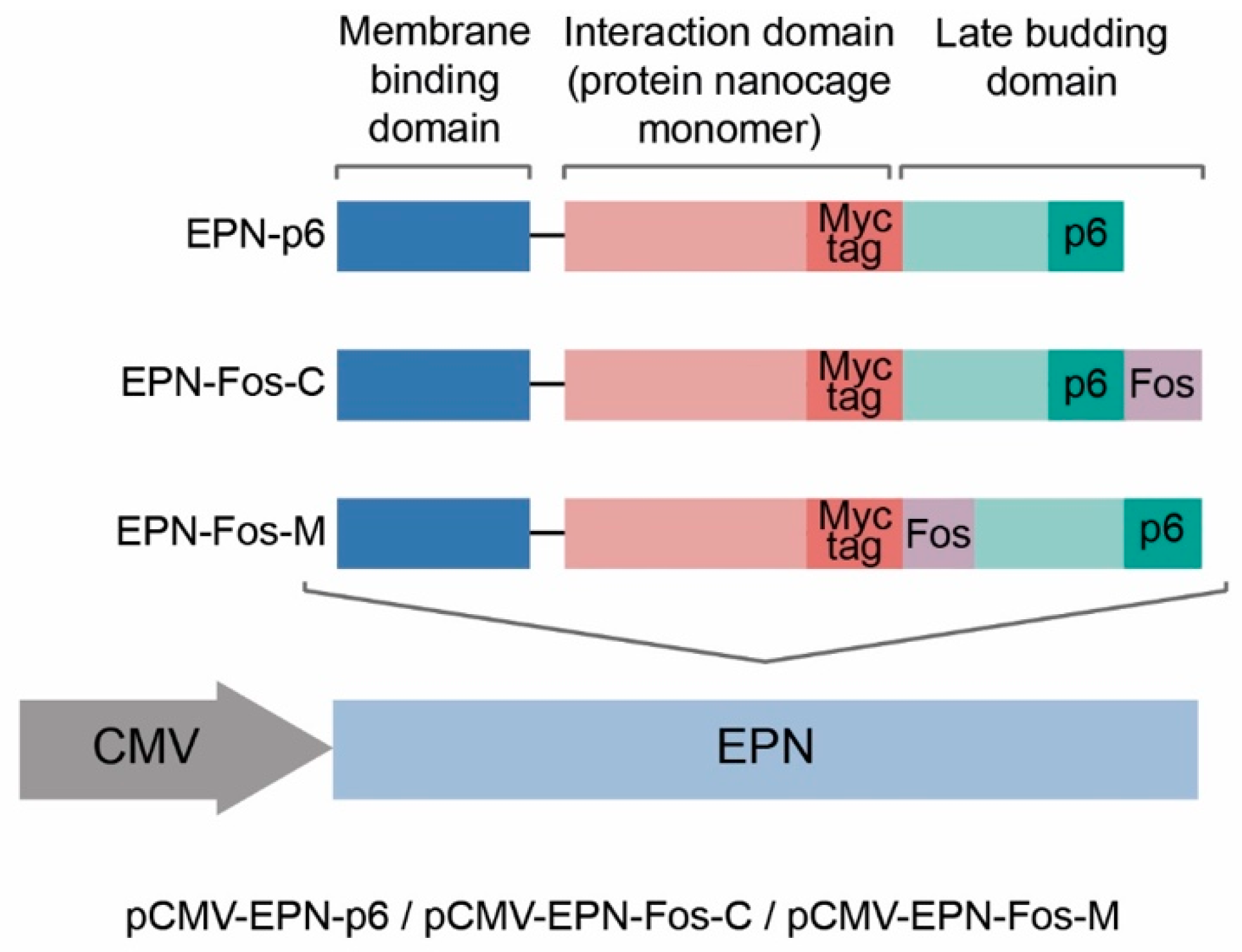
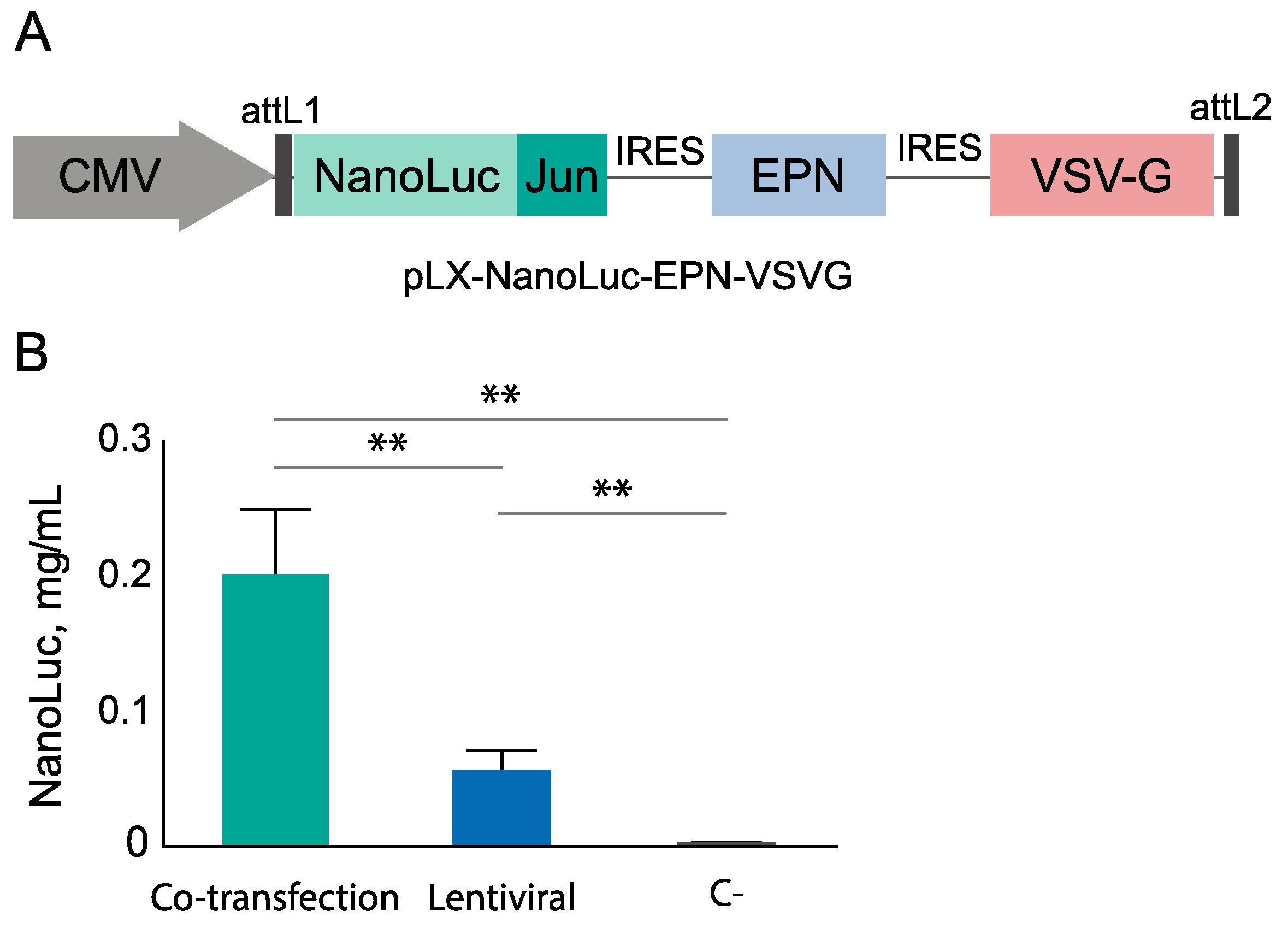
2.2. Plasmids
2.3. Protein Expression in E. coli and Purification
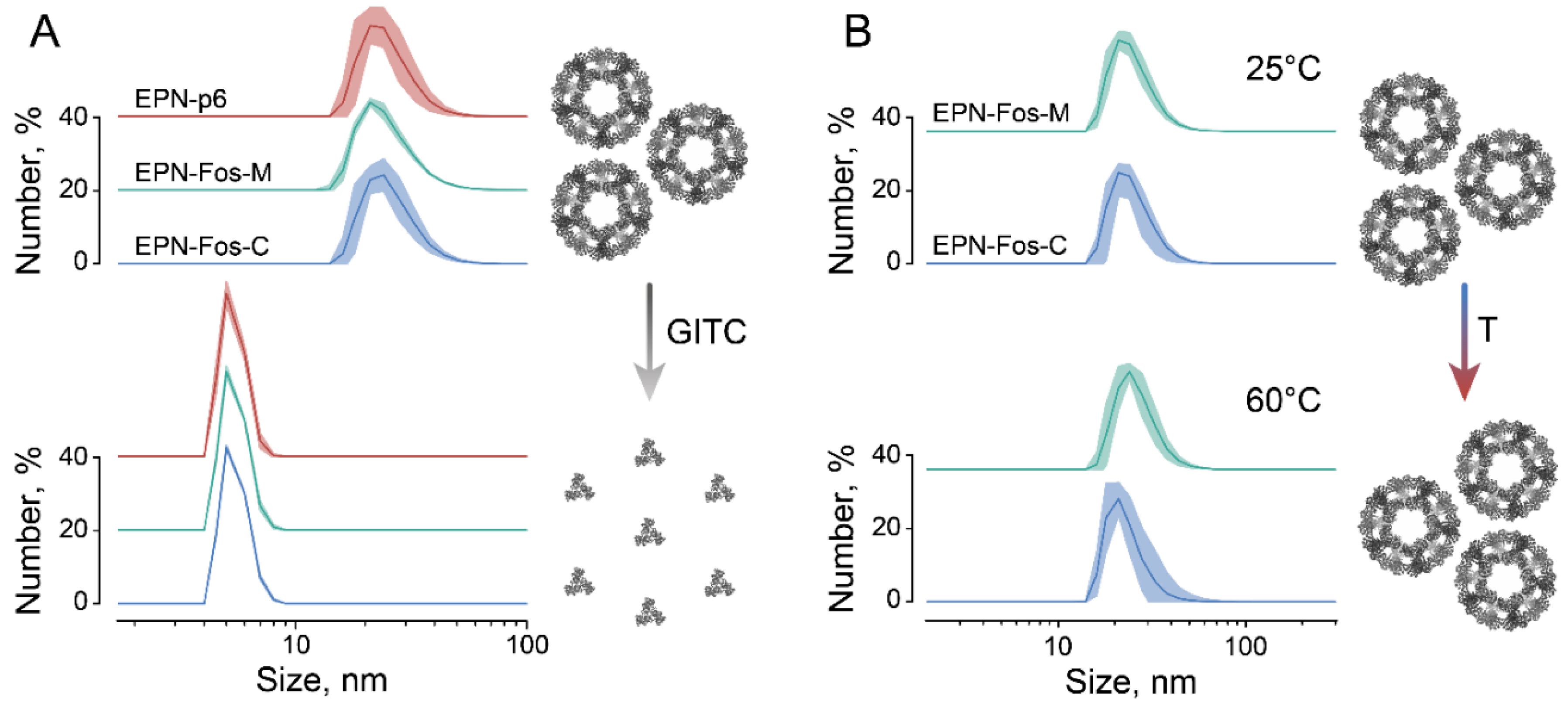
2.4. Dynamic Light Scattering
2.5. Isolation and Purification of EVs
2.6. Western Blot Analysis
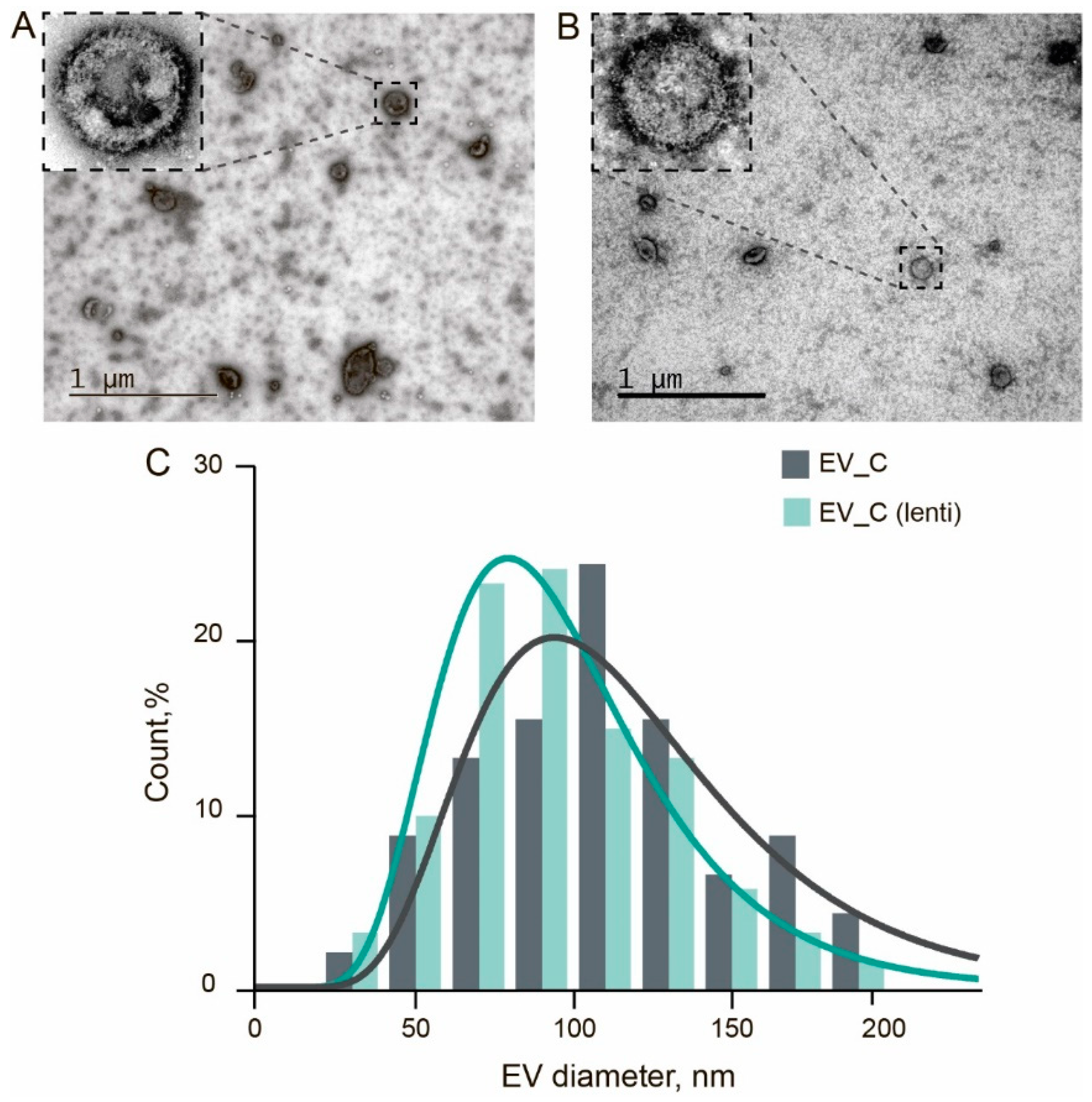
2.7. Transmission Electron Microscopy (TEM)
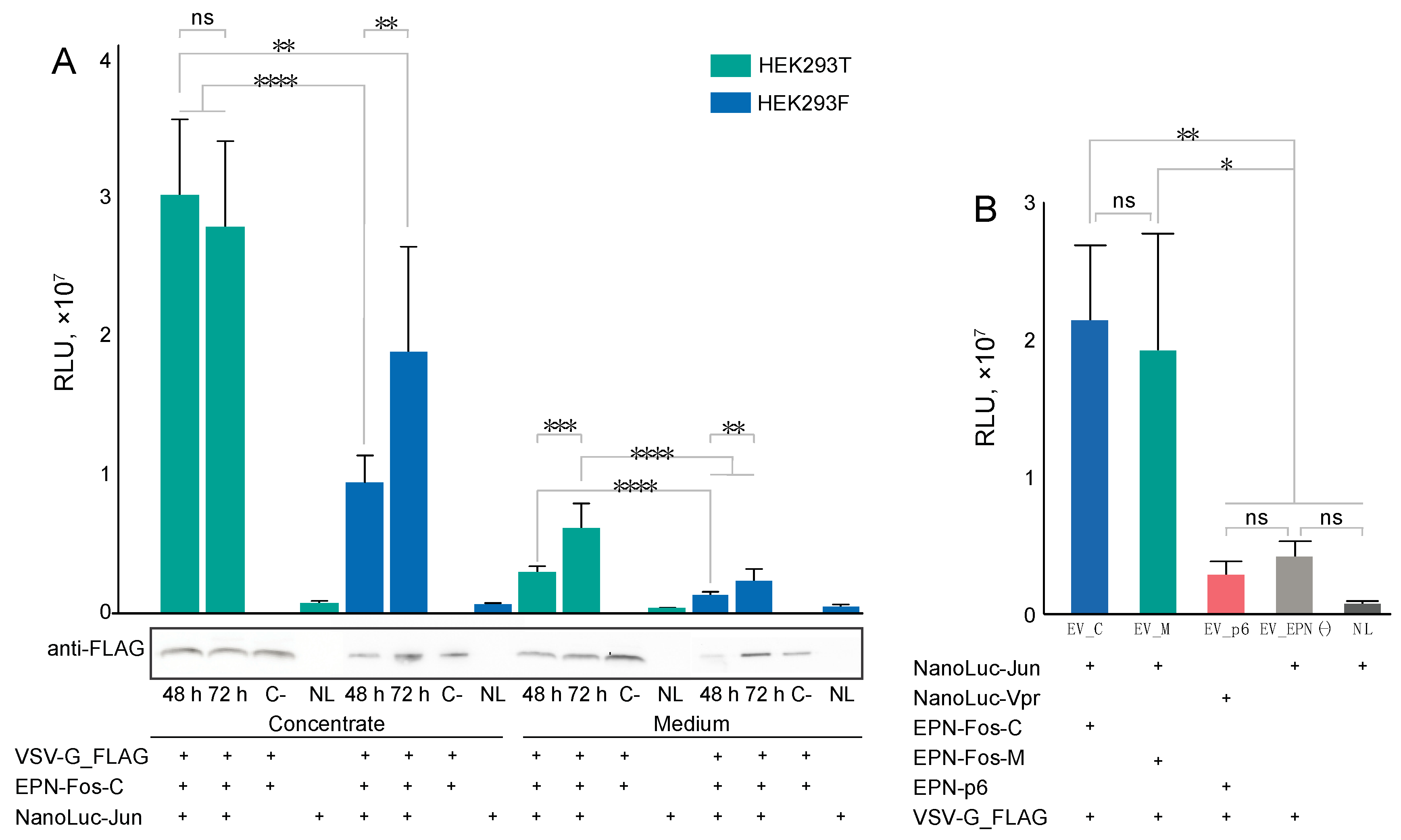
2.8. Protein Delivery of EV Cargoes to Target Cells
2.9. LC-MS/MS and Bioinformatics
2.10. Molecular Modeling
2.11. Estimation of Protein Numbers in EVs
2.12. Quantification and Statistical Analysis
3. Results
3.1. Improving Loading Capacity of Protein Nanocages Preserves Their Capacity to Self-Assemble
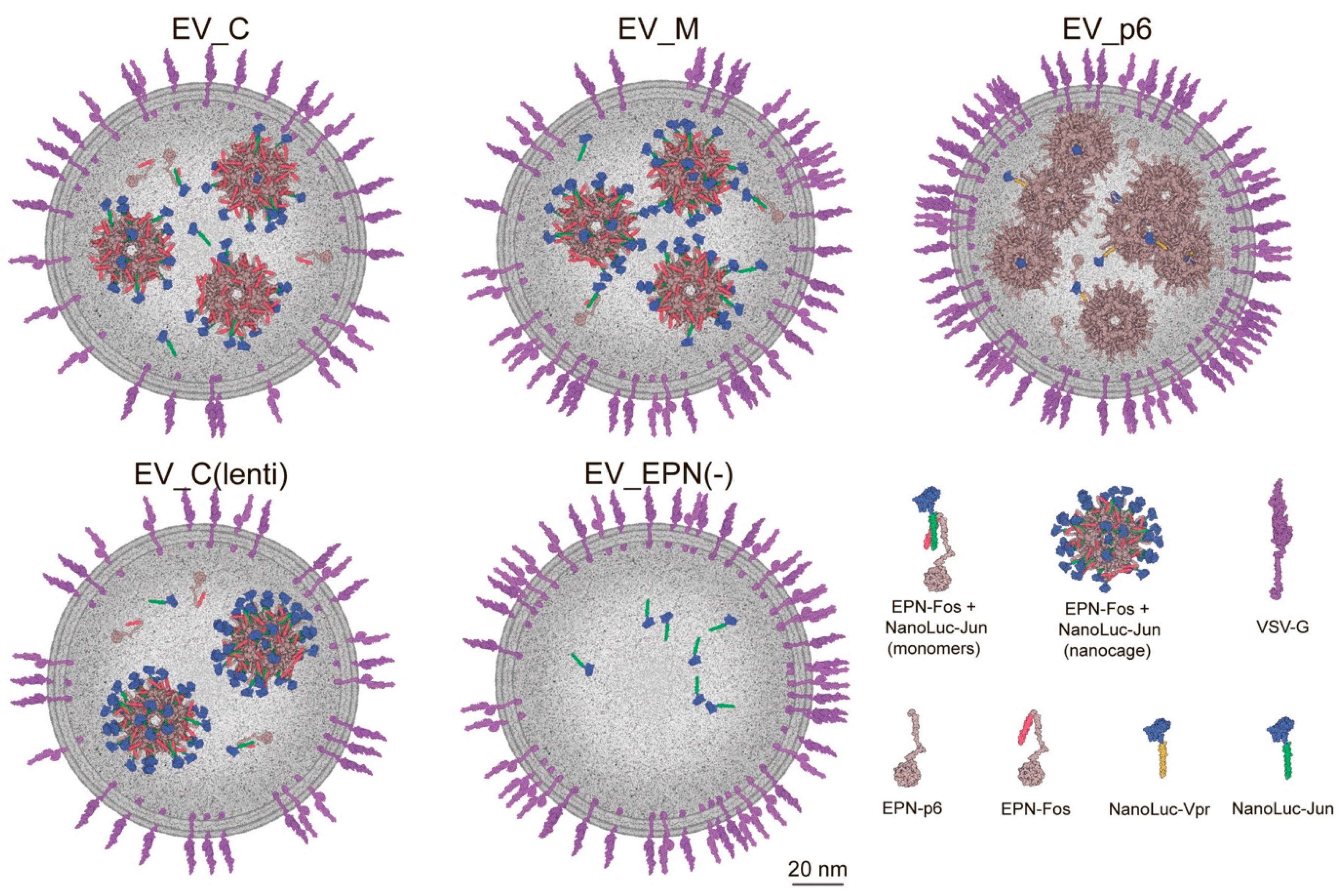
3.2. Production of Engineered EVs in Mammalian Cells
3.3. EV Production by Lentiviral Construction
3.4. Isolation and Characterization of Genetically Encoded EVs
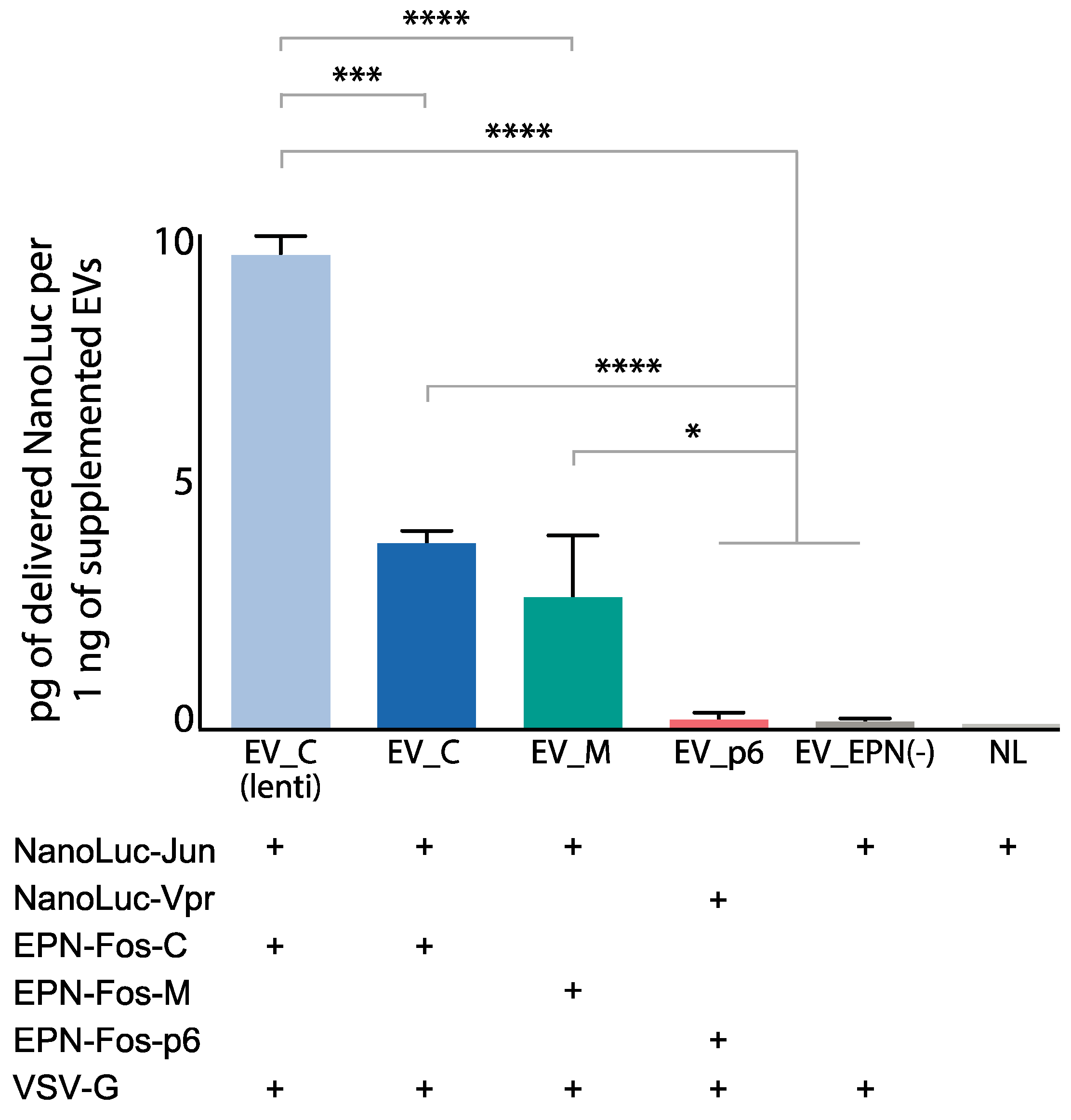
3.5. Delivery of Protein Cargo to Target Cells
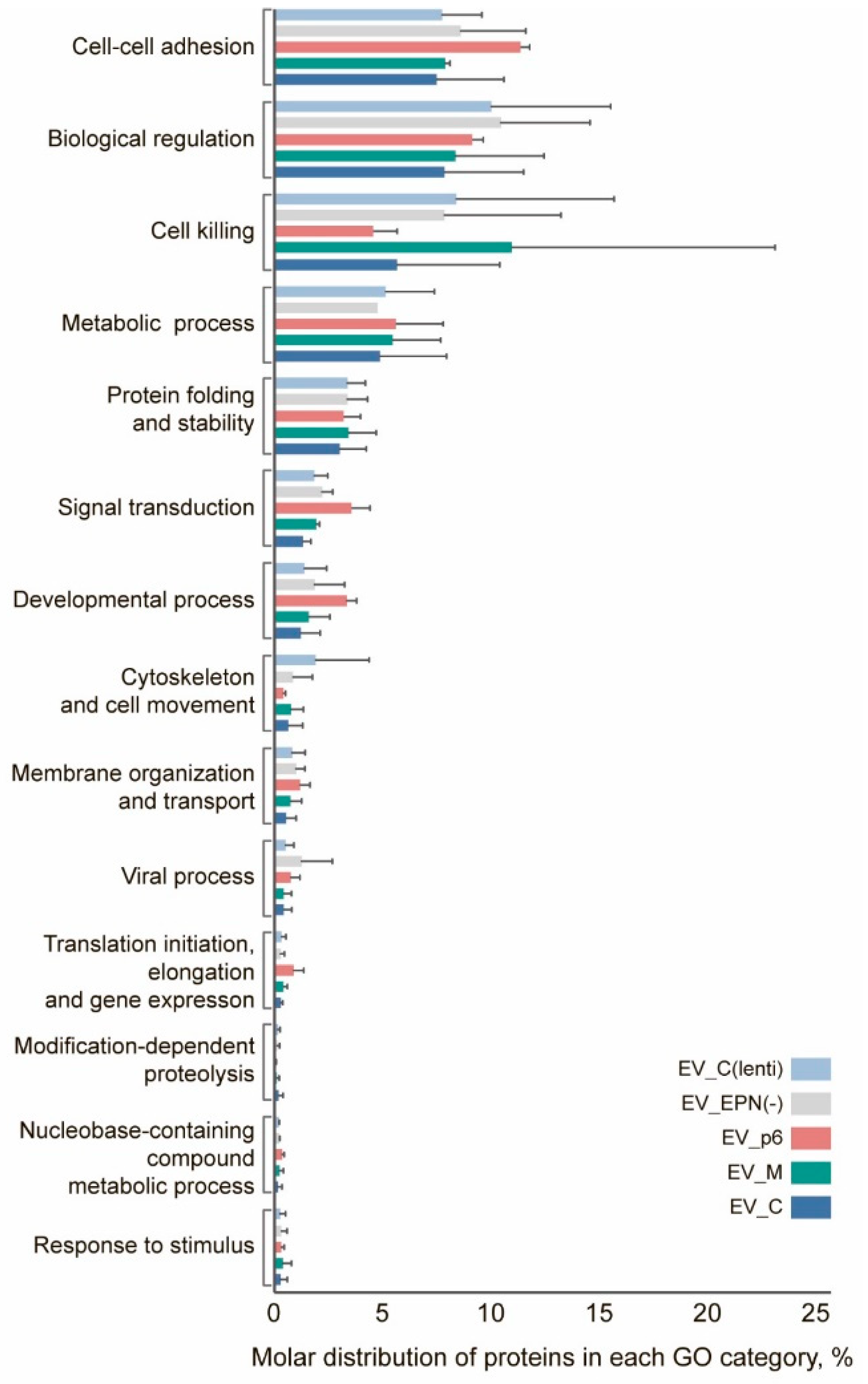
3.6. Proteome Profiling of Engineered EVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, W.; Von Roemeling, C.A.; Chen, Y.; Qie, Y.; Liu, X.; Chen, J.; Kim, B.Y.S. Designing nanomedicine for immuno-oncology. Nat. Biomed. Eng. 2017, 1, 0029. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Ratajczak, J. Extracellular microvesicles/exosomes: Discovery, disbelief, acceptance, and the future? Leukemia 2020, 34, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30. [Google Scholar] [CrossRef]
