
Enhanced Bioavailability and Efficacy of Silymarin Solid Dispersion in Rats with Acetaminophen-Induced Hepatotoxicity


Abstract
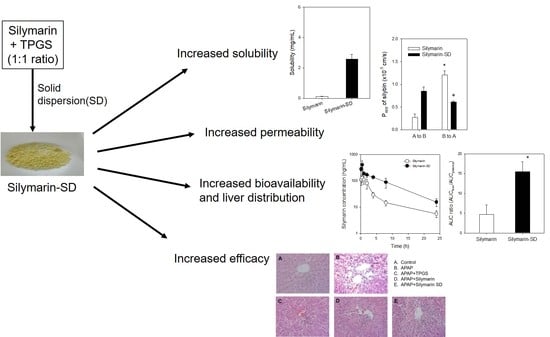
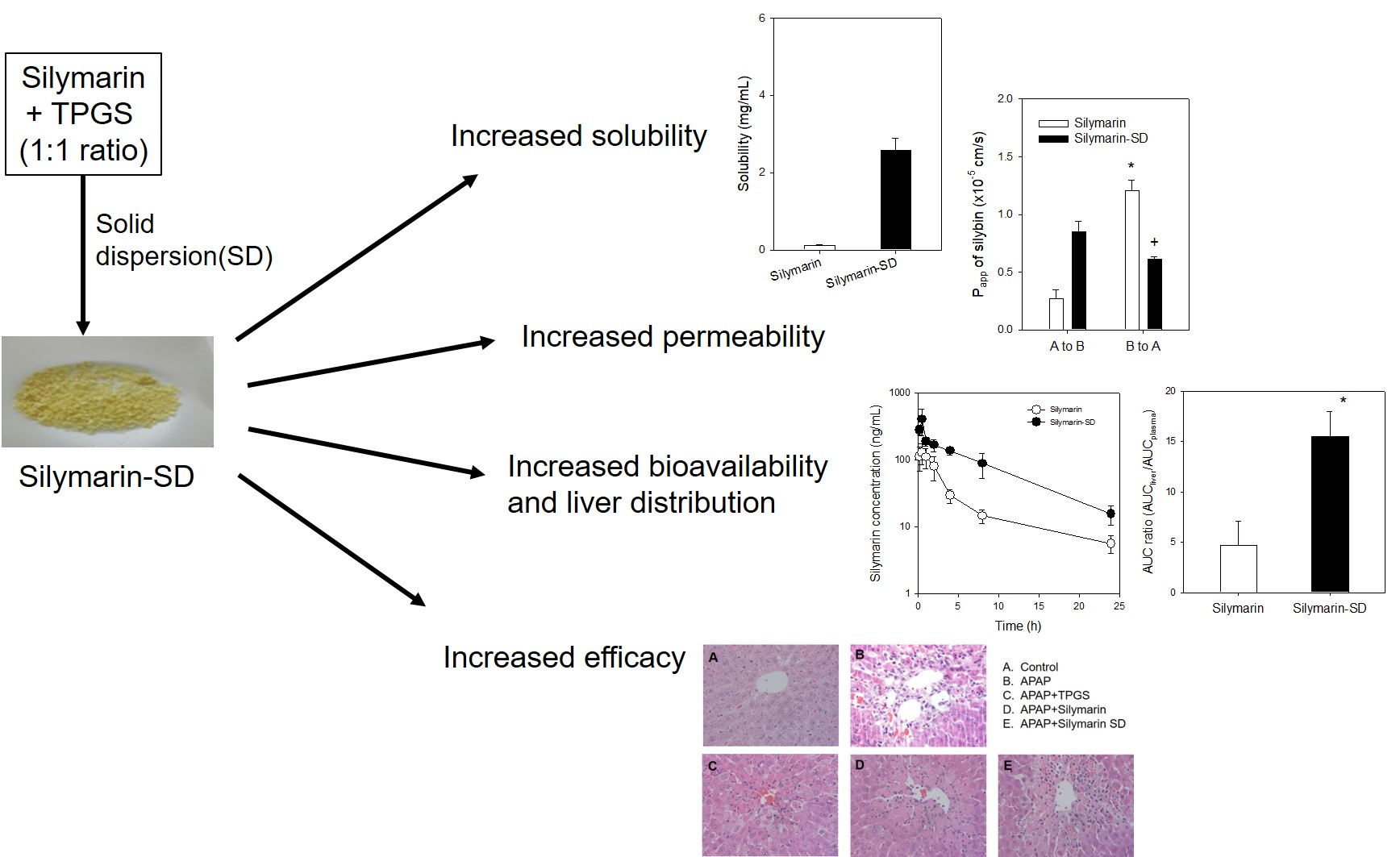
:
1. Introduction
2. Materials and Methods
2.1. Reagents
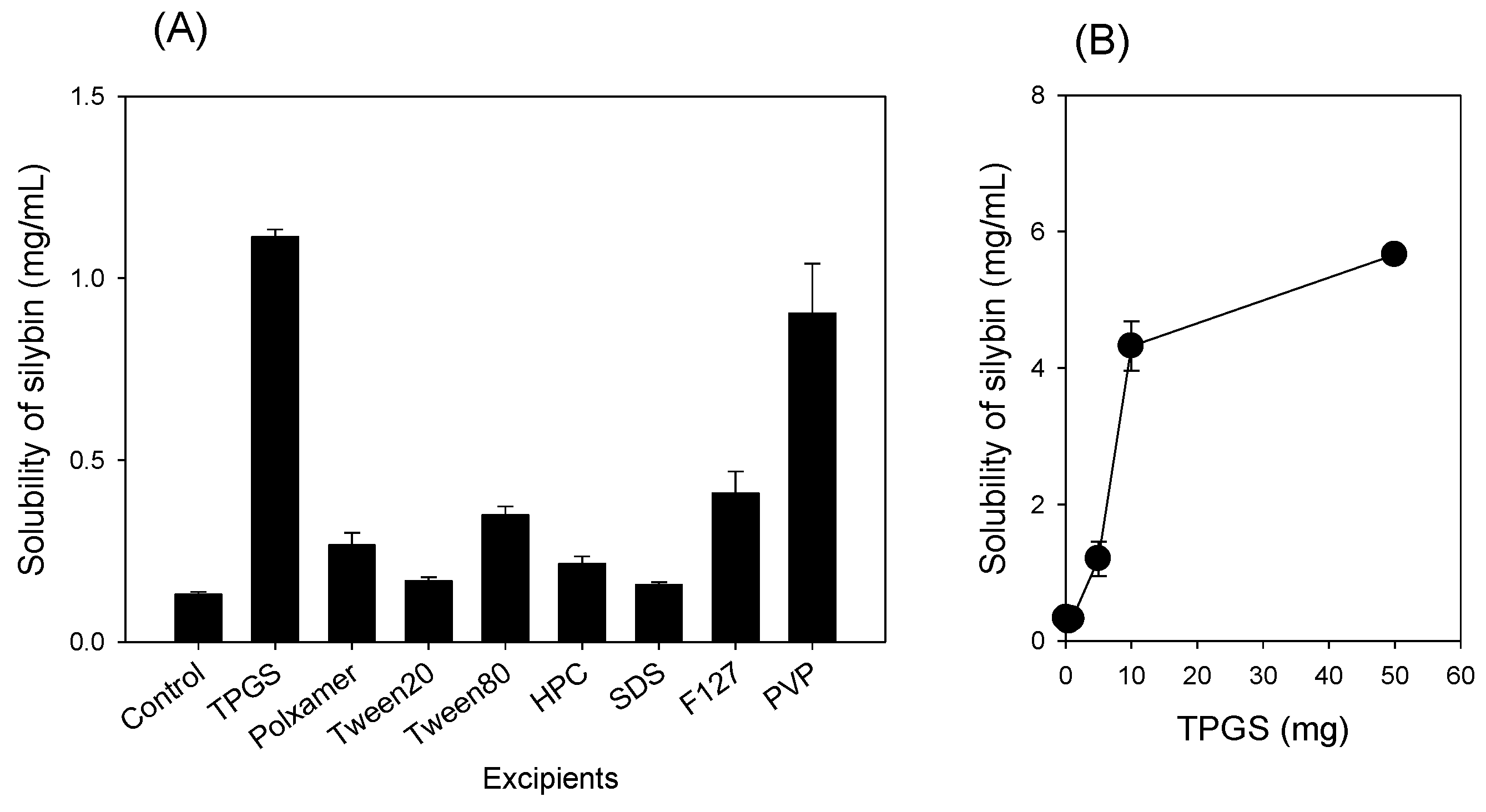
2.2. Selection of Excipients
2.3. Preparation of Silymarin-SD
2.4. Characterization of Silymarin-SD
2.5. Intestinal Permeability Study
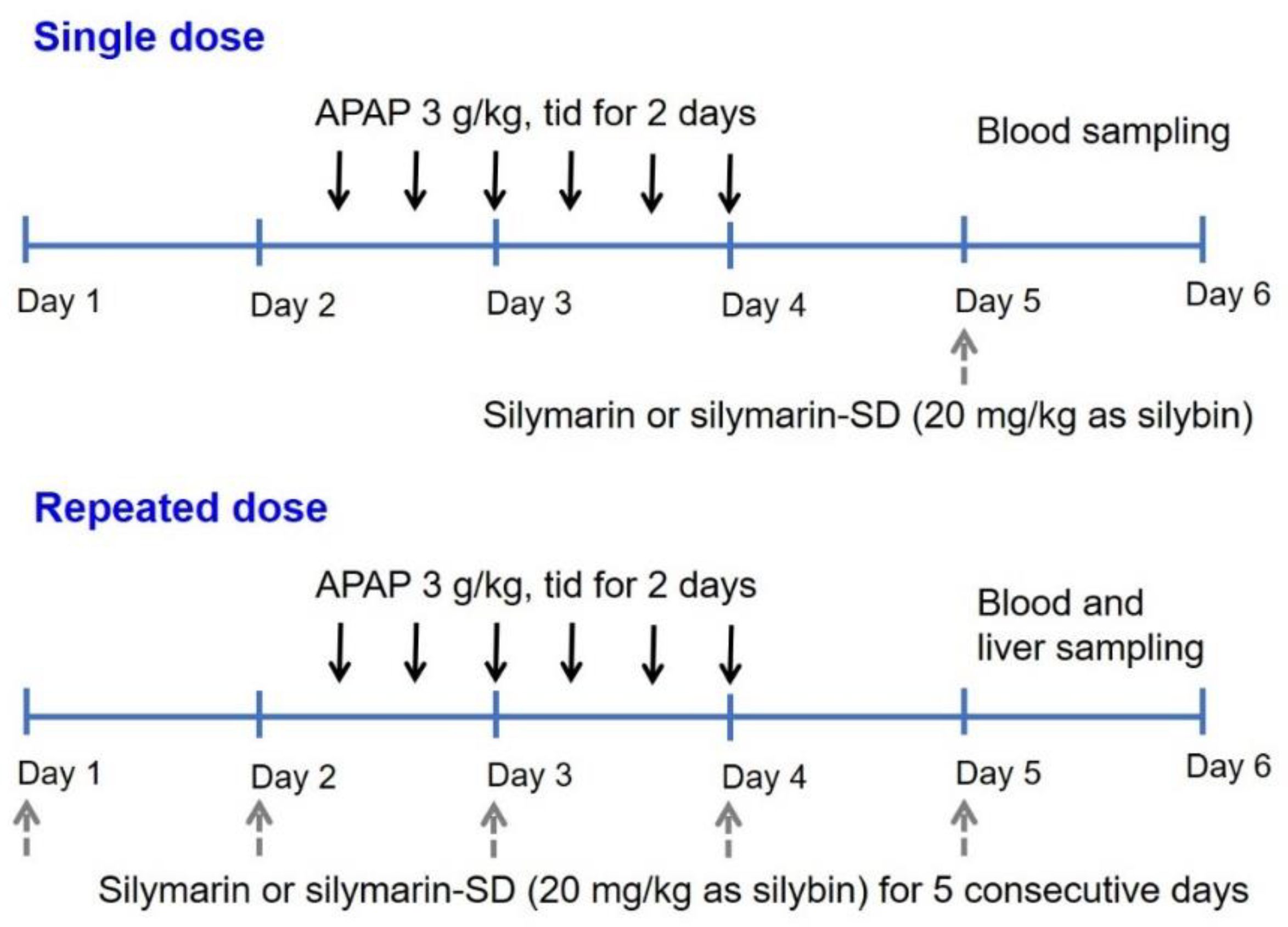
2.6. Pharmacokinetics of Silymarin or Silymarin-SD in APAP-Induced Hepatotoxic Rats
2.7. LC-MS/MS Analysis of Silybin
2.8. Efficacy of Silymarin or Silymarin-SD in APAP-Induced Hepatotoxic Rats
2.9. Data Analysis
3. Results
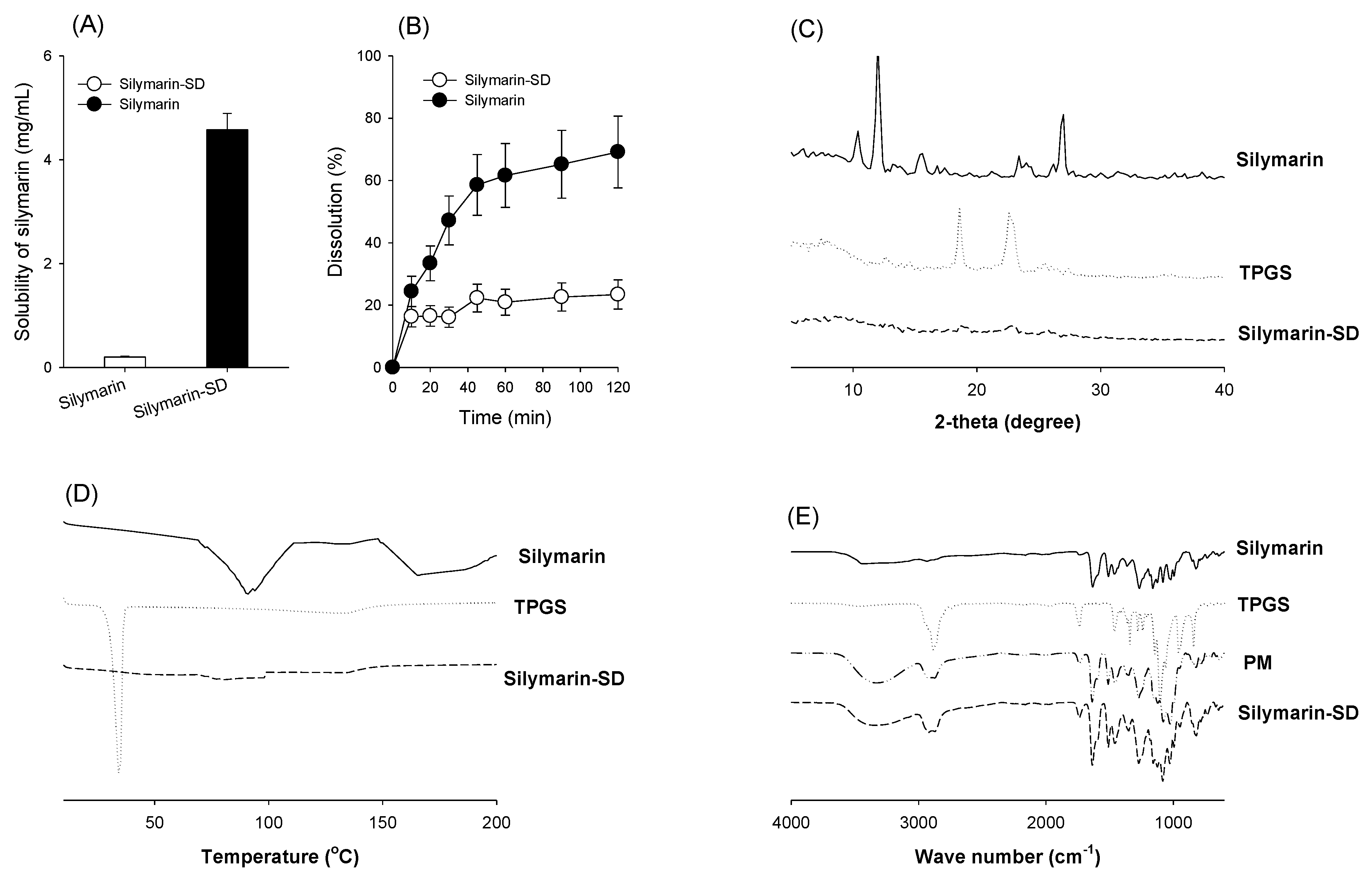
3.1. Preparation of Silymarin-SD
3.2. Characterization of Silymarin-SD
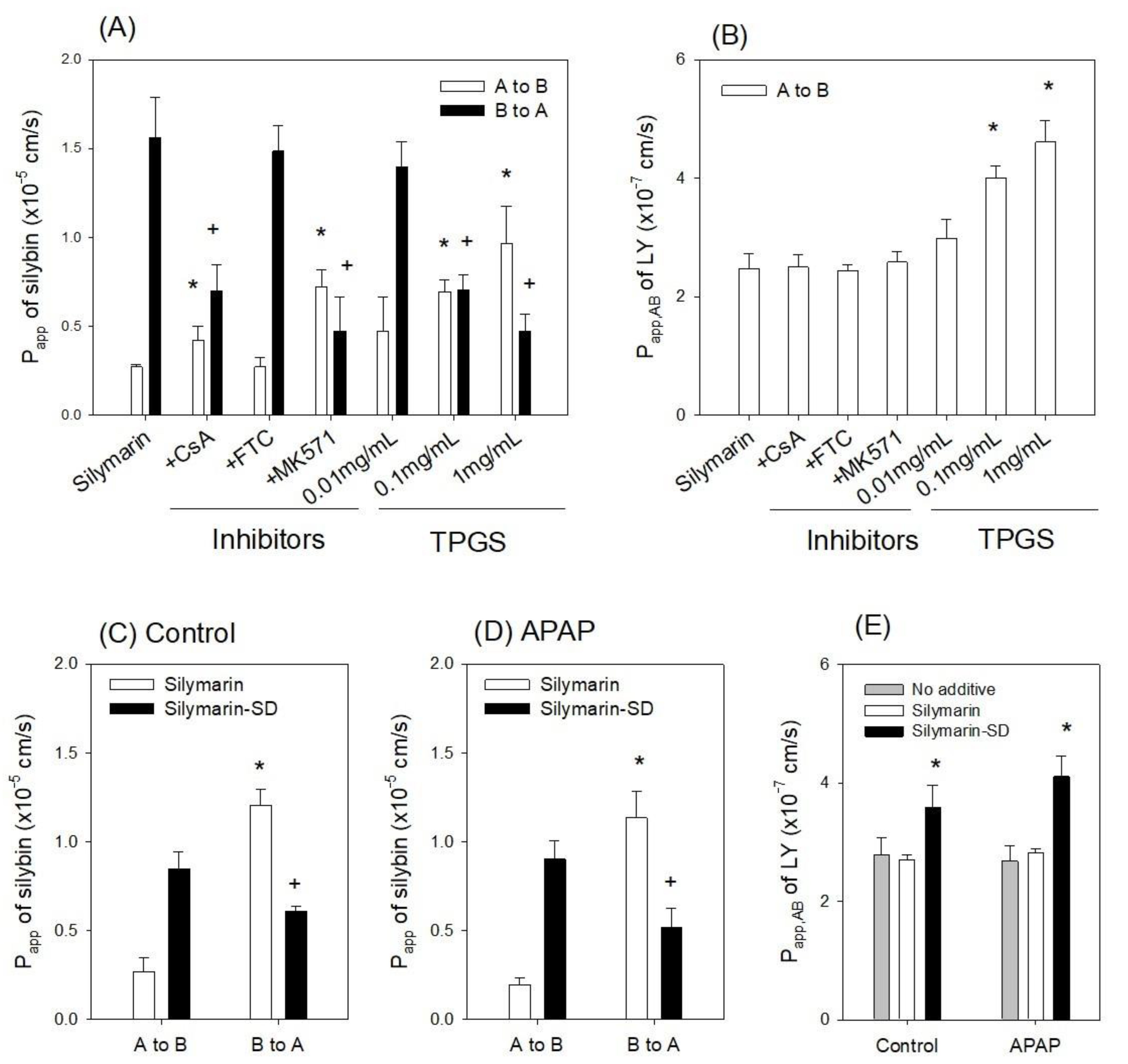
3.3. Enhanced Intestinal Permeability of Silymarin-SD
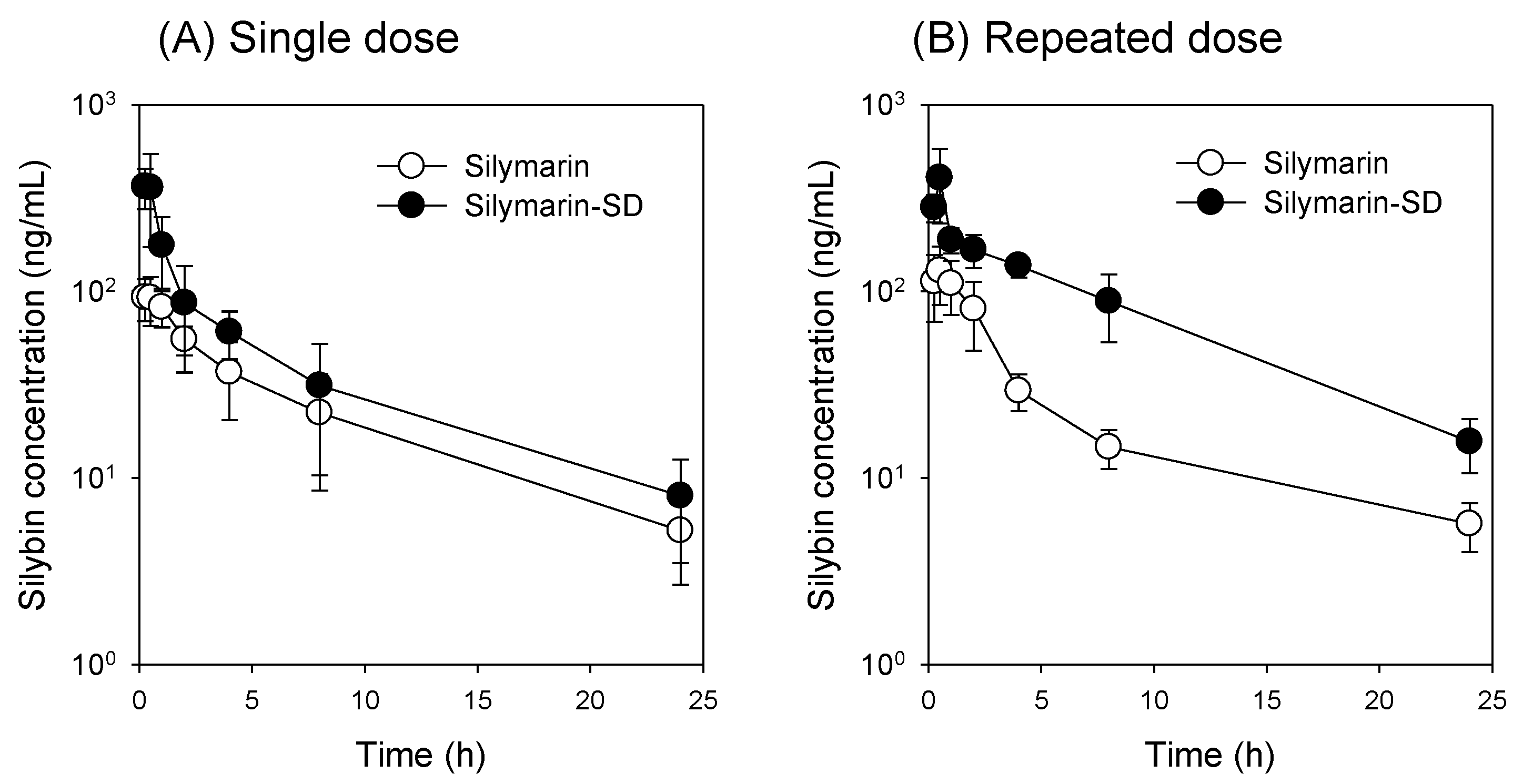
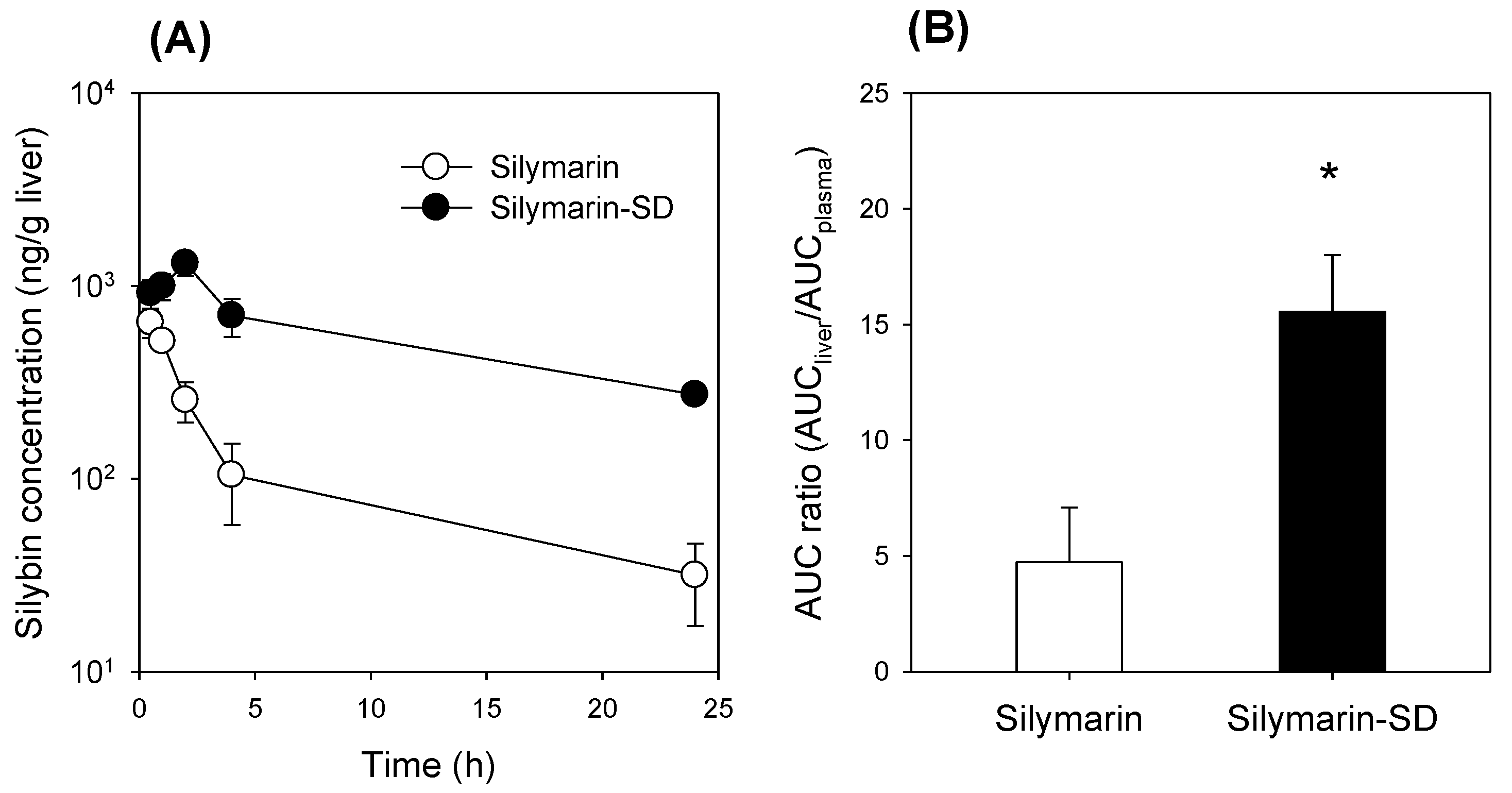
3.4. Increased Bioavailability of Silymarin-SD
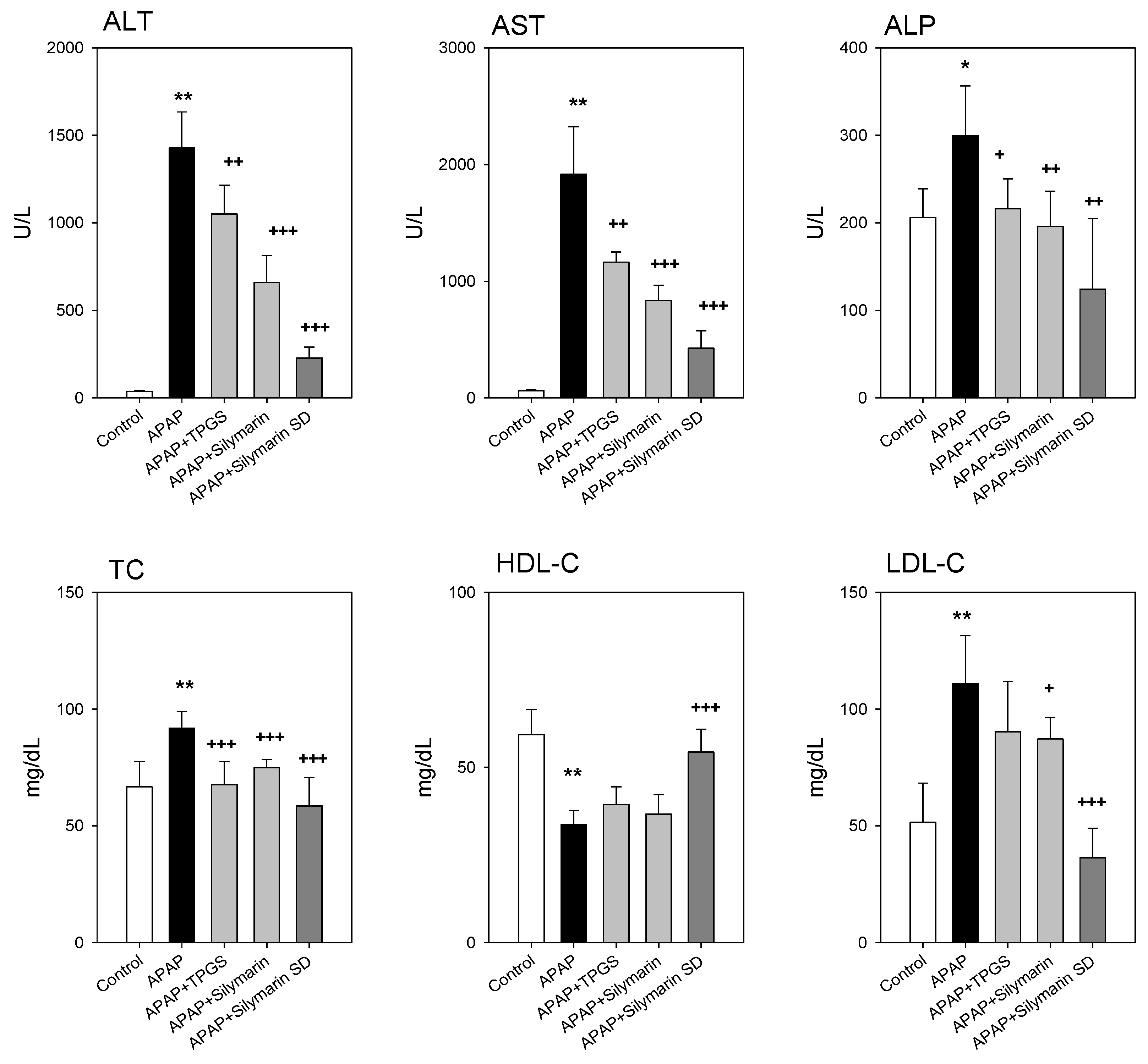
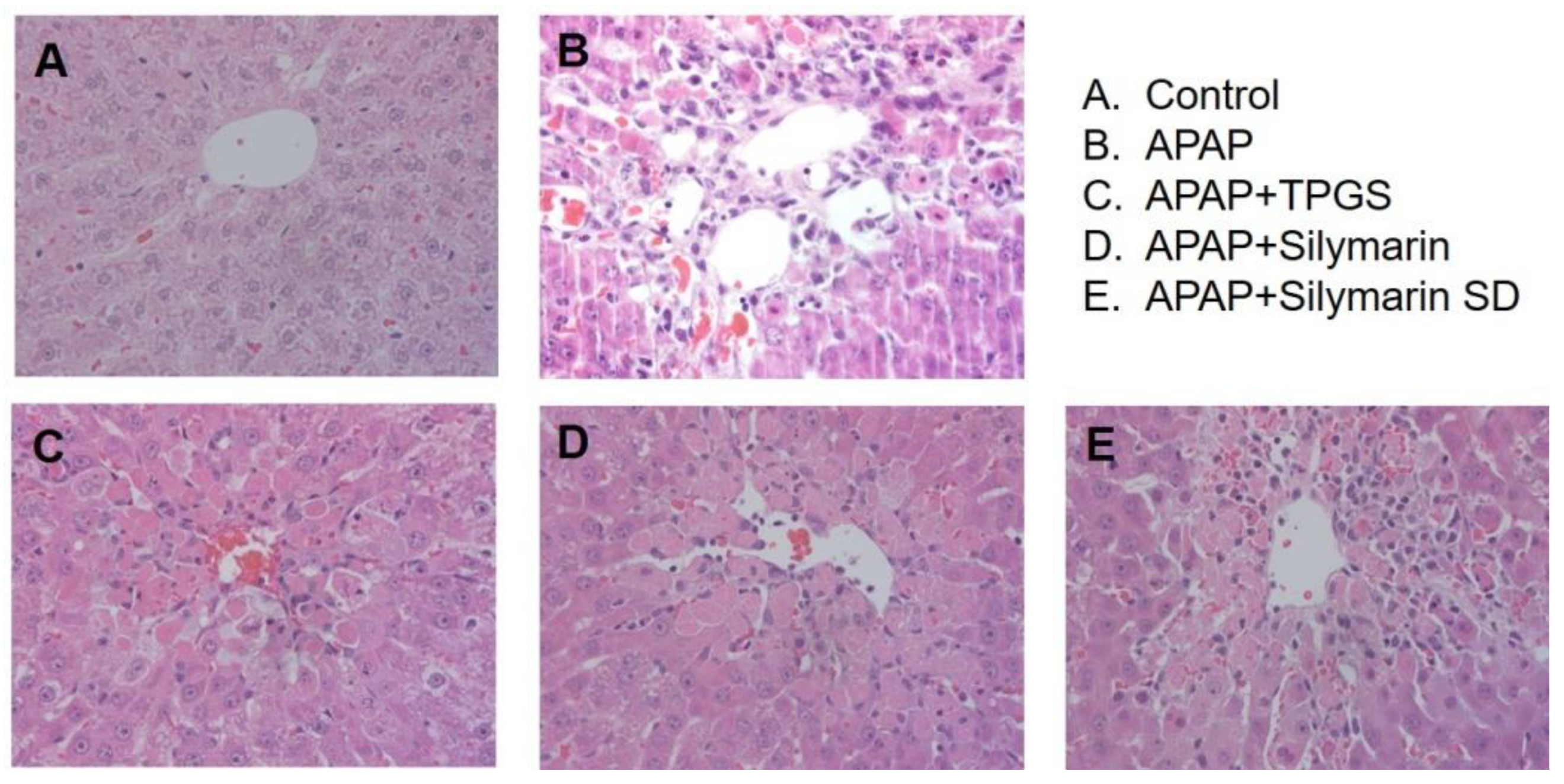
3.5. Effect of Silymarin-SD on the APAP-Induced Hepatotoxicity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schuppan, D.; Jia, J.D.; Brinkhaus, B.; Hahn, E.G. Herbal products for liver diseases: A therapeutic challenge for the new millennium. Hepatology 1999, 30, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Flora, K.; Hahn, M.; Rosen, H.; Benner, K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am. J. Gastroenterol. 1998, 93, 139–143. [Google Scholar] [CrossRef]
