
Dual Inhibition of P-gp and BCRP Improves Oral Topotecan Bioavailability in Rodents

 , ,
, ,
Abstract
:
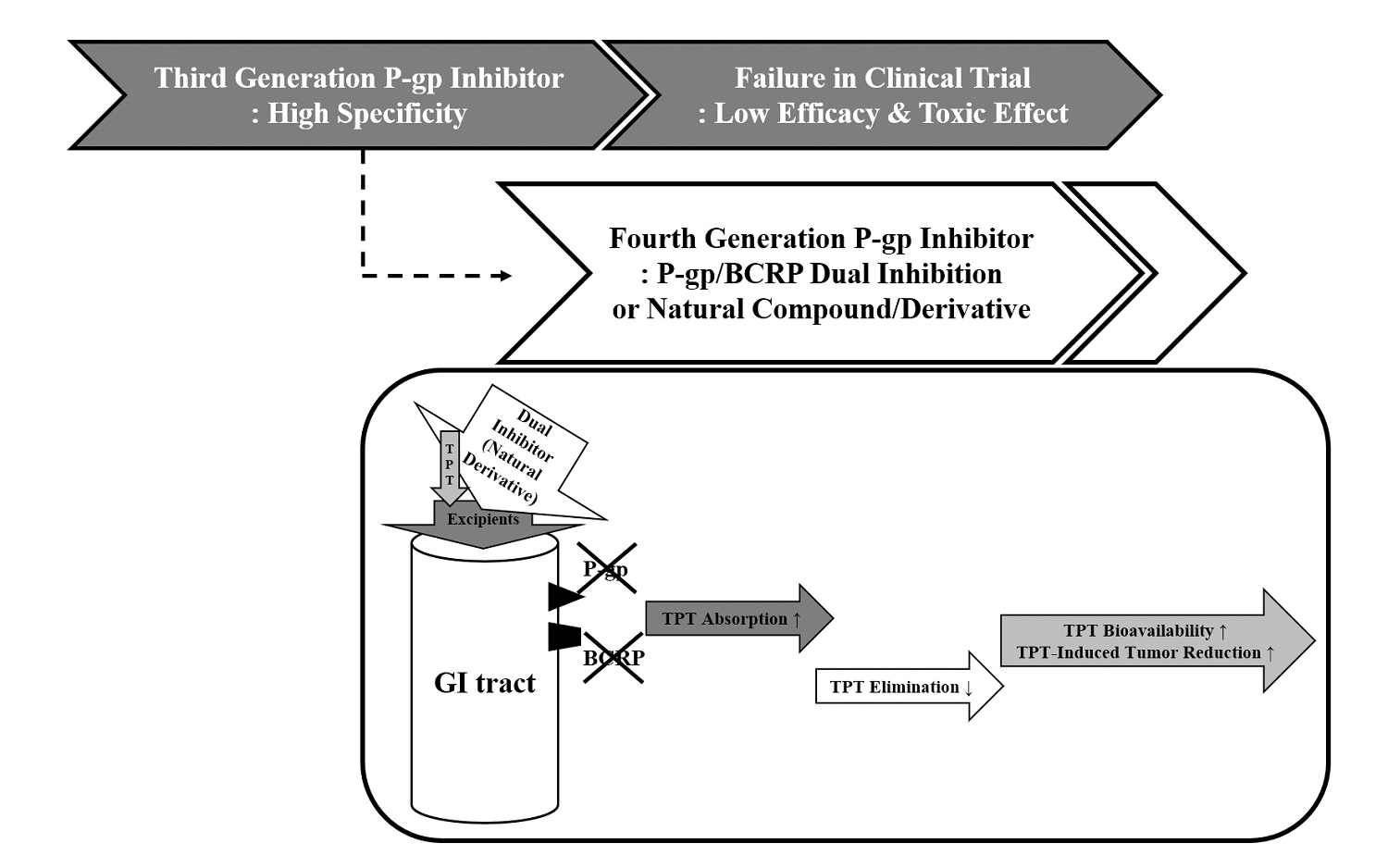
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Western Blotting Analysis
2.4. Cell Survival Study
2.5. Substrate Drug Accumulation Study by Flow Cytometry
2.6. Animal Experiments
2.6.1. Pharmacokinetic (PK) Studies
Drug Formulations
Oral Administration and Plasma Sampling
High Performance Liquid Chromatography-Fluorescence (HPLC-FC) Analysis
PK Analysis
2.6.2. Xenograft Trial
2.7. Statistical Analysis
3. Results
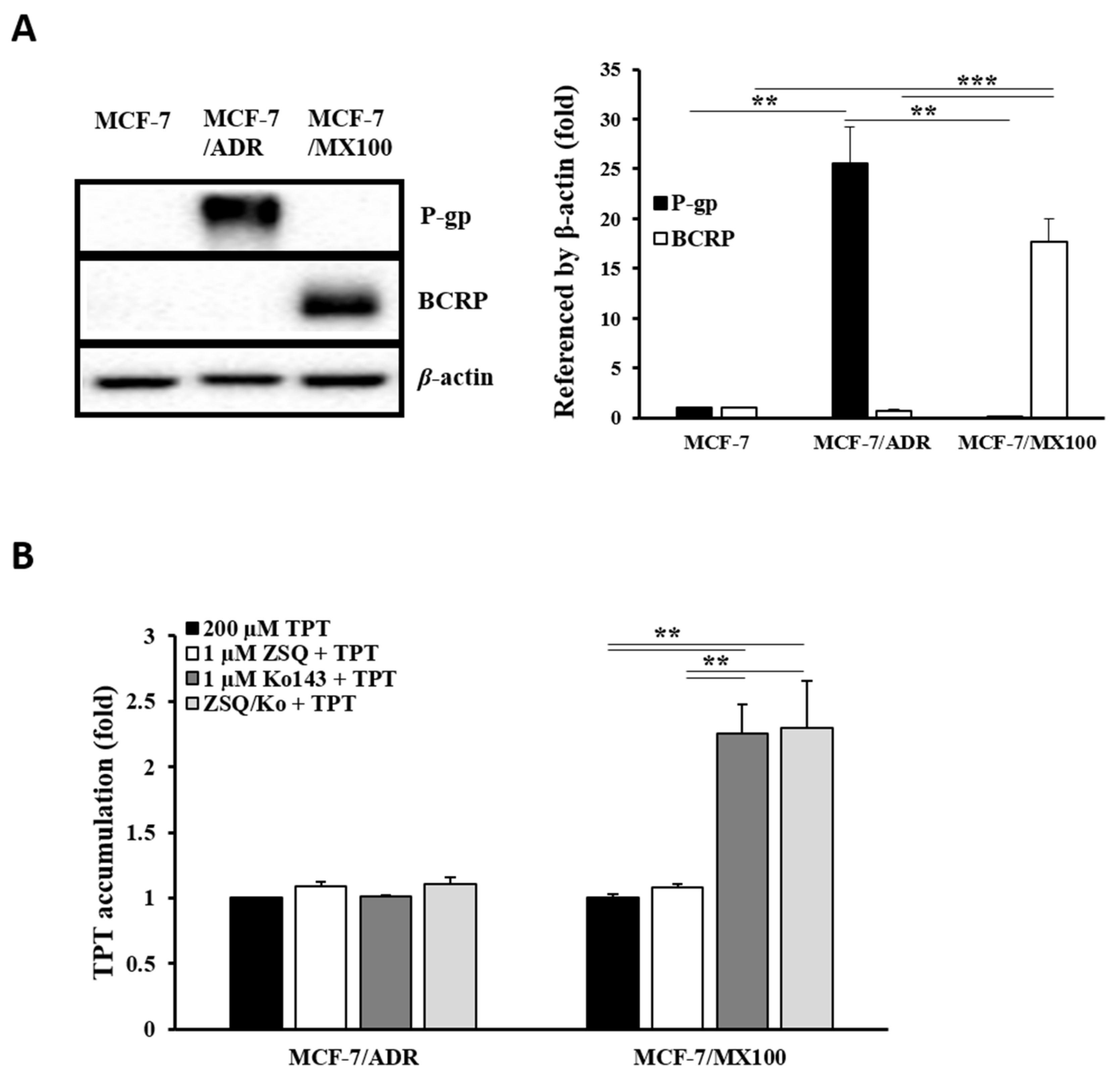
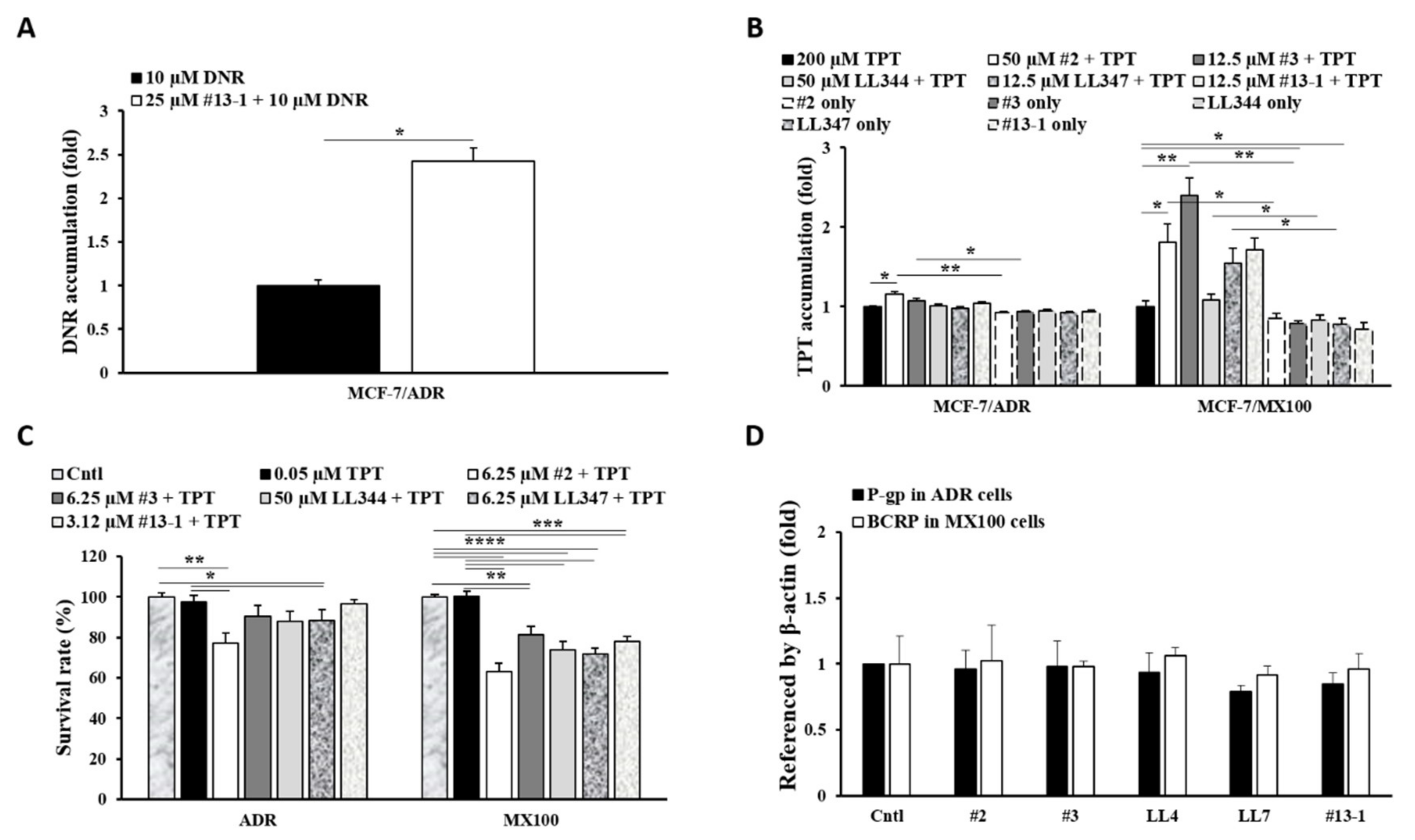
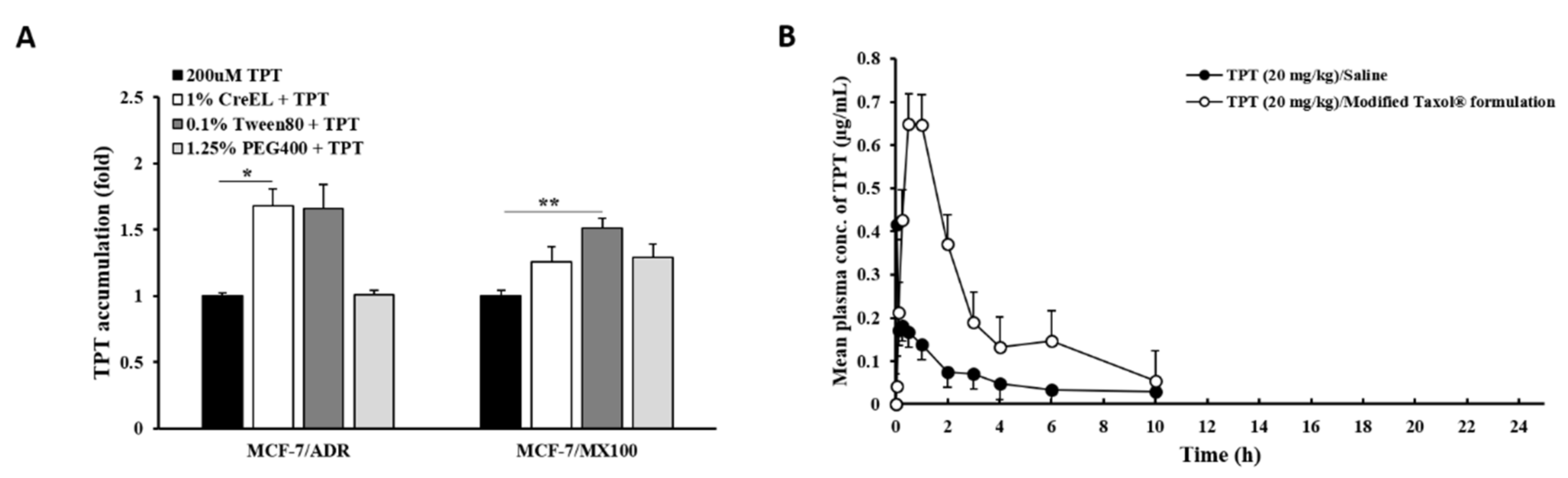
3.1. Effects of P-gp Inhibitors on BCRP In Vitro
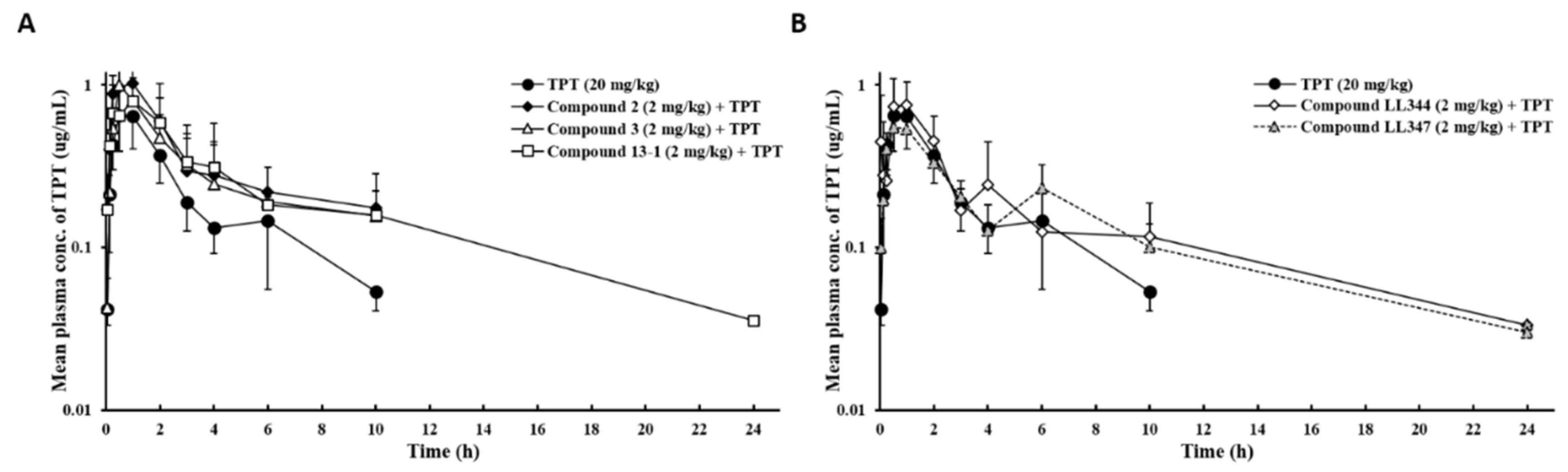
3.2. Effects of Dual Inhibitor Candidates In Vivo: Pharmacokinetics (PK) of TPT Following Oral Co-Administration
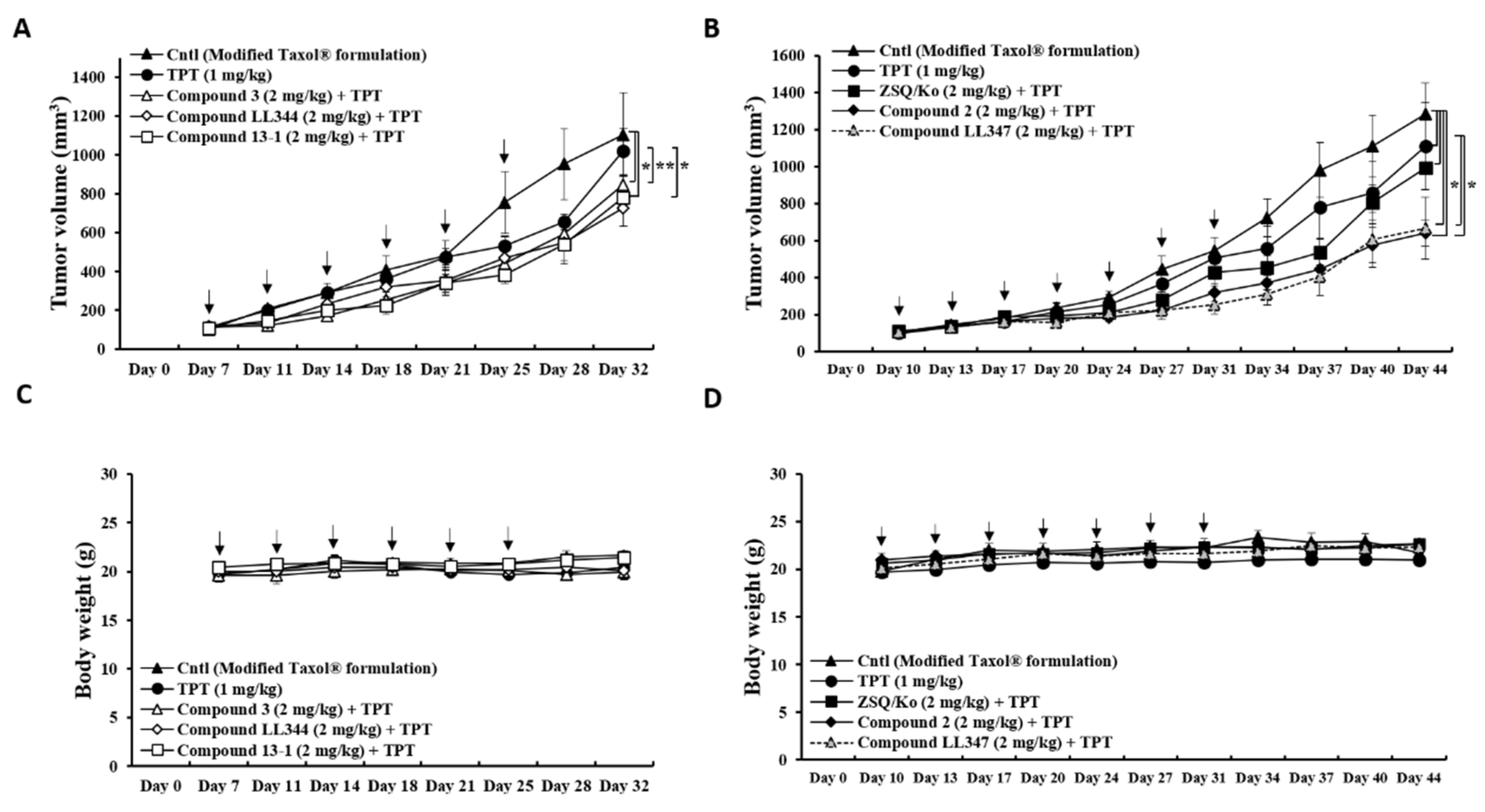
3.3. Effects of Dual Inhibitor Candidates In Vivo: Tumor Growth after Oral Co-Administration with TPT