- Ståhl, A.-L.; Johansson, K.; Mossberg, M.; Kahn, R.; Karpman, D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019, 34, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Zebrowska, A.; Skowronek, A.; Wojakowska, A.; Widlak, P.; Pietrowska, M. Metabolome of Exosomes: Focus on Vesicles Released by Cancer Cells and Present in Human Body Fluids. Int. J. Mol. Sci. 2019, 20, 3461. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef]
- Nieuwland, R.; Sturk, A. Why do cells release vesicles? Thromb. Res. 2010, 125, S49–S51. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Kostin, N.N.; Ovchinnikova, L.A.; Lomakin, Y.A.; Kudriaeva, A.A. Targeted Drug Delivery in Lipid-like Nanocages and Extracellular Vesicles. Acta Nat. 2019, 11, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.L.; Rood, I.M.; Deegens, J.K.J.; Klein, J.B. Isolation and characterization of urinary extracellular vesicles: Implications for biomarker discovery. Nat. Rev. Nephrol. 2017, 13, 731–749. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.B.; Ray, R.B. Extracellular vesicles: Novel mediator for cell to cell communications in liver pathogenesis. Mol. Asp. Med. 2018, 60, 115–122. [Google Scholar] [CrossRef]
- Chong, S.Y.; Lee, C.K.; Huang, C.; Ou, Y.H.; Charles, C.J.; Richards, A.M.; Neupane, Y.R.; Pavon, M.V.; Zharkova, O.; Pastorin, G.; et al. Extracellular Vesicles in Cardiovascular Diseases: Alternative Biomarker Sources, Therapeutic Agents, and Drug Delivery Carriers. Int. J. Mol. Sci. 2019, 20, 3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroz-Baez, R.; Hernández-Ortega, K.; Martínez-Martínez, E. Insights Into the Proteomic Profiling of Extracellular Vesicles for the Identification of Early Biomarkers of Neurodegeneration. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Cho, Y.-E.; Song, B.-J.; Akbar, M.; Baek, M.-C. Extracellular vesicles as potential biomarkers for alcohol- and drug-induced liver injury and their therapeutic applications. Pharmacol. Ther. 2018, 187, 180–194. [Google Scholar] [CrossRef]
- Yee, N.S.; Zhang, S.; He, H.-Z.; Zheng, S.-Y. Extracellular Vesicles as Potential Biomarkers for Early Detection and Diagnosis of Pancreatic Cancer. Biomedicines 2020, 8, 581. [Google Scholar] [CrossRef]
- Akuma, P.; Okagu, O.D.; Udenigwe, C.C. Naturally Occurring Exosome Vesicles as Potential Delivery Vehicle for Bioactive Compounds. Front. Sustain. Food Syst. 2019, 3. [Google Scholar] [CrossRef]
- Hood, J.L.; Wickline, S.A. A systematic approach to exosome-based translational nanomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2012, 4, 458–467. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilligan, K.E.; Dwyer, R.M. Engineering Exosomes for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hong, Y.; Cho, E.; Kim, G.B.; Kim, I.-S. Extracellular vesicles as a platform for membrane-associated therapeutic protein delivery. J. Extracell. Vesicles 2018, 7, 1440131. [Google Scholar] [CrossRef] [Green Version]
