
Drug-Like Small Molecule HSP27 Functional Inhibitor Sensitizes Lung Cancer Cells to Gefitinib or Cisplatin by Inducing Altered Cross-Linked Hsp27 Dimers

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals for Pharmacokinetic and Toxicity Experiments
2.3. HPLC Analysis
2.4. Pharmacokinetic Study
2.5. Single-Dose Toxicity and Cytotoxicity
2.6. Physicochemical Properties and In Vitro Metabolic Stability
2.7. Cell Culture
2.8. Cell Transfection
2.9. Viability Assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay)
2.10. Western Blot
2.11. Antibodies
2.12. Flow Cytometry Analysis (FACS)
2.13. Tumor Xenografts in Mice
2.14. Immunohistochemical Staining
2.15. Statistics
3. Results
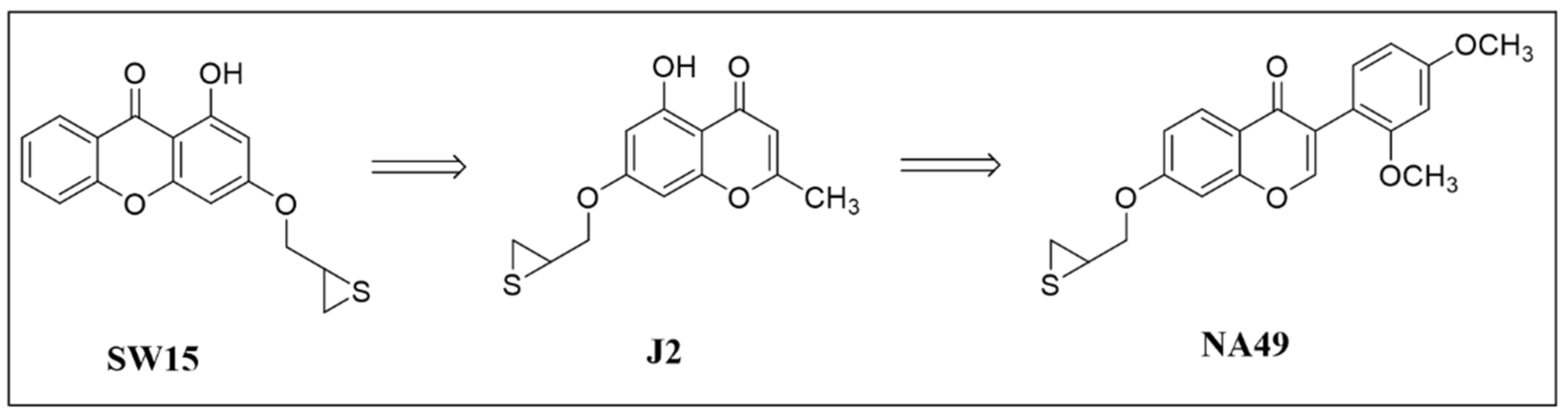
3.1. Synthesis of NA49 and J2
3.2. Characterization of Cytoxicity, Physicochemical Properties, and Metabolic Stability of NA49 and J2
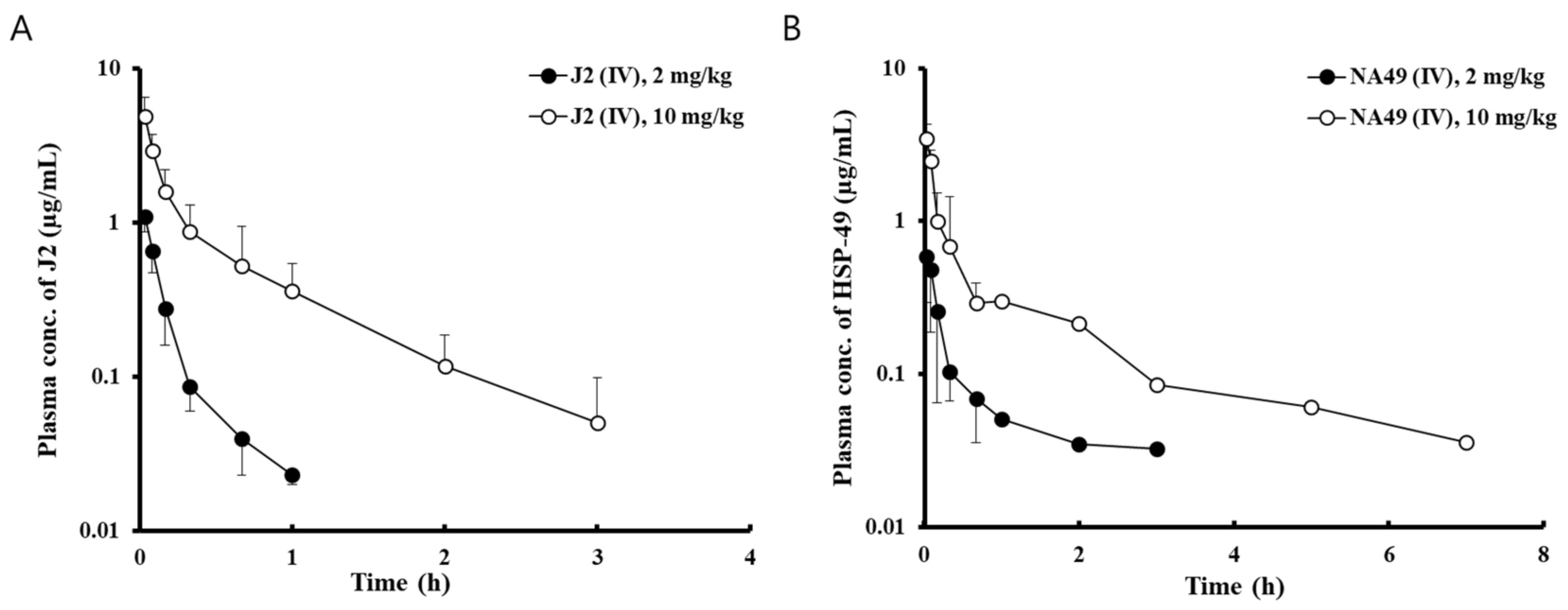
3.3. Analytical method Validation and Pharmacokinetics of J2 and NA49
3.4. Single-Dose Toxicity of J2 and NA49
3.5. NA49 Showed Similar HSP27 Cross-Linking Activity to J2
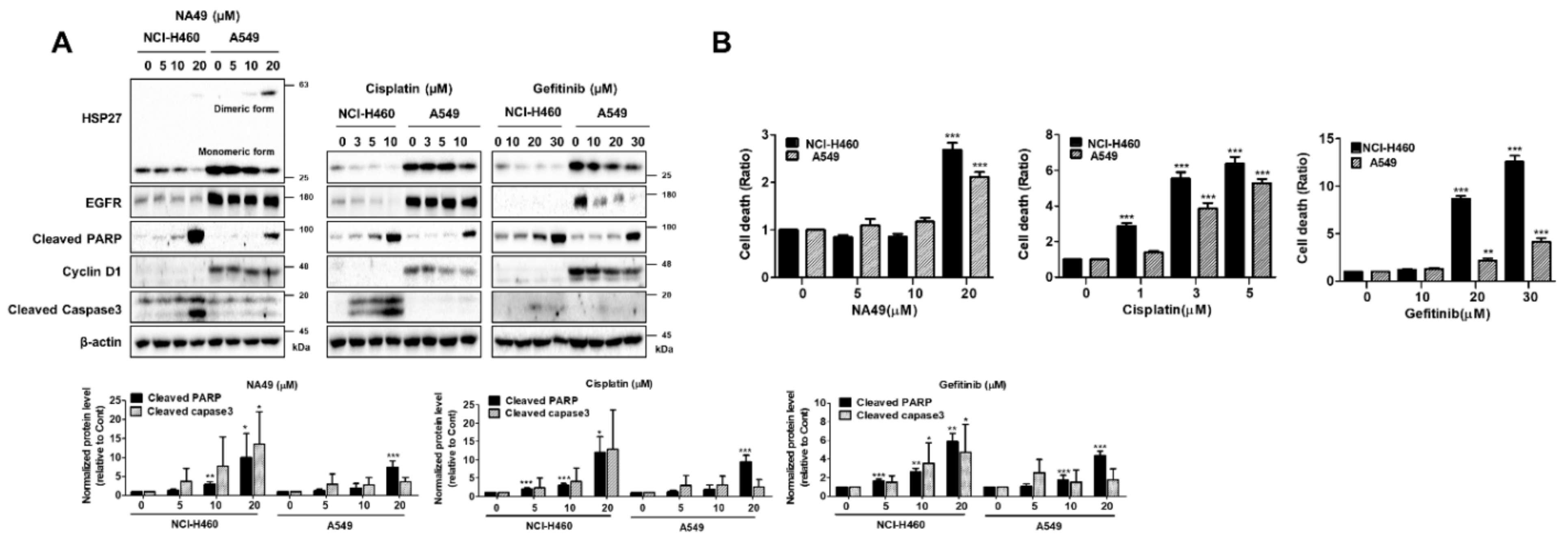
3.6. Cytotoxicity of Cisplatin, Gefitinib, and NA49 in EGFR WT and Mut Lung Cancer Cell Lines
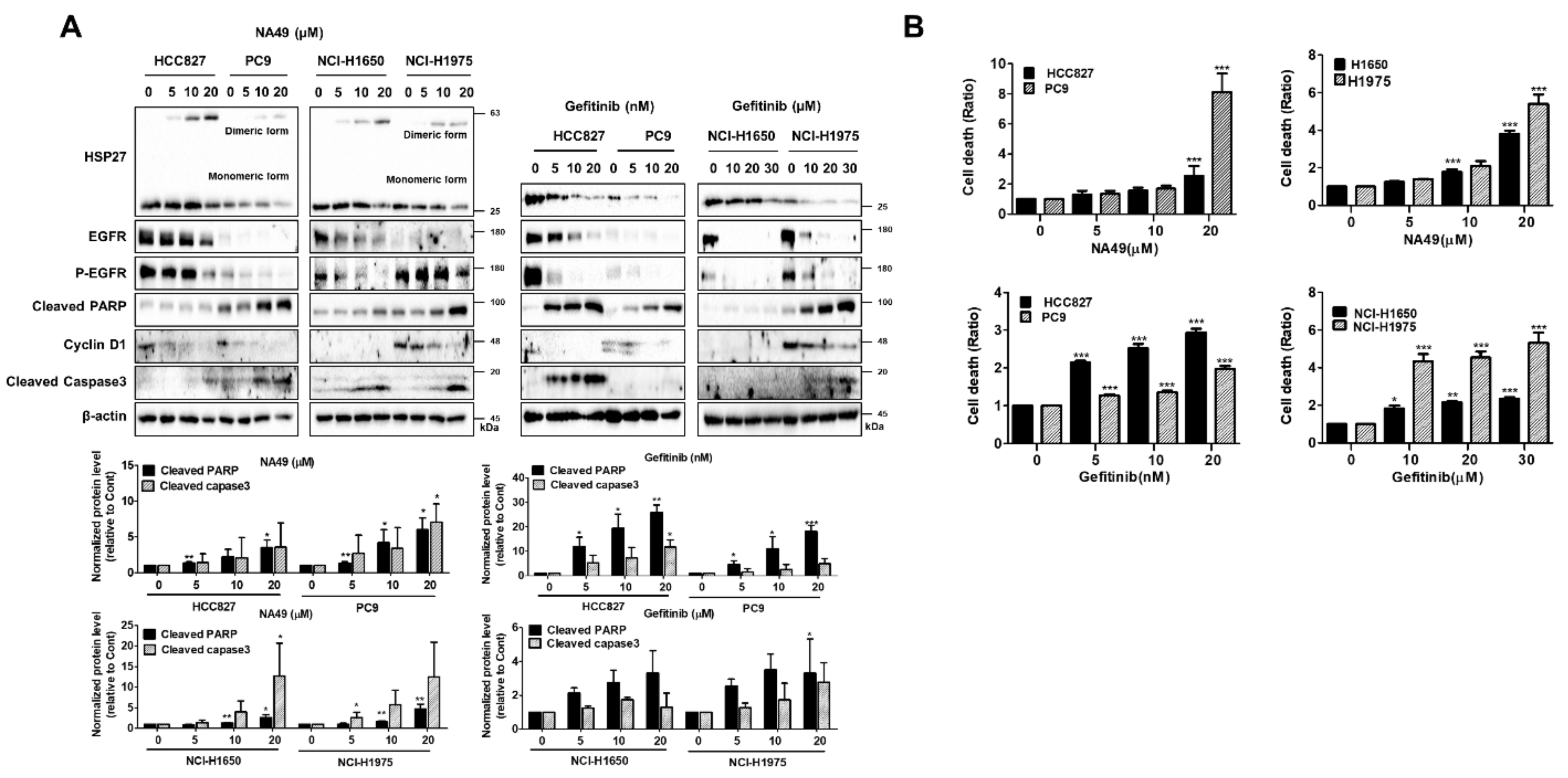
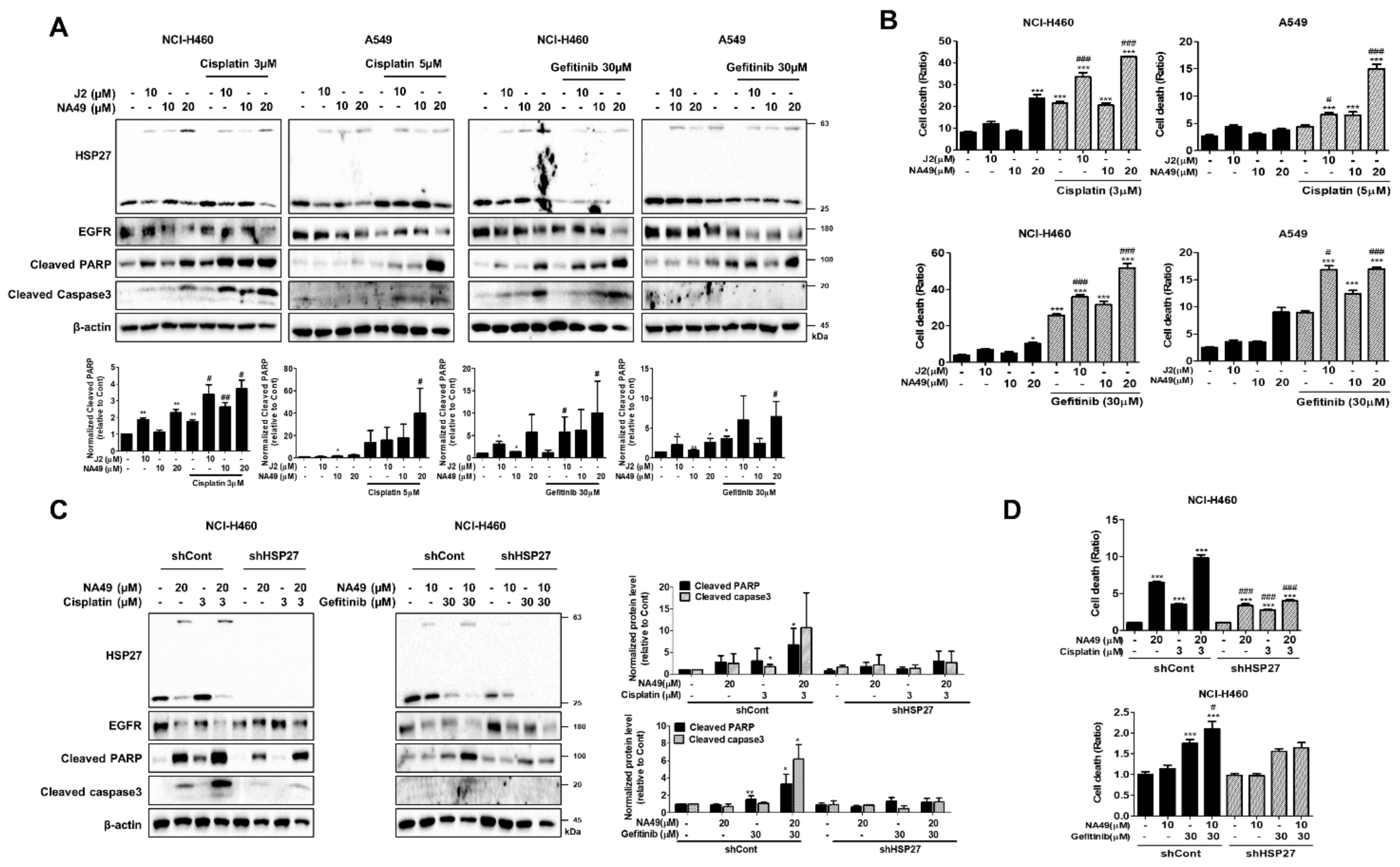
3.7. NA49 Showed Sensitization Effects in Combination with Cisplatin or Gefitinib in Both EGFR Wild Type and Mutant Lung Cancer Cell Lines
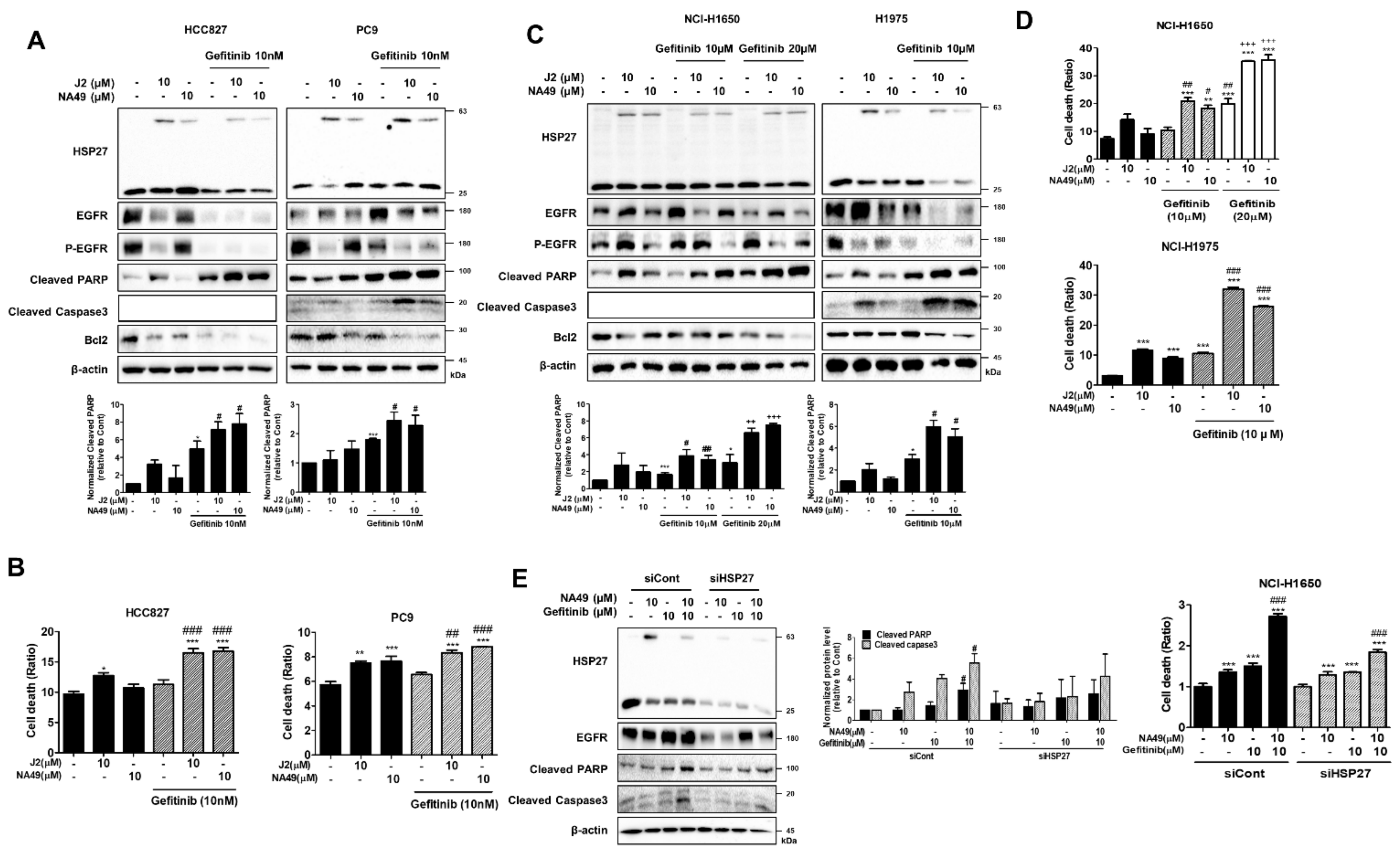
3.8. NA49 Showed Sensitization Effects in Combination with Gefitinib in EGFR Mutant Cells
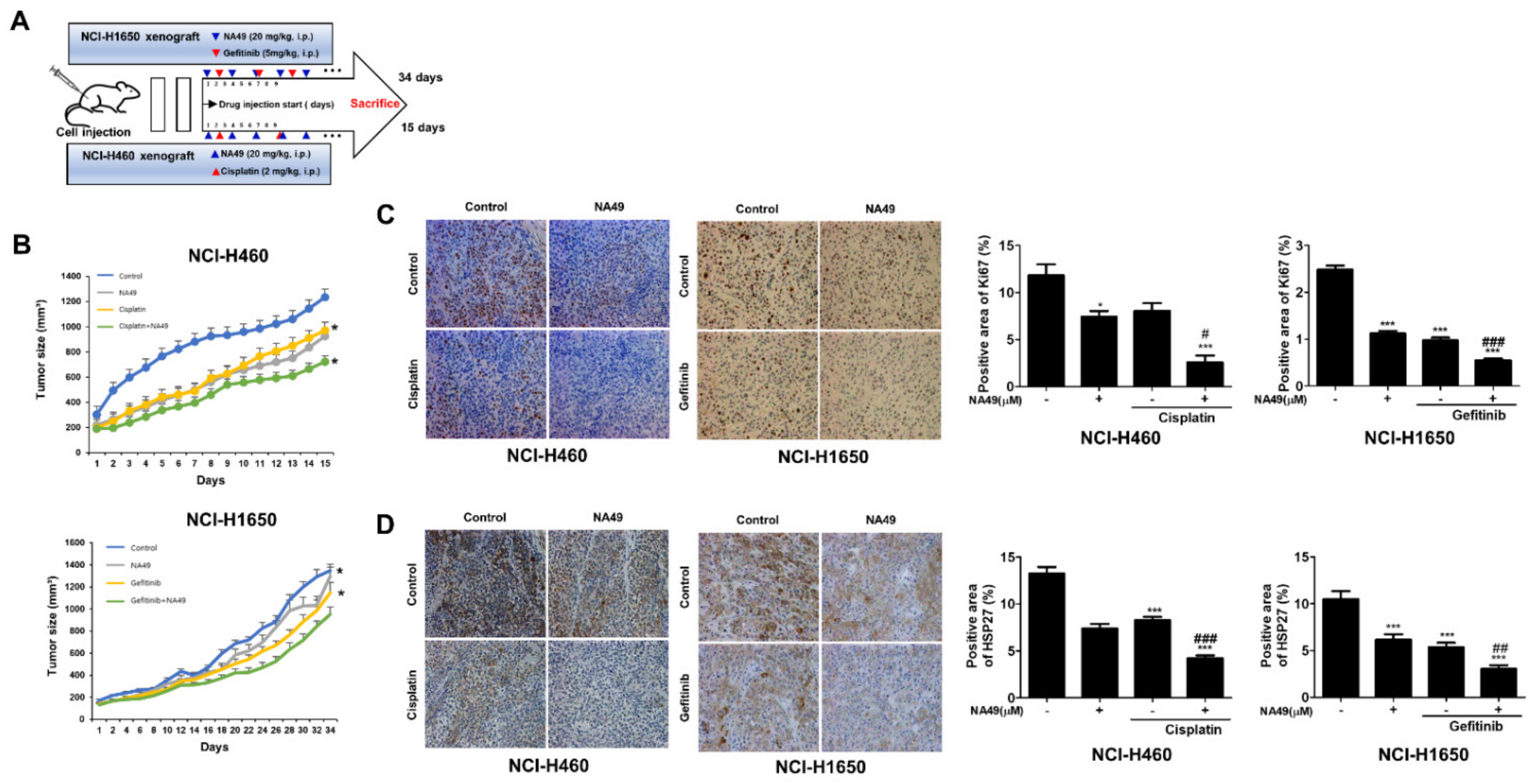