3.4. Effects of Excipients on P-gp and BCRP Dual Inhibition In Vitro and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trowitzsch, S.; Tampe, R. ABC transporters in dynamic macromolecular assemblies. J. Mol. Biol. 2018, 430, 4481–4495. [Google Scholar]
- Liu, X. ABC Family Transporters. Adv. Exp. Med. Biol 2019, 1141, 13–100. [Google Scholar]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta Biomembr. 1976, 455, 152–162. [Google Scholar]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar]
- Dei, S.; Braconi, L.; Romanelli, M.N.; Teodori, E. Recent advances in the search of BCRP-and dual P-gp/BCRP-based multidrug resistance modulators. Cancer Drug Resist. 2019, 2, 710–743. [Google Scholar]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar]
- Durmus, S.; Hendrikx, J.J.; Schinkel, A.H. Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv. Cancer Res. 2015, 125, 1–41. [Google Scholar]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar]
- Hee Choi, Y.; Yu, A.-M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Currt. Pharm. Des. 2014, 20, 793–807. [Google Scholar]
- U.S. Food and Drug Administration, In Vitro Drug Interaction Studies-Cytochrome P450 Enzymes- and Transporter-Mediated Drug Interactions Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 24 August 2020).
- Palmeira, A.; Sousa, E.; H. Vasconcelos, M.; M. Pinto, M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar]
- Chung, F.S.; Santiago, J.S.; De Jesus, M.F.M.; Trinidad, C.V.; See, M.F.E. Disrupting P-glycoprotein function in clinical settings: What can we learn from the fundamental aspects of this transporter? Am. J. Cancer Res. 2016, 6, 1583. [Google Scholar]
- Waghray, D.; Zhang, Q. Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar]
- Peña-Solórzano, D.; Stark, S.A.; König, B.; Sierra, C.A.; Ochoa-Puentes, C. ABCG2/BCRP: Specific and nonspecific modulators. Med. Res. Rev. 2017, 37, 987–1050. [Google Scholar]