- Hsia, Y.; Bale, J.B.; Gonen, S.; Shi, D.; Sheffler, W.; Fong, K.K.; Nattermann, U.; Xu, C.; Huang, P.S.; Ravichandran, R.; et al. Corrigendum: Design of a hyperstable 60-subunit protein icosahedron. Nature 2016, 540, 150, Erratum in 2016, 535, 136–139. [Google Scholar] [CrossRef]
- Votteler, J.; Ogohara, C.; Yi, S.; Hsia, Y.; Nattermann, U.; Belnap, D.M.; King, N.P.; Sundquist, W.I. Designed proteins induce the formation of nanocage-containing extracellular vesicles. Nature 2016, 540, 292–295. [Google Scholar] [CrossRef]
- Planelles, V.; Jowett, J.B.; Li, Q.X.; Xie, Y.; Hahn, B.; Chen, I.S. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 1996, 70, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Chiang, S.-F.; Lin, T.-Y.; Chiou, S.-H.; Chow, K.-C. HIV-1 Vpr Triggers Mitochondrial Destruction by Impairing Mfn2-Mediated ER-Mitochondria Interaction. PLoS ONE 2012, 7, e33657. [Google Scholar] [CrossRef] [Green Version]
- Urquiza-García, U.; Millar, A.J. Expanding the bioluminescent reporter toolkit for plant science with NanoLUC. Plant Methods 2019, 15, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikishin, I.; Dulimov, R.; Skryabin, G.; Galetsky, S.; Tchevkina, E.; Bagrov, D. ScanEV—A neural network-based tool for the automated detection of extracellular vesicles in TEM images. Micron 2021, 145, 103044. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, S.I.; Jensen, O.N.; Rogowska-Wrzesinska, A. FlashPack: Fast and Simple Preparation of Ultrahigh-performance Capillary Columns for LC-MS. Mol. Cell. Proteom. 