- Brinda, B.J.; Zhu, H.J.; Markowitz, J.S. A sensitive LC-MS/MS assay for the simultaneous analysis of the major active components of silymarin in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 902, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Saller, R.; Meier, R.; Brignoli, R. The use of silymarin in the treatment of liver diseases. Drugs 2001, 61, 2035–2063. [Google Scholar] [CrossRef]
- Dixit, N.; Baboota, S.; Kohli, K.; Ahmad, S.; Ali, J. Silymarin: A review of pharmacological aspects and bioavailability enhancement approaches. Indian J. Pharmacol. 2007, 39, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.W.; Shon, J.H.; Shin, H.J.; Park, S.J.; Yeo, C.W.; Zhou, H.H.; Song, I.S.; Shin, J.G. Effect of silymarin supplement on the pharmacokinetics of rosuvastatin. Pharm. Res. 2008, 25, 1807–1814. [Google Scholar] [CrossRef]
- Abenavoli, L.; Izzo, A.A.; Milic, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Raskovic, A.; Stilinovic, N.; Kolarovic, J.; Vasovic, V.; Vukmirovic, S.; Mikov, M. The protective effects of silymarin against doxorubicin-induced cardiotoxicity and hepatotoxicity in rats. Molecules 2011, 16, 8601–8613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, S.; Kohli, K.; Ali, M. Reassessing bioavailability of silymarin. Altern. Med. Rev. 2011, 16, 239–249. [Google Scholar] [PubMed]
- Schulz, H.U.; Schurer, M.; Krumbiegel, G.; Wachter, W.; Weyhenmeyer, R.; Seidel, G. The solubility and bioequivalence of silymarin preparations. Arzneimittelforschung 1995, 45, 61–64. [Google Scholar] [PubMed]
- Morazzoni, P.; Montalbetti, A.; Malandrino, S.; Pifferi, G. Comparative pharmacokinetics of silipide and silymarin in rats. Eur. J. Drug Metab. Pharmacokinet. 1993, 18, 289–297. [Google Scholar] [CrossRef]