3.9. NA49 in Combination with Cisplatin or Gefitinib Sensitized Lung Cancer Cells in a Xenograft Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.S.; Lin, J.H.; Huang, W.C.; Hsu, T.W.; Su, K.; Chiou, S.H.; Tsai, Y.T.; Hung, S.C. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer 2011, 117, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Kang, K.W.; Kim, K.H.; Kwon, B.; Kim, S.K.; Lee, H.Y.; Kong, S.Y.; Lee, E.S.; Jang, S.G.; Yoo, B.C. Upregulated HSP27 in human breast cancer cells reduces Herceptin susceptibility by increasing Her2 protein stability. BMC Cancer 2008, 8, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramitsu, Y.; Wang, Y.; Taba, K.; Suenaga, S.; Ryozawa, S.; Kaino, S.; Sakaida, I.; Nakamura, K. Heat-shock protein 27 plays the key role in gemcitabine-resistance of pancreatic cancer cells. Anticancer Res. 2012, 32, 2295–2299. [Google Scholar]
- Xia, Y.; Rocchi, P.; Iovanna, J.L.; Peng, L. Targeting heat shock response pathways to treat pancreatic cancer. Drug Discov. Today 2012, 17, 35–43. [Google Scholar] [CrossRef]
- Singh, M.K.; Sharma, B.; Tiwari, P.K. The small heat shock protein Hsp27: Present understanding and future prospects. J. Therm. Biol. 2017, 69, 149–154. [Google Scholar] [CrossRef]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-Coll, A.; Yamanaka, K.; Gleave, M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res. 2004, 64, 6595–6602. [Google Scholar] [CrossRef] [Green Version]
- Baylot, V.; Andrieu, C.; Katsogiannou, M.; Taieb, D.; Garcia, S.; Giusiano, S.; Acunzo, J.; Iovanna, J.; Gleave, M.; Garrido, C.; et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011, 2, e221. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Gurbuxani, S.; Ravagnan, L.; Kroemer, G. Heat shock proteins: Endogenous modulators of apoptotic cell death. Biochem. Biophys. Res. Commun. 