- Dong, J.; Qin, Z.; Zhang, W.D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R., Jr.; Chen, Z.S.; Cheng, X.D.; Qin, J.J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updat. 2020, 49, 100681. [Google Scholar]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved brain penetration and antitumor efficacy of temozolomide by inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar]
- Wu, C.-P.; Lusvarghi, S.; Wang, J.-C.; Hsiao, S.-H.; Huang, Y.-H.; Hung, T.-H.; Ambudkar, S.V. The selective class IIa histone deacetylase inhibitor TMP195 resensitizes ABCB1- and ABCG2-overexpressing multidrug-resistant cancer cells to cytotoxic anticancer drugs. Int. J. Mol. Sci. 2019, 21, 238. [Google Scholar]
- Al-Ali, A.A.A.; Nielsen, R.B.; Steffansen, B.; Holm, R.; Nielsen, C.U. Nonionic surfactants modulate the transport activity of ATP-binding cassette (ABC) transporters and solute carriers (SLC): Relevance to oral drug absorption. Int. J. Pharm. 2019, 566, 410–433. [Google Scholar]
- Zou, L.; Pottel, J.; Khuri, N.; Ngo, H.X.; Ni, Z.; Tsakalozou, E.; Warren, M.S.; Huang, Y.; Shoichet, B.K.; Giacomini, K.M. Interactions of oral molecular excipients with breast cancer resistance protein, BCRP. Mol. Pharm. 2020, 17, 748–756. [Google Scholar]
- Saraf, S.; Jain, A.; Hurkat, P.; Jain, S.K. Topotecan liposomes: A visit from a molecular to a therapeutic platform. Crit Rev. Ther Drug Carr. Syst. 2016, 33, 401–432. [Google Scholar]
- Herben, V.M.; ten Bokkel Huinink, W.W.; Beijnen, J.H. Clinical pharmacokinetics of topotecan. Clin. Pharm. 1996, 31, 85–102. [Google Scholar]
- Breedveld, P.; Beijnen, J.H.; Schellens, J.H. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol. Sci. 2006, 27, 17–24. [Google Scholar]
- Yamagata, T.; Kusuhara, H.; Morishita, M.; Takayama, K.; Benameur, H.; Sugiyama, Y. Improvement of the oral drug absorption of topotecan through the inhibition of intestinal xenobiotic efflux transporter, breast cancer resistance protein, by excipients. Drug Metab. Dispos. 2007, 35, 1142–1148. [Google Scholar]