- Fenclova, M.; Novakova, A.; Viktorova, J.; Jonatova, P.; Dzuman, Z.; Ruml, T.; Kren, V.; Hajslova, J.; Vitek, L.; Stranska-Zachariasova, M. Poor chemical and microbiological quality of the commercial milk thistle-based dietary supplements may account for their reported unsatisfactory and non-reproducible clinical outcomes. Sci. Rep. 2019, 9, 11118. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, D.Q.; Zhang, J.; Yuan, J.L. Metabolism, transport and drug-drug interactions of silymarin. Molecules 2019, 24, 3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Costanzo, A.; Angelico, R. Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art. Molecules 2019, 24, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrol, S.; Trehan, A.; Katare, O.P. Formulation, characterization, and in vitro evaluation of silymarin-loaded lipid microspheres. Drug Deliv. 2004, 11, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.S.; Kim, T.S.; Park, J.H.; Chi, S.C. Formulation and biopharmaceutical evaluation of silymarin using SMEDDS. Arch. Pharm. Res. 2007, 30, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, Q.; Huang, Y.; Zhou, Y.; Liu, Y. Development of silymarin self-microemulsifying drug delivery system with enhanced oral bioavailability. AAPS PharmSciTech 2010, 11, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.Y.; Hwang du, H.; Yousaf, A.M.; Kim, D.W.; Shin, Y.J.; Bae, O.N.; Kim, Y.I.; Kim, J.O.; Yong, C.S.; Choi, H.G. Silymarin-loaded solid nanoparticles provide excellent hepatic protection: Physicochemical characterization and in vivo evaluation. Int. J. Nanomed. 2013, 8, 3333–3343. [Google Scholar]
- Calligaris, S.; Comuzzo, P.; Bot, F.; Lippe, G.; Zironi, R.; Anese, M.; Nicoli, M.C. Nanoemulsions as delivery systems of hydrophobic silybin from silymarin extract: Effect of oil type on silybin solubility, in vitro bioaccessibility and stability. LWT Food Sci. Technol. 2015, 63, 77–84. [Google Scholar] [CrossRef]