2001, 286, 433–442. [Google Scholar] [CrossRef]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.K.; Parra, I.; Lemieux, P.; Oesterreich, S.; Hilsenbeck, S.G.; Fuqua, S.A. Hsp27 overexpression inhibits doxorubicin-induced apoptosis in human breast cancer cells. Breast Cancer Res. Treat. 1999, 56, 187–196. [Google Scholar] [CrossRef]
- Paul, C.; Simon, S.; Gibert, B.; Virot, S.; Manero, F.; Arrigo, A.P. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp. Cell Res. 2010, 316, 1535–1552. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nappi, L.; Aguda, A.H.; Nakouzi, N.A.; Lelj-Garolla, B.; Beraldi, E.; Lallous, N.; Thi, M.; Moore, S.; Fazli, L.; Battsogt, D.; et al. Ivermectin inhibits HSP27 and potentiates efficacy of oncogene targeting in tumor models. J. Clin. Investig. 2020, 130, 699–714. [Google Scholar] [CrossRef]
- McConnell, J.R.; McAlpine, S.R. Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorg. Med. Chem. Lett. 2013, 23, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, J.C.; Tuukkanen, A.; Schroeder, M.; Fahrig, T.; Fahrig, R. RP101 (brivudine) binds to heat shock protein HSP27 (HSPB1) and enhances survival in animals and pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2011, 137, 1349–1361. [Google Scholar] [CrossRef]
- Heinrich, J.C.; Donakonda, S.; Haupt, V.J.; Lennig, P.; Zhang, Y.; Schroeder, M. New HSP27 inhibitors efficiently suppress drug resistance development in cancer cells. Oncotarget 2016, 7, 68156–68169. [Google Scholar] [CrossRef] [Green Version]
