
Liposomal Nanosystems in Rheumatoid Arthritis

 , , , and
, , , and
Abstract
: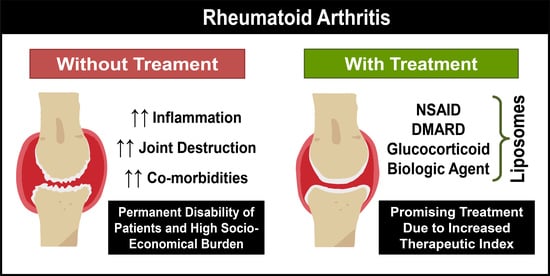
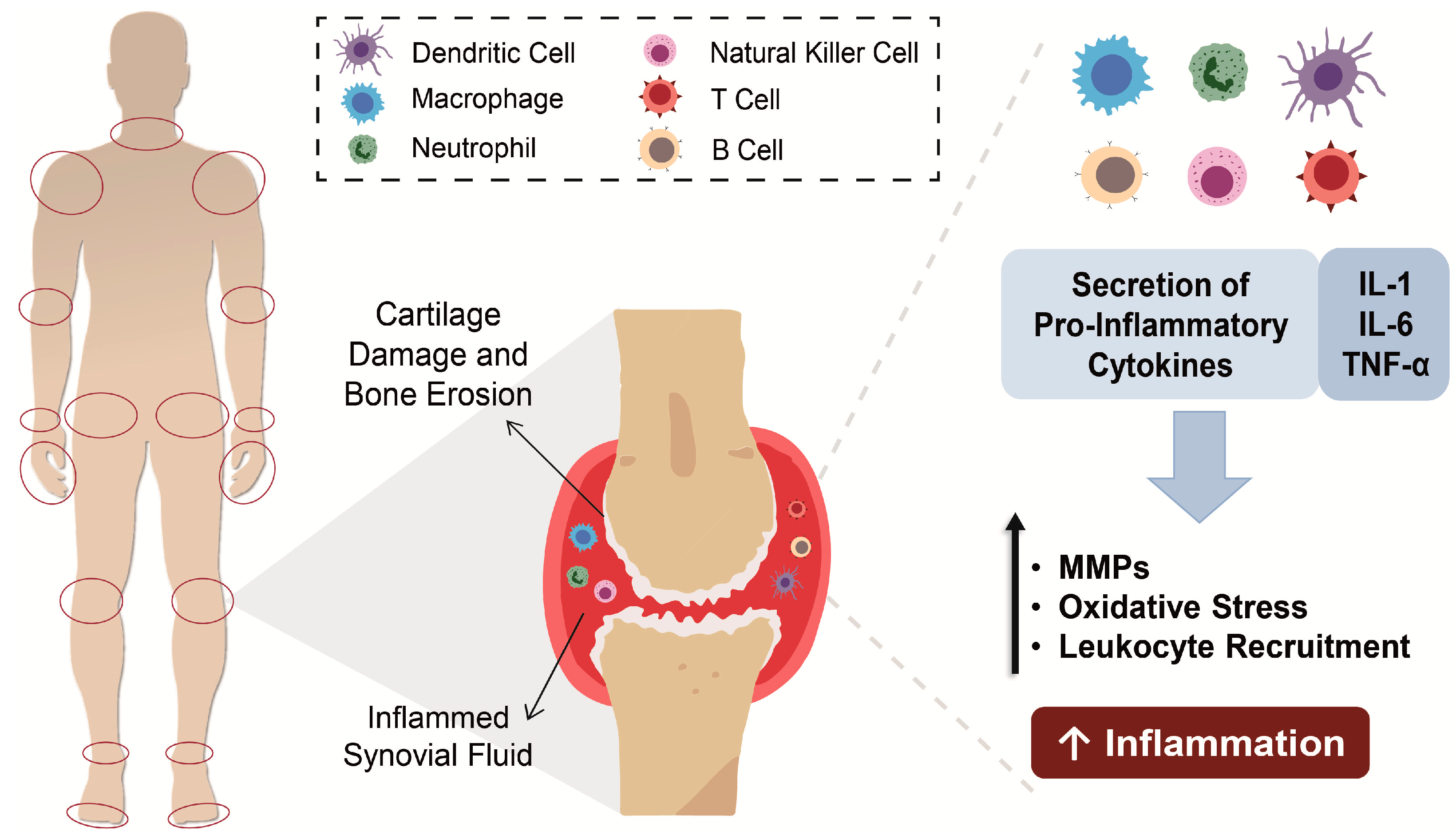
1. Rheumatoid Arthritis
Pathophysiology
2. Therapeutic Strategies Used in the Clinic
Tapering Therapy in Remission
3. Biomarkers for Active Targeting in Rheumatoid Arthritis
4. Drug Delivery Nanosystems
4.1. Liposomal Formulations Developed for Rheumatoid Arthritis Treatment
4.1.1. Liposomes Containing Nonsteroidal Anti-Inflammatory Drugs
4.1.2. Liposomes Containing Glucocorticoids
4.1.3. Liposomes Containing Disease-Modifying Antirheumatic Drugs
4.1.4. Liposomes Containing Biologic Agents
4.1.5. Liposomes Containing a Combination of Distinct Therapeutic Compounds
4.1.6. Liposomes Containing Nonconventional Compounds
5. Translation to the Clinic
5.1. Clinical Trials with Drug Delivery Nanosystems in Rheumatoid Arthritis
5.2. Transition of Drug-Delivery Nanosystems to the Market
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ulbrich, W.; Lamprecht, A. Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases. J. R. Soc. Interface 2010, 7, S55–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.C.; Moore, A.; Vasilescu, R.; Alvir, J.; Tarallo, M. A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: A current perspective. Rheumatol. Int. 2016, 36, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.; Carville, S.; McKenna, F. Diagnosis and management of rheumatoid arthritis in adults: Summary of updated NICE guidance. BMJ 2018, 362, k3015. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Crielaard, B.J.; Lammers, T.; Schiffelers, R.M.; Storm, G. Drug targeting systems for inflammatory disease: One for all, all for one. J. Control. Release 2012, 161, 225–234. [Google Scholar] [CrossRef]
- Sparks, J.A. Rheumatoid Arthritis. Ann. Intern. Med. 2019, 170, ITC1. [Google Scholar] [CrossRef]
- Rein, P.; Mueller, R.B. Treatment with Biologicals in Rheumatoid Arthritis: An Overview. Rheumatol. Ther. 2017, 4, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Nerurkar, L.; Siebert, S.; McInnes, I.B.; Cavanagh, J. Rheumatoid arthritis and depression: An inflammatory perspective. Lancet Psychiatry 2019, 6, 164–173. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Veigas, B.; Matias, A.; Calmeiro, T.; Fortunato, E.; Fernandes, A.R.; Baptista, P.V. Antibody modified gold nanoparticles for fast colorimetric screening of rheumatoid arthritis. Analyst 2019, 144, 3613–3619. [Google Scholar] [CrossRef]