- Nagi, A.; Iqbal, B.; Kumar, S.; Sharma, S.; Ali, J.; Baboota, S. Quality by design based silymarin nanoemulsion for enhancement of oral bioavailability. J. Drug Del. Sci. Technol. 2017, 40, 35–44. [Google Scholar] [CrossRef]
- Xiao, Y.Y.; Song, Y.M.; Chen, Z.P.; Ping, Q.N. Preparation of silymarin proliposome: A new way to increase oral bioavailability of silymarin in beagle dogs. Int. J. Pharm. 2006, 319, 162–168. [Google Scholar]
- Chu, C.; Tong, S.-S.; Xu, Y.; Wang, L.; Fu, M.; Ge, Y.-R.; Yu, J.-N.; Xu, X.-M. Proliposomes for oral delivery of dehydrosilymarin: Preparation and evaluation in vitro and in vivo. Acta Pharmacol. Sin. 2011, 32, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Elmowafy, M.; Viitala, T.; Ibrahim, H.M.; Abu-Elyazid, S.K.; Samy, A.; Kassem, A.; Yliperttula, M. Silymarin loaded liposomes for hepatic targeting: In vitro evaluation and HepG2 drug uptake. Eur. J. Pharm. Sci. 2013, 50, 161–171. [Google Scholar] [CrossRef]
- He, J.; Hou, S.X.; Lu, W.G.; Zhu, L.; Feng, J.F. Preparation, pharmacokinetics and body distribution of silymarin-loaded solid lipid nanoparticles after oral administration. J. Biomed. Nanotechnol. 2007, 3, 195–202. [Google Scholar] [CrossRef]
- Saller, R.; Melzer, J.; Reichling, J.; Brignoli, R.; Meier, R. An updated systematic review of the pharmacology of silymarin. Forsch Komplementmed. 2007, 14, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.W.; Li, Y.Z.; Liu, Z.Q.; Feng, S.L.; Zhou, H.; Liu, C.X.; Liu, L.; Xie, Y. Role of tangeretin as a potential bioavailability enhancer for silybin: Pharmacokinetic and pharmacological studies. Pharmacol. Res. 2018, 128, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal permeability study of clinically relevant formulations of silibinin in Caco-2 cell monolayers. Int. J. Mol. Sci. 2019, 20, 1606. [Google Scholar] [CrossRef] [Green Version]