2019, 18, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-B.; Krey, J.F.; Hassan, A.; Metlagel, Z.; Tauscher, A.N.; Pagana, J.M.; Sherman, N.E.; Jeffery, E.D.; Spinelli, K.J.; Zhao, H.; et al. Molecular architecture of the chick vestibular hair bundle. Nat. Neurosci. 2013, 16, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, A.; Autin, L.; Fuentes, D.; Maritan, M.; Barad, B.A.; Medina, M.; Olson, A.J.; Grotjahn, D.A.; Goodsell, D.S. CellPAINT: Turnkey Illustration of Molecular Cell Biology. Front. Bioinform. 2021, 1. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Autin, L.; Olson, A.J. Illustrate: Software for Biomolecular Illustration. Structure 2019, 27, 1716–1720. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Mangeot, P.-E.; Dollet, S.; Girard, M.; Ciancia, C.; Joly, S.; Peschanski, M.; Lotteau, V. Protein Transfer into Human Cells by VSV-G-induced Nanovesicles. Mol. Ther. 2011, 19, 1656–1666. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, Q.; Zi, Z.; Liu, Z.; Wan, C.; Crisman, L.; Shen, J.; Liu, X. Programmable Extracellular Vesicles for Macromolecule Delivery and Genome Modifications. Dev. Cell 2020, 55, 784–801. [Google Scholar] [CrossRef]
- Tungaturthi, P.K.; Sawaya, B.E.; Singh, S.P.; Tomkowicz, B.; Ayyavoo, V.; Khalili, K.; Collman, R.G.; Amini, S.; Srinivasan, A. Role of HIV-1 Vpr in AIDS pathogenesis: Relevance and implications of intravirion, intracellular and free Vpr. Biomed. Pharmacother. 2003, 57, 20–24. [Google Scholar] [CrossRef]
- Liu, H.; Wu, X.; Xiao, H.; Kappes, J.C. Targeting Human Immunodeficiency Virus (HIV) Type 2 Integrase Protein into HIV Type. J. Virol. 1999, 73, 8831–8836. [Google Scholar] [CrossRef] [Green Version]
