
Developing pH-Modulated Spray Dried Amorphous Solid Dispersion of Candesartan Cilexetil with Enhanced In Vitro and In Vivo Performance


Abstract
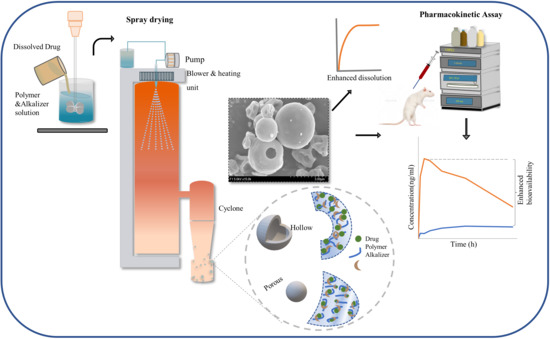
:
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Saturation Solubility Studies of CC
2.3. Screening of Polymer Carrier and Alkalizers
2.4. Supersaturation Stabilization Assessment
2.5. Preparation of ASD
2.6. UV-VIS Spectroscopy
2.7. Optimization of CCSDpM
2.7.1. Aqueous Saturated Solubility and In Vitro Dissolution Study
2.7.2. Moisture Content Measurement
2.8. Characterization of CCSDpM
2.8.1. Drug Content Assay
2.8.2. External Morphology
2.8.3. Differential Scanning Calorimetry (DSC)
2.8.4. X-ray Diffraction (XRD)
2.8.5. Fourier Transform Infrared Spectroscopy (FTIR)
2.9. Stability Assessment
2.10. Pharmacokinetic Study
2.10.1. Animal Handling and Blood Sampling
2.10.2. Plasma Sample Analysis
2.10.3. Statistical Evaluation
3. Results and Discussion
3.1. Saturation Solubility and Screening Studies
3.2. Optimization of CCSDpM Formulations
3.3. Assessment of Selected CCSDpM Formulations
3.4. Physiochemical Characterization
3.5. Stability Studies
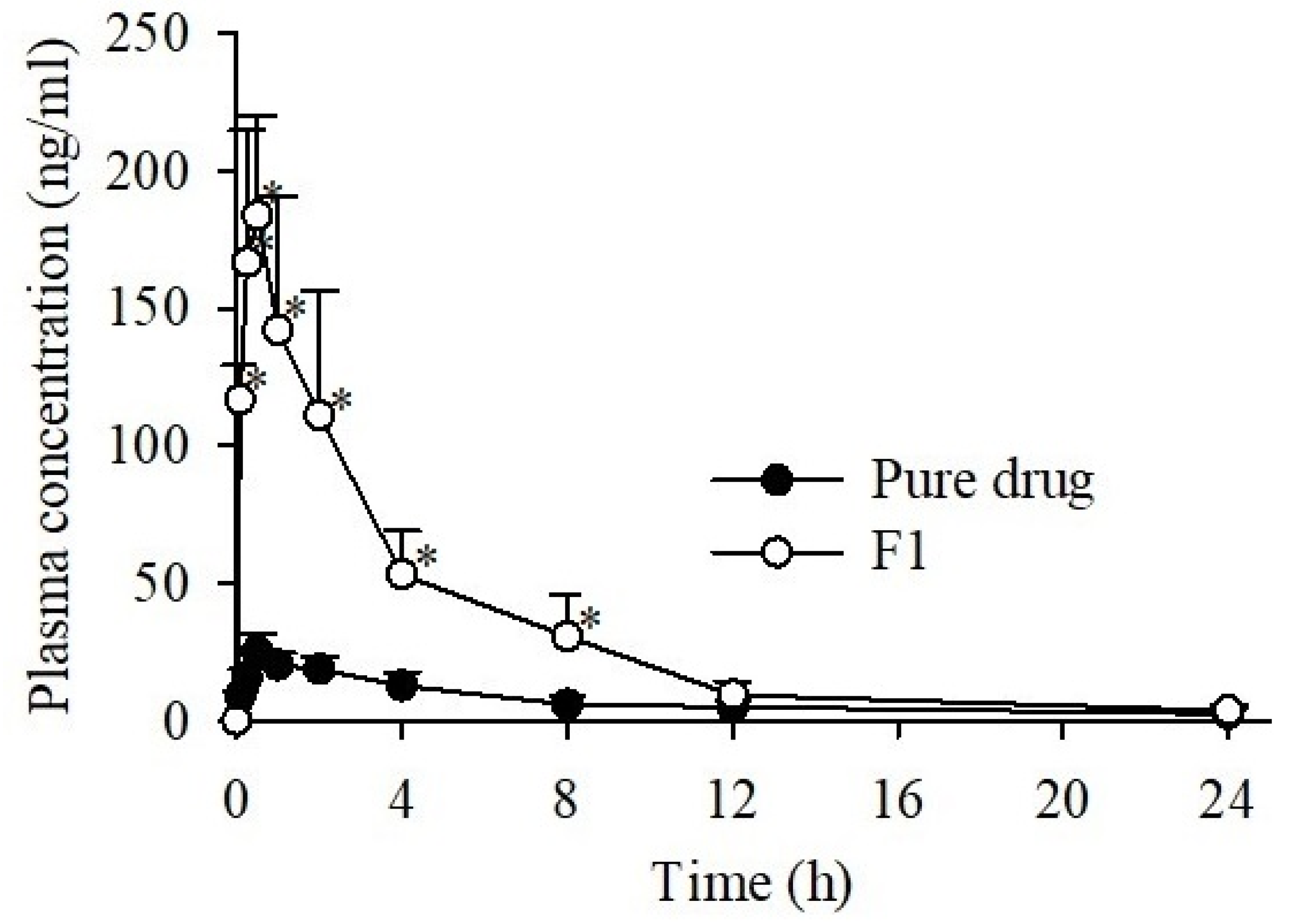
3.6. Pharmacokinetic Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Hauss, D.J. Oral lipid-based formulations. Adv. Drug Deliv. Rev. 2007, 59, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, P.; Sandhu, H.; Singhal, D.; Malick, W.; Shah, N.; Serpil Kislalioglu, M. Nanoparticles in the Pharmaceutical Industry and the Use of Supercritical Fluid Technologies for Nanoparticle Production. Curr. Drug Deliv. 2012, 9, 269–284. [Google Scholar] [CrossRef]
- Ali, H.H.; Hussein, A.A. Oral solid self-nanoemulsifying drug delivery systems of candesartan citexetil: Formulation, characterization and in vitro drug release studies. AAPS Open 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Cho, J.H.; Kim, D.S.; Kim, J.S.; Din, F.U.; Kim, J.O.; Yong, C.S.; Youn, Y.S.; Oh, K.T.; Kim, D.W.; et al. Revaprazan-loaded surface-modified solid dispersion: Physicochemical characterization and in vivo evaluation. Pharm. Dev. Technol. 2019, 24, 788–793. [Google Scholar] [CrossRef]