- Sahibzada, M.U.K.; Sadiq, A.; Khan, S.; Faidah, H.S.; Naseemullah; Khurram, M.; Amin, M.U.; Haseeb, A. Fabrication, characterization and in vitro evaluation of silibinin nanoparticles: An attempt to enhance its oral bioavailability. Drug Des. Dev. Ther. 2017, 11, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousaf, A.M.; Malik, U.R.; Shahzad, Y.; Hussain, T.; Khan, I.U.; Din, F.U.; Mahmood, T.; Ahsan, H.M.; Syed, A.S.; Akram, M.R. Silymarin-laden PVP-nanocontainers prepared via the electrospraying technique for improved aqueous solubility and dissolution rate. Braz. Arch. Biol. Technol. 2019, 62, e19170754. [Google Scholar] [CrossRef]
- Song, I.S.; Cha, J.S.; Choi, M.K. Characterization, in vivo and in vitro evaluation of solid dispersion of curcumin containing d-alpha-tocopheryl polyethylene glycol 1000 succinate and mannitol. Molecules 2016, 21, 1386. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.A.; Yoon, Y.H.; Choi, K.; Kwon, M.; Goo, S.H.; Cha, J.S.; Choi, M.K.; Lee, H.S.; Song, I.S. Enhanced oral bioavailability of morin administered in mixed micelle formulation with PluronicF127 and Tween80 in rats. Biol. Pharm. Bull. 2015, 38, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Lim, D.Y.; Lee, C.H.; Jeon, J.H.; Choi, M.K.; Song, I.S. Enhanced intestinal absorption and pharmacokinetic modulation of berberine and its metabolites through the inhibition of P-glycoprotein and intestinal metabolism in rats using a berberine mixed micelle formulation. Pharmaceutics 2020, 12, 882. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, Y.-I.; Cho, K.H.; Poudel, B.K.; Choi, J.Y.; Kim, D.-W.; Shin, Y.-J.; Bae, O.-N.; Yousaf, A.M.; Yong, C.S.; et al. A novel solid dispersion system for natural product-loaded medicine: Silymarin-loaded solid dispersion with enhanced oral bioavailability and hepatoprotective activity. J. Microencapsul. 2014, 31, 619–626. [Google Scholar] [CrossRef]