- Shen, J.; Carcaboso, A.M.; Hubbard, K.E.; Tagen, M.; Wynn, H.G.; Panetta, J.C.; Waters, C.M.; Elmeliegy, M.A.; Stewart, C.F. Compartment-specific roles of ATP-binding cassette transporters define differential topotecan distribution in brain parenchyma and cerebrospinal fluid. Cancer Res. 2009, 69, 5885–5892. [Google Scholar]
- Ling, X.; Liu, X.; Zhong, K.; Smith, N.; Prey, J.; Li, F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am. J. Transl. Res. 2015, 7, 1765–1781. [Google Scholar]
- Lee, K.; Chae, S.W.; Xia, Y.; Kim, N.H.; Kim, H.J.; Rhie, S.; Lee, H.J. Effect of coumarin derivative-mediated inhibition of P-glycoprotein on oral bioavailability and therapeutic efficacy of paclitaxel. Eur. J. Pharmacol. 2014, 723, 381–388. [Google Scholar]
- Chae, S.W.; Han, A.R.; Park, J.H.; Rhie, J.Y.; Lim, H.J.; Seo, E.K.; Lee, H.J. In vitro and in vivo evaluation of phenylbutenoid dimers as inhibitors of P-glycoprotein. J. Nat. Prod. 2013, 76, 2277–2281. [Google Scholar]