- Guenzel, C.A.; Hérate, C.; Benichou, S. HIV-1 Vpr-a still “enigmatic multitasker”. Front. Microbiol. 2014, 5, 127. [Google Scholar] [CrossRef] [Green Version]
- Bolton, D.L.; Lenardo, M.J. Vpr Cytopathicity Independent of G2/M Cell Cycle Arrest in Human Immunodeficiency Virus Type 1-Infected CD4+ T Cells. J. Virol. 2007, 81, 8878–8890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K.; Minamisawa, T.; Suga, K.; Yajima, Y.; Shiba, K. Isolation of human salivary extracellular vesicles by iodixanol density gradient ultracentrifugation and their characterizations. J. Extracell. Vesicles 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittig, S.; Ganzella, M.; Barth, M.; Kostmann, S.; Riedel, D.; Pérez-Lara, Á.; Jahn, R.; Schmidt, C. Cross-linking mass spectrometry uncovers protein interactions and functional assemblies in synaptic vesicle membranes. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Munis, A.M.; Mattiuzzo, G.; Bentley, E.M.; Collins, M.K.; Eyles, J.E.; Takeuchi, Y. Use of Heterologous Vesiculovirus G Proteins Circumvents the Humoral Anti-envelope Immunity in Lentivector-Based In Vivo Gene Delivery. Mol. Ther. Nucleic Acids 2019, 17, 126–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonaro-Sarracino, D.A.; Tarantal, A.F.; Lee, C.C.I.; Kaufman, M.L.; Wandro, S.; Jin, X.; Martinez, M.; Clark, D.N.; Chun, K.; Koziol, C.; et al. Dosing and Re-Administration of Lentiviral Vector for In Vivo Gene Therapy in Rhesus Monkeys and