- Johnson, B.M.; Charman, W.N.; Porter, C.J. An in vitro examination of the impact of polyethylene glycol 400, Pluronic P85, and vitamin E d-alpha-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS PharmSci 2002, 4, E40. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, L.Y.; Wang, B.; Ding, M.H.; Bao, Y.L.; Tan, S.W. Recent advances of D-alpha-tocopherol polyethylene glycol 1000 succinate based stimuli-responsive nanomedicine for cancer treatment. Curr. Med. Sci. 2020, 40, 218–231. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, S.; Feng, S.S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012, 33, 4889–4906. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Bridgers, A.; Polli, J.; Vickers, A.; Long, S.; Roy, A.; Winnike, R.; Coffin, M. Vitamin E-TPGS increases absorption flux of an HIV protease inhibitor by enhancing its solubility and permeability. Pharm. Res. 1999, 16, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Luiz, M.T.; Di Filippo, L.D.; Alves, R.C.; Araujo, V.H.S.; Duarte, J.L.; Marchetti, J.M.; Chorilli, M. The use of TPGS in drug delivery systems to overcome biological barriers. Eur. Polym. J. 2021, 142, 110129. [Google Scholar] [CrossRef]
- Collnot, E.M.; Baldes, C.; Schaefer, U.F.; Edgar, K.J.; Wempe, M.F.; Lehr, C.M. Vitamin E TPGS P-glycoprotein inhibition mechanism: Influence on conformational flexibility, intracellular ATP levels, and role of time and site of access. Mol. Pharm. 