- Chae, S.W.; Woo, S.; Park, J.H.; Kwon, Y.; Na, Y.; Lee, H.J. Xanthone analogues as potent modulators of intestinal P-glycoprotein. Eur. J. Med. Chem. 2015, 93, 237–245. [Google Scholar]
- Chae, S.W.; Lee, J.; Park, J.H.; Kwon, Y.; Na, Y.; Lee, H.J. Intestinal P-glycoprotein inhibitors, benzoxanthone analogues. J. Pharm. Pharmacol 2018, 70, 234–241. [Google Scholar]
- Lee, J.; Chae, S.W.; Oh, A.R.; Yoo, J.H.; Park Choo, H.Y.; Rhie, S.J.; Lee, H.J. Effects of piperazine derivative on paclitaxel pharmacokinetics. Pharmaceutics 2019, 11, 23–31. [Google Scholar]
- Lee, J.; Chae, S.W.; Ma, L.; Lim, S.Y.; Alnajjar, S.; Park Choo, H.Y.; Lee, H.J.; Rhie, S.J. Pharmacokinetic ateration of paclitaxel by ferulic acid derivative. Pharmaceutics 2019, 11, 593–603. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar]
- US Department of Health and Human Services, Food and Drug Administration Center, Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.dov/media/70858/download (accessed on 24 August 2020).