- Lamoureux, F.; Thomas, C.; Yin, M.J.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Suppression of heat shock protein 27 using OGX-427 induces endoplasmic reticulum stress and potentiates heat shock protein 90 inhibitors to delay castrate-resistant prostate cancer. Eur. Urol. 2014, 66, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Lee, Y.J.; Seo, W.D.; Lee, H.J.; Nam, J.W.; Lee, Y.J.; Kim, J.; Seo, E.K.; Lee, Y.S. Altered cross-linking of HSP27 by zerumbone as a novel strategy for overcoming HSP27-mediated radioresistance. Int. J. Radiat Oncol. Biol. Phys. 2011, 79, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jung, Y.J.; Choi, B.; Lee, N.L.; Lee, H.J.; Kwak, S.Y.; Kwon, Y.; Na, Y.; Lee, Y.S. Overcoming HSP27-mediated resistance by altered dimerization of HSP27 using small molecules. Oncotarget 2016, 7, 53178–53190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.; Choi, S.K.; Park, Y.N.; Kwak, S.Y.; Lee, H.J.; Kwon, Y.; Na, Y.; Lee, Y.S. Sensitization of lung cancer cells by altered dimerization of HSP27. Oncotarget 2017, 8, 105372–105382. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.G.; Lee, J.; Kim, K.M.; Lee, K.I.; Bae, Y.S.; Lee, H.J. Pharmacokinetic Study of NADPH Oxidase Inhibitor Ewha-18278, a Pyrazole Derivative. Pharmaceutics 2019, 11, 482. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, K.; Tanimoto, A.; Abe, T.; Ogawa, M.; Yutsudo, T.; Kashimura, M.; Yoshida, S. Shiga toxin 1 and 2 induce apoptosis in the amniotic cell line WISH. J. Soc. Gynecol. Investig. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Suzuki, A.K.; Sagai, M. The cytotoxic effects of diesel exhaust particles on human pulmonary artery endothelial cells in vitro: Role of active oxygen species. Free Radic. Biol. Med. 2001, 30, 555–562. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. Environ. Mutagenesis Relat. Subj. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Kim, I.; Boyle, K.M.; Carroll, J.L. Postnatal development of E-4031-sensitive potassium current in rat carotid chemoreceptor cells. J. Appl. Physiol. 2005, 98, 1469–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, T.J.; Loniello, K.R.; Beebe, J.A.; Ervin, K.M. Development and application of fluorescence polarization assays in drug discovery. Comb. Chem. High Throughput Screen. 2003, 6, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Weinsberg, F.; Bauer, C.K.; Schwarz, J.R. The class III antiarrhythmic agent E-4031 selectively blocks the inactivating inward-rectifying potassium current in rat anterior pituitary tumor cells (GH3/B6 cells). Pflügers Arch. 1997, 434, 1–10. [Google Scholar] [CrossRef]
- Avdeef, A.; Artursson, P.; Neuhoff, S.; Lazorova, L.; Grasjo, J.; Tavelin, S. Caco-2 permeability of weakly basic drugs predicted with the double-sink PAMPA pKa(flux) method. Eur. J. Pharm. Sci. 2005, 24, 333–349. [Google Scholar] [CrossRef]
- Masimirembwa, C.M.; Bredberg, U.; Andersson, T.B. Metabolic stability for drug discovery and development: Pharmacokinetic and biochemical challenges. Clin. Pharm. 2003, 42, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.J.; McGinnity, D.F.; Austin, R.P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos. 2005, 33, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perryman, A.L.; Stratton, T.P.; Ekins, S.; Freundlich, J.S. Predicting Mouse Liver Microsomal Stability with “Pruned” Machine Learning Models and Public Data. Pharm. Res. 2016, 33, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.; Oskamp, A.; Holschbach, M.; Neumaier, B.; Bauer, A.; Bier, D. Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data. Pharmaceuticals 2019, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Andersson, T.B.; Bredberg, E.; Ericsson, H.; Sjoberg, H. An evaluation of the in vitro metabolism data for predicting the clearance and drug-drug interaction potential of CYP2C9 substrates. Drug Metab. Dispos. 2004, 32, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalczyk, A.; Klüter, S.; Rode, H.B.; Simard, J.R.; Grütter, C.; Rabiller, M.; Rauh, D. Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg. Med. Chem. 2008, 16, 3482–3488. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [Green Version]
- Scheff, R.J.; Schneider, B.J. Non-small-cell lung cancer: Treatment of late stage disease: Chemotherapeutics and new frontiers. Semin. Interv. Radiol. 2013, 30, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Kulkarni, P.; Salgia, R. Combined Checkpoint Inhibition and Chemotherapy: New Era of 1(st)-Line Treatment for Non-Small-Cell Lung Cancer. Mol. Ther. Oncolytics 2019, 13, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.Q.; da Cunha Santos, G.; Ding, K.; Sakurada, A.; Cutz, J.C.; Liu, N.; Zhang, T.; Marrano, P.; Whitehead, M.; Squire, J.A.; et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J. Clin. Oncol. 2008, 26, 4268–4275. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | J2 | NA49 | |||
|---|---|---|---|---|---|
| IC50 (μM) of compounds in various normal mammalian cells a | HFL-1 | >100 | 94.5 | ||
| L929 | >100 | 91.9 | |||
| NIH3T3 | >100 | 96.5 | |||
| CHO-K1 | >100 | 90.4 | |||
| VERO | 91.8 | 83.6 | |||
| Ames test | Amount treated (μg/plate) | Tester strain | TA98 | 40 | 200 |
| TA100 | 40 | 200 | |||
| Revertant colonies/plate b (Mean ± S.D.) [Factor] # | Without S-9 mix | TA98 | 57 ± 7 [3.6] | 19 ± 3 [1.2] | |