2010, 7, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Collnot, E.M.; Baldes, C.; Wempe, M.F.; Kappl, R.; Huttermann, J.; Hyatt, J.A.; Edgar, K.J.; Schaefer, U.F.; Lehr, C.M. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: Influence on ATPase activity and membrane fluidity. Mol. Pharm. 2007, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Dumas, T.E.; Schrieber, S.J.; Hawke, R.L.; Fried, M.W.; Smith, P.C. Pharmacokinetics and metabolic profile of free, conjugated, and total silymarin flavonolignans in human plasma after oral administration of milk thistle extract. Drug Metab. Dispos. 2008, 36, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548, vi. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.R.; Kurtz, A.; Bajt, M.L.; Hinson, J.A.; Jaeschke, H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: Role of mitochondrial oxidant stress. Toxicol. Sci. 2001, 62, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Shafey, M.M.; Abd-Allah, G.M.; Mohamadin, A.M.; Harisa, G.I.; Mariee, A.D. Quercetin protects against acetaminophen-induced hepatorenal toxicity by reducing reactive oxygen and nitrogen species. Pathophysiology 2015, 22, 49–55. [Google Scholar] [CrossRef]
- Kwon, M.; Ji, H.K.; Goo, S.H.; Nam, S.J.; Kang, Y.J.; Lee, E.; Liu, K.H.; Choi, M.K.; Song, I.S. Involvement of intestinal efflux and metabolic instability in the pharmacokinetics of platycodin D in rats. Drug Metab. Pharmacokinet. 2017, 32, 248–254. [Google Scholar] [CrossRef]
- Choi, M.K.; Kwon, M.; Ahn, J.H.; Kim, N.J.; Bae, M.A.; Song, I.S. Transport characteristics and transporter-based drug-drug interactions of TM-25659, a novel TAZ modulator. Biopharm. Drug Dispos. 2014, 35, 183–194. [Google Scholar] [CrossRef]
- Song, I.S.; Jeong, H.U.; Choi, M.K.; Kwon, M.; Shin, Y.; Kim, J.H.; Lee, H.S. Interactions between cyazofamid and human drug transporters. J. Biochem. Mol. Toxic. 2020, 34, e22459. [Google Scholar] [CrossRef]