- Hu, T.; Li, Z.; Gao, C.-Y.; Cho, C.H. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J. Gastroenterol 2016, 22, 6876–6889. [Google Scholar]
- Takano, M.; Yumoto, R.; Murakmi, T. Expression and function of efflux drug transporters in the intestine. Pharmacol. Ther. 2006, 109, 137–161. [Google Scholar]
- Matsuda, Y.; Konno, Y.; Hashimoto, T.; Nagai, M.; Taguchi, T.; Satsukawa, M.; Yamashita, S. In Vivo assessment of the impact of efflux transporter on oral drug absorption using portal vein–cannulated rats. Drug Metab. Dispos. 2013, 41, 1514–1521. [Google Scholar]
- Park, J.H.; Park, J.H.; Hur, H.J.; Woo, J.S.; Lee, H.J. Effects of silymarin and formulation on the oral bioavailability of paclitaxel in rats. Eur. J. Pharm. Sci. 2012, 45, 296–301. [Google Scholar]
- Thelen, K.; Dressman, J.B. Cytochrome P450-mediated metabolism in the human gut wall. J. Pharm. Pharmacol. 2009, 61, 541–558. [Google Scholar]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar]
- Prescribing Information of HYCAMTIN® (Topotecan) Capsule. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/020981lbl.pdf (accessed on 12 August 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PK Parameters | Oral Administration | |||||
|---|---|---|---|---|---|---|
| TPT | #2 + TPT | #3 + TPT | LL344 + TPT | LL347 + TPT | #13-1 + TPT | |
| Cmax (μg/mL) | 0.727 ± 0.265 | 1.14 ± 0.215 | 1.02 ± 0.266 | 0.832 ± 0.266 | 0.604 ± 0.189 | 0.938 ± 0.375 |
| Tmax (h) | (0.5–1.0)) | (0.5–1.0) | (0.5–1.0) | (0.5–1.0) | (0.5–2.0) | (0.25–2.0) |
| AUCINF (μg·h/mL) | 2.36 ± 0.543 | 4.86 ± 2.02 | 3.97 ± 1.71 | 3.82 ± 2.01 | 3.33 ± 0.365 | 4.90 ± 2.63 |
| t1/2 (h) | 3.39 ± 1.09 | 4.88 ± 2.70 | 4.06 ± 2.41 | 4.40 ± 1.91 | 5.95 ± 1.63 | 6.80 ± 5.61 |
| Vd/F (L) | 42.1 ± 14.1 | 29.9 ± 14.8 | 27.2 ± 8.93 | 37.2 ± 16.6 | 51.1 ± 11.1 | 41.3 ± 30.3 |
| Clt/F (L/h) | 8.94 ± 2.58 | 4.85 ± 2.22 | 6.52 ± 4.71 | 6.34 ± 3.01 | 6.06 ± 0.671 | 5.14 ± 2.87 |
| RB (%) | 100.0 | 205.7 ± 86.7 | 168.2 ± 70.5 | 161.6 ± 85.2 | 140.8 ± 15.4 | 207.2 ± 99.4 |
| PK Parameters | Oral Administration | |
|---|---|---|
| TPT in Saline | TPT in Modified Taxol® Formulation 1 | |
| Cmax (μg/mL) | 0.214 ± 0.141 | 0.727 ± 0.265 ** |
| Tmax (h) | (0.05–0.5) | (0.5–1.0) |
| AUCINF (μg·h/mL) | 0.611 ± 0.209 | 2.36 ± 0.543 ** |
| t1/2 (h) | 3.06 ± 1.34 | 3.39 ± 1.09 |
| Vd/F (L) | 155.2 ± 79.9 | 42.1 ± 14.1 ** |
| Clt/F (L/h) | 35.9 ± 11.4 | 8.94 ± 2.58 ** |
| RB (%) | 100.0 | 387.0 ± 88.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kang, J.; Kwon, N.-Y.; Sivaraman, A.; Naik, R.; Jin, S.-Y.; Oh, A.R.; Shin, J.-H.; Na, Y.; Lee, K.; et al. Dual Inhibition of P-gp and BCRP Improves Oral Topotecan Bioavailability in Rodents. Pharmaceutics 2021, 13, 559. https://doi.org/10.3390/pharmaceutics13040559
Lee J, Kang J, Kwon N-Y, Sivaraman A, Naik R, Jin S-Y, Oh AR, Shin J-H, Na Y, Lee K, et al. Dual Inhibition of P-gp and BCRP Improves Oral Topotecan Bioavailability in Rodents. Pharmaceutics. 2021; 13(4):559. https://doi.org/10.3390/pharmaceutics13040559
Chicago/Turabian StyleLee, Jaeok, Jiyeon Kang, Na-Yun Kwon, Aneesh Sivaraman, Ravi Naik, So-Young Jin, A. Reum Oh, Jae-Ho Shin, Younghwa Na, Kyeong Lee, and et al. 2021. "Dual Inhibition of P-gp and BCRP Improves Oral Topotecan Bioavailability in Rodents" Pharmaceutics 13, no. 4: 559. https://doi.org/10.3390/pharmaceutics13040559