| TA100 | 643 ± 70 [6.0] | 229 ± 2 [2.0] | |||
| With S-9 mix | TA98 | 65 ± 2 [3.8] | 22 ± 2 [1.3] | ||
| TA100 | 418 ± 9 [3.1] | 262 ± 11 [1.9] | |||
| hERG K+ Channel Binding Assay c | Concentration treated (μM) | 10 | 10 | ||
| % Inhibition of hERG ligand binding | 8.7 ± 2.22 | 39.0 ± 3.9 | |||
| Compound | J2 | NA49 | |
|---|---|---|---|
| Solubility (pH 7.0) a | 73.2 ± 1.1 μM (19.5 ± 0.3 μg/mL) | 90.2 ± 2.6 μM (33.4 ± 1.0 μg/mL) | |
| Log P a | 2.78 | 3.04 | |
| Cell Permeability b Log Papp (cm/s) | −4.90 ± 0.047 | −4.49 ± 0.152 | |
| In vitro Metabolic Stability c (% of remaining after 30 min) | Mouse | 6.2 ± 2.55 | 2.4 ± 0.95 |
| Rat | 2.6 ± 0.45 | 5.0 ± 1.33 | |
| Human | 47.8 ± 4.50 | 24.9 ± 0.77 | |
| In vitro Human Plasma Stability at 37 °C (% of remaining after 1 h) | When treated with 0.5 μg/mL | 76.2 ± 0.72 | 146.6 ± 0.01 |
| When treated with 5 μg/mL | 82.7 ± 0.73 | 101.4 ± 0.13 | |
| Isotype of CYP450 /Compound | J2 | NA49 |
|---|---|---|
| 1A2 | 0.1 | >50 |
| 2C9 | >50 | 19.2 |
| 2C19 | >50 | >50 |
| 2D6 | >50 | >50 |
| 3A4 | >50 | >50 |
| Pharmacokinetic Parameters | Drug | 2 mg/kg | 10 mg/kg |
|---|---|---|---|
| C0 (µg/mL) | J2 | 1.64 ± 0.73 | 6.96 ± 2.60 *** |
| NA49 | 0.72 ± 0.40 | 4.96 ± 1.12 *** | |
| AUC0–last (µg·h/mL) | J2 | 0.18 ± 0.05 | 1.48 ± 0.75 *** |
| NA49 | 0.21 ± 0.07 | 1.24 ± 0.59 *** | |
| t1/2 (h) | J2 | 0.19 ± 0.07 | 0.45 ± 0.30 * |
| NA49 | 1.40 ± 0.84 | 1.55 ± 0.48 | |
| Vd (L) | J2 | 0.77 ± 0.27 | 1.05 ± 0.72 |
| NA49 | 3.35 ± 1.69 | 4.13 ± 1.87 | |
| Cl (L/h) | J2 | 2.79 ± 0.69 | 1.90 ± 0.76 |
| NA49 | 1.84 ± 0.49 | 1.80 ± 0.56 |
| Group/Compound | Survival Rate (%) | |
|---|---|---|
| J2 | NA49 | |
| Control | 100 | 100 |
| Vehicle | 100 | 100 |