ADA-Deficient Mice. Mol. Ther. Methods Clin. Dev. 2020, 16, 78–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonaro-Sarracino, D.A.; Chun, K.; Clark, D.N.; Kaufman, M.L.; Jin, X.; Wang, X.; Kohn, D.B. Gene delivery using AAV8 in vivo for disease stabilization in a bimodal gene therapy approach for the treatment of ADA-deficient SCID. Mol. Ther. Methods Clin. Dev. 2021, 20, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.-E.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [Green Version]
- Bohmann, D.; Bos, T.; Admon, A.; Nishimura, T.; Vogt, P.K.; Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 1987, 238, 1386–1392. [Google Scholar] [CrossRef]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [Green Version]
- Topping, L.M.; Thomas, B.L.; Rhys, H.I.; Tremoleda, J.L.; Foster, M.; Seed, M.; Voisin, M.-B.; Vinci, C.; Law, H.L.; Perretti, M.; et al. Targeting Extracellular Vesicles to the Arthritic Joint Using a Damaged Cartilage-Specific Antibody. Front. Immunol. 2020, 11, 10. [Google Scholar] [CrossRef]
- Cui, G.-H.; Guo, H.-D.; Li, H.; Zhai, Y.; Gong, Z.-B.; Wu, J.; Liu, J.-S.; Dong, Y.-R.; Hou, S.-X. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer’s disease. Immun. Ageing 2019, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Schlehuber, L.D.; Rose, J.K. Prediction and Identification of a Permissive Epitope Insertion Site in the Vesicular Stomatitis Virus Glycoprotein. J. Virol. 2004, 78, 5079–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 2015, 34, 474–490. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | EV_C | EV_M | EV_p6 | EV_EPN(-) | EV_C(lenti) |