- Kwon, J.; Giri, B.R.; Song, E.S.; Bae, J.; Lee, J.; Kim, D.W. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.Q.M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Weuts, I.; Kempen, D.; Verreck, G.; Decorte, A.; Heymans, K.; Peeters, J.; Brewster, M.; Mooter, G. Van Den Study of the physicochemical properties and stability of solid dispersions of loperamide and PEG6000 prepared by spray drying. Eur. J. Pharm. Biopharm. 2005, 59, 119–126. [Google Scholar] [CrossRef]
- Ali, H.H.; Hussein, A.A. Oral nanoemulsions of candesartan cilexetil: Formulation, characterization and in vitro drug release studies. AAPS Open 2017, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Loftsson, T. Drug solubilization by complexation. Int. J. Pharm. 2017, 531, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-D.; Kim, H.-K.; Seo, K.-H.; Lee, W.S.; Lee, G.-S.; Woo, J.-S.; Yong, C.-S.; Choi, H.-G. The Physicochemical Properties, in Vitro Metabolism and Pharmacokinetics of a Novel Ester Prodrug of EXP3174. Mol. Pharm. 2010, 7, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, Part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Van den Mooter, G. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef]
- Marsac, P.J.; Li, T.; Taylor, L.S. Estimation of drug-polymer miscibility and solubility in amorphous solid dispersions using experimentally determined interaction parameters. Pharm. Res. 2009, 26, 139–151. [Google Scholar] [CrossRef]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. i. non-crystalline copolymers. J. Appl. Chem. 2007, 2, 493–500. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids Below Their Glass Transition Temperatures. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 799–806. [Google Scholar]
- BASF Kollidon. Polyvinylpyrrolidone Excipients; BASF: Ludwigshafen, Germany, 2008; pp. 1–331. [Google Scholar]
- Taniguchi, C.; Kawabata, Y.; Wada, K.; Yamada, S.; Onoue, S. Microenvironmental pH-modification to improve dissolution behavior and oral absorption for drugs with pH-dependent solubility. Expert Opin. Drug Deliv. 2014, 11, 505–516. [Google Scholar] [CrossRef]
- Farag Badawy, S.I.; Hussain, M.A. Microenvironmental pH modulation in solid dosage forms. J. Pharm. Sci. 2007, 96, 948–959. [Google Scholar] [CrossRef]
- McClellan, K.J.; Goa, K.L. Candesartan cilexetil. A review of its use in essential hypertension. Drugs 1998, 56, 847–869. [Google Scholar] [CrossRef]