| 2.5 mg/kg | 100 | 100 |
| 7.5 mg/kg | 100 | 100 |
| 15 mg/kg | 100 | 100 |
| 30 mg/kg | 100 | 75 |
| Cell Lines | EGFR | EGFR Expression | HSP27 Expression | Cisplatin IC50 (µM) | NA49 IC50 (µM) |
|---|---|---|---|---|---|
| NCI-H460 | Wild-type | + | + | 11.7 | 38.05 ± 0.69 |
| A549 | Wild-type | +++ | ++ | 14.4 | 62.48 ± 0.90 |
| HCC827 | Mutant-type (Exon19 del) | +++++ | +++ | 64.42 ± 2.31 | |
| PC9 | Mutant-type (Exon19 del) | +++ | +++ | 72.99 ± 2.66 | |
| NCI-H1650 | Mutant-type (Exon19 del) | ++ | +++++ | 62.94 ± 1.58 | |
| NCI-H1975 | Mutant-type (L858R, T790M) | +++ | ++++ | 89.44 ± 2.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, H.; Choi, S.-K.; Lee, J.; Park, S.H.; Park, Y.N.; Hwang, S.-Y.; Shin, J.-H.; Na, Y.; Kwon, Y.; Lee, H.J.; et al. Drug-Like Small Molecule HSP27 Functional Inhibitor Sensitizes Lung Cancer Cells to Gefitinib or Cisplatin by Inducing Altered Cross-Linked Hsp27 Dimers. Pharmaceutics 2021, 13, 630. https://doi.org/10.3390/pharmaceutics13050630
Yoo H, Choi S-K, Lee J, Park SH, Park YN, Hwang S-Y, Shin J-H, Na Y, Kwon Y, Lee HJ, et al. Drug-Like Small Molecule HSP27 Functional Inhibitor Sensitizes Lung Cancer Cells to Gefitinib or Cisplatin by Inducing Altered Cross-Linked Hsp27 Dimers. Pharmaceutics. 2021; 13(5):630. https://doi.org/10.3390/pharmaceutics13050630
Chicago/Turabian StyleYoo, Hawon, Seul-Ki Choi, Jaeok Lee, So Hyeon Park, You Na Park, Soo-Yeon Hwang, Jae-Ho Shin, Younghwa Na, Youngjoo Kwon, Hwa Jeong Lee, and et al. 2021. "Drug-Like Small Molecule HSP27 Functional Inhibitor Sensitizes Lung Cancer Cells to Gefitinib or Cisplatin by Inducing Altered Cross-Linked Hsp27 Dimers" Pharmaceutics 13, no. 5: 630. https://doi.org/10.3390/pharmaceutics13050630