- Choi, M.K.; Song, I.S. Interactions of ginseng with therapeutic drugs. Arch. Pharm. Res. 2019, 42, 862–878. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The applications of Vitamin E TPGS in drug delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Single Administration | Repeated Administration | ||
|---|---|---|---|---|
| Silymarin | Silymarin-SD | Silymarin | Silymarin-SD | |
| Cmax (ng/mL) | 106 ± 14.9 | 427 ± 147 * | 146 ± 48.1 | 412 ± 168 * |
| Tmax (h) | 0.56 ± 0.31 | 0.40 ± 0.14 | 0.45 ± 0.11 | 0.44 ± 0.13 |
| AUC24h (ng∙h/mL) | 578 ± 225 | 957 ± 350 * | 547 ± 131 | 2040 ± 435 *,+ |
| AUC∞ (n∙h/mL) | 634 ± 239 | 1060 ± 406 * | 587 ± 138 | 2190 ± 374 *,+ |
| T1/2 (h) | 6.77 ± 2.06 | 7.98 ± 2.80 | 6.74 ± 1.11 | 7.36 ± 1.32 |
| MRT (h) | 5.16 ± 0.47 | 7.66 ± 1.86 * | 4.88 ± 0.72 | 8.32 ± 1.82 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, I.-S.; Nam, S.-J.; Jeon, J.-H.; Park, S.-J.; Choi, M.-K. Enhanced Bioavailability and Efficacy of Silymarin Solid Dispersion in Rats with Acetaminophen-Induced Hepatotoxicity. Pharmaceutics 2021, 13, 628. https://doi.org/10.3390/pharmaceutics13050628
Song I-S, Nam S-J, Jeon J-H, Park S-J, Choi M-K. Enhanced Bioavailability and Efficacy of Silymarin Solid Dispersion in Rats with Acetaminophen-Induced Hepatotoxicity. Pharmaceutics. 2021; 13(5):628. https://doi.org/10.3390/pharmaceutics13050628
Chicago/Turabian StyleSong, Im-Sook, So-Jeong Nam, Ji-Hyeon Jeon, Soo-Jin Park, and Min-Koo Choi. 2021. "Enhanced Bioavailability and Efficacy of Silymarin Solid Dispersion in Rats with Acetaminophen-Induced Hepatotoxicity" Pharmaceutics 13, no. 5: 628. https://doi.org/10.3390/pharmaceutics13050628