- Sharma, G.; Beg, S.; Thanki, K.; Katare, O.P.; Jain, S.; Kohli, K.; Singh, B. Systematic development of novel cationic self-nanoemulsifying drug delivery systems of candesartan cilexetil with enhanced biopharmaceutical performance. RSC Adv. 2015, 5, 71500–71513. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, F.; Bu, H.; Xiao, J.; Li, Y. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: In vitro characteristics and absorption mechanism in rats. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 740–747. [Google Scholar] [CrossRef]
- Surampalli, G.; Nanjwade, B.K.; Patil, P.A.; Chilla, R. Novel tablet formulation of amorphous candesartan cilexetil solid dispersions involving P-gp inhibition for optimal drug delivery: In vitro and in vivo evaluation. Drug Deliv. 2014, 23, 1–15. [Google Scholar]
- AboulFotouh, K.; Allam, A.A.; El-Badry, M.; El-Sayed, A.M. A Self-Nanoemulsifying Drug Delivery System for Enhancing the Oral Bioavailability of Candesartan Cilexetil: Ex Vivo and In Vivo Evaluation. J. Pharm. Sci. 2019, 108, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, Z.; Bu, H.; Huang, Y.; Gao, Z.; Shen, J.; Zhao, C.; Li, Y. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: Performance and mechanism. J. Control. Release 2011, 149, 168–174. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining supersaturation in aqueous drug solutions: Impact of different polymers on induction times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Trasi, N.S.; Taylor, L.S. Effect of polymers on nucleation and crystal growth of amorphous acetaminophen. CrystEngComm 2012, 14, 5188–5197. [Google Scholar] [CrossRef]
- Vandecruys, R.; Peeters, J.; Verreck, G.; Brewster, M.E. Use of a screening method to determine excipients which optimize the extent and stability of supersaturated drug solutions and application of this system to solid formulation design. Int. J. Pharm. 2007, 342, 168–175. [Google Scholar] [CrossRef]
- Blaabjerg, L.I.; Grohganz, H.; Lindenberg, E.; Löbmann, K.; Müllertz, A.; Rades, T. The influence of polymers on the supersaturation potential of poor and good glass formers. Pharmaceutics 2018, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4544148/ (accessed on 4 March 2021).