|---|---|---|---|---|---|
| Size determined by DLS, nm | 174 ± 45 | 176 ± 59 | 171 ± 57 | 150 ± 38 | 168 ± 45 |
| EV Samples | EV_C | EV_M | EV_p6 | EV_(EPN-) | EV_C(lenti) |
|---|---|---|---|---|---|
| VSV-G, molecules | 450 ± 50 | 700 ± 80 | 1000 ± 90 | 850 ± 80 | 450 ± 40 |
| NanoLuc, molecules | 430 ± 50 | 520 ± 50 | 110 ± 30 | 70 ± 30 | 1000 ± 80 |
| EPN, molecules | 1700 ± 600 | 1500 ± 500 | 4300 ± 1000 | - | 1500 ± 400 |
| Assembled nanocages | 28 ± 10 | 25 ± 8 | 71 ± 16 | - | 25 ± 7 |
| NanoLuc, molecules in a single nanocage | 15 ± 6 | 21 ± 7 | 1.5 ± 0.5 | - | 40 ± 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovchinnikova, L.A.; Terekhov, S.S.; Ziganshin, R.H.; Bagrov, D.V.; Filimonova, I.N.; Zalevsky, A.O.; Lomakin, Y.A. Reprogramming Extracellular Vesicles for Protein Therapeutics Delivery. Pharmaceutics 2021, 13, 768. https://doi.org/10.3390/pharmaceutics13060768
Ovchinnikova LA, Terekhov SS, Ziganshin RH, Bagrov DV, Filimonova IN, Zalevsky AO, Lomakin YA. Reprogramming Extracellular Vesicles for Protein Therapeutics Delivery. Pharmaceutics. 2021; 13(6):768. https://doi.org/10.3390/pharmaceutics13060768
Chicago/Turabian StyleOvchinnikova, Leyla A., Stanislav S. Terekhov, Rustam H. Ziganshin, Dmitriy V. Bagrov, Ioanna N. Filimonova, Arthur O. Zalevsky, and Yakov A. Lomakin. 2021. "Reprogramming Extracellular Vesicles for Protein Therapeutics Delivery" Pharmaceutics 13, no. 6: 768. https://doi.org/10.3390/pharmaceutics13060768