- Figueroa-Campos, A.; Sánchez-Dengra, B.; Merino, V.; Dahan, A.; González-Álvarez, I.; García-Arieta, A.; González-Álvarez, M.; Bermejo, M. Candesartan Cilexetil In Vitro–In Vivo Correlation: Predictive Dissolution as a Development Tool. Pharmaceutics 2020, 12, 633. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Chavan, R.B.; Thipparaboina, R.; Kumar, D.; Shastri, N.R. Evaluation of the inhibitory potential of HPMC, PVP and HPC polymers on nucleation and crystal growth. RSC Adv. 2016, 6, 77569–77576. [Google Scholar] [CrossRef]
- Kim, D.W.; Kwon, M.S.; Yousaf, A.M.; Balakrishnan, P.; Park, J.H.; Kim, D.S.; Lee, B.J.; Park, Y.J.; Yong, C.S.; Kim, J.O.; et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydr. Polym. 2014, 114, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.L.; Tran, T.T.D.; Lee, K.H.; Kim, D.J.; Lee, B.J. Dissolution-modulating mechanism of pH modifiers in solid dispersion containing weakly acidic or basic drugs with poor water solubility. Expert Opin. Drug Deliv. 2010, 7, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Buda, V.; Baul, B.; Andor, M.; Man, D.E.; Ledeţi, A.; Vlase, G.; Vlase, T.; Danciu, C.; Matusz, P.; Peter, F.; et al. Solid state stability and kinetics of degradation for candesartan—Pure compound and pharmaceutical formulation. Pharmaceutics 2020, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Xu, W.; Yang, Y.; Yang, Z.; Lv, H.; Yang, S.; Lin, Z.; Li, J.; Gu, J.; Hill, C.L.; et al. Three Candesartan Salts with Enhanced Oral Bioavailability. Cryst. Growth Des. 2015, 15, 3707–3714. [Google Scholar] [CrossRef]
- Matsunaga, H.; Eguchi, T.; Nishijima, K.; Enomoto, T.; Sasaoki, K.; Nakamura, N. Solid-State Characterization of Candesartan Cilexetil (TCV-116): Crystal Structure and Molecular Mobility. Chem. Pharm. Bull. (Tokyo) 1999, 47, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.; Gilmor, T.; Lane, P.; Ranweiler, J.S. 12-Stability Studies. In Handbook of Modern Pharmaceutical Analysis; Ahuja, S., Scypinski, S.B.T.S., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 10, pp. 459–505. ISBN 1877-1718. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | CC (g) | PVPK30 (g) | Na2CO3 (g) |
|---|---|---|---|
| F1 | 1 | 0.5 | 1 |
| F2 | 1 | 1 | 1 |
| F3 | 1 | 2 | 1 |
| F4 | 1 | 4 | 1 |
| F5 | 1 | 8 | 1 |
| Medium | CC (μg/mL) | PM (μg/mL) | F1 (μg/mL) |
|---|---|---|---|
| Water | 1.03 ± 0.06 | 7613.11 ± 6480.31 | 31,156.05 ± 4552.69 |
| pH 1.2 | 0.89 ± 0.13 | 10,462.60 ± 6492.75 | 32,212.37± 4785.75 |
| pH 4.0 | 0.48 ± 0.03 | 103.78 ± 76.71 | 31,095.11 ± 5395.98 |
| pH 6.8 | 2.30 ± 0.37 | 822.71 ± 171.25 | 38,424.75 ± 7539.25 |
| Weeks | Drug Content (μg/mL) | |
|---|---|---|
| Real-Time Stability Condition | Accelerated Stability Condition | |
| 0 | 99.99 ± 2.55 | |
| 1 | 96.45 ± 2.98 | 96.08 ± 6.75 |
| 4 | 94.90 ± 1.75 | 95.75 ± 3.54 |
| 8 | 95.61 ± 3.06 | - |
| 12 | 96.38 ± 1.04 | - |
| Formulations | CC | F1 |
|---|---|---|
| AUC (h·ng/mL) | 173.29 ± 30.27 | 771.87 ± 227.63 * |
| Cmax (ng/mL) | 25.44 ± 6.28 | 188.75 ± 41.06 * |
| Tmax (h) | 0.83 ± 0.60 | 0.50 ± 0.27 |
| t1/2 (h) | 7.47 ± 2.81 | 4.93 ± 1.42 |
| Kel (h−1) | 0.10 ± 0.03 | 0.15 ± 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poudel, S.; Kim, D.W. Developing pH-Modulated Spray Dried Amorphous Solid Dispersion of Candesartan Cilexetil with Enhanced In Vitro and In Vivo Performance. Pharmaceutics 2021, 13, 497. https://doi.org/10.3390/pharmaceutics13040497
Poudel S, Kim DW. Developing pH-Modulated Spray Dried Amorphous Solid Dispersion of Candesartan Cilexetil with Enhanced In Vitro and In Vivo Performance. Pharmaceutics. 2021; 13(4):497. https://doi.org/10.3390/pharmaceutics13040497
Chicago/Turabian StylePoudel, Surendra, and Dong Wuk Kim. 2021. "Developing pH-Modulated Spray Dried Amorphous Solid Dispersion of Candesartan Cilexetil with Enhanced In Vitro and In Vivo Performance" Pharmaceutics 13, no. 4: 497. https://doi.org/10.3390/pharmaceutics13040497