- Hui, A.Y.; McCarty, W.J.; Masuda, K.; Firestein, G.S.; Sah, R.L. A systems biology approach to synovial joint lubrication in health, injury, and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 4, 15–37. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Feng, X.; Chen, Y. Drug delivery targets and systems for targeted treatment of rheumatoid arthritis. J. Drug Target. 2018, 26, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnäker, E.; Tarner, I.H.; Robbins, P.D.; et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Gremese, E.; Salaffi, F.; Bosello, S.L.; Ciapetti, A.; Bobbio-Pallavicini, F.; Caporali, R.; Ferraccioli, G. Very early rheumatoid arthritis as a predictor of remission: A multicentre real life prospective study. Ann. Rheum. Dis. 2013, 72, 858–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.A.; Saag, K.G.; Bridges, S.L.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2016, 68, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.A.; McWilliams, D.F. Mechanisms, impact and management of pain in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 581–592. [Google Scholar] [CrossRef]
- Koning, G.A.; Schiffelers, R.M.; Wauben, M.H.M.; Kok, R.J.; Mastrobattista, E.; Molema, G.; ten Hagen, T.L.M.; Storm, G. Targeting of angiogenic endothelial cells at sites of inflammation by dexamethasone phosphate-containing RGD peptide liposomes inhibits experimental arthritis. Arthritis Rheum. 2006, 54, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; van der Heijde, D.; Machold, K.P.; Aletaha, D.; Landewé, R. Proposal for a new nomenclature of disease-modifying antirheumatic drugs. Ann. Rheum. Dis. 2014, 73, 3–5. [Google Scholar] [CrossRef]
- Crofford, L.J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 2013, 15 (Suppl. S3), S2. [Google Scholar] [CrossRef] [Green Version]
- Solomon, D.H.; Husni, M.E.; Libby, P.A.; Yeomans, N.D.; Lincoff, A.M.; Lϋscher, T.F.; Menon, V.; Brennan, D.M.; Wisniewski, L.M.; Nissen, S.E.; et al. The Risk of Major NSAID Toxicity with Celecoxib, Ibuprofen, or Naproxen: A Secondary Analysis of the PRECISION Trial. Am. J. Med. 2017, 130, 1415–1422. [Google Scholar] [CrossRef]
- Pillai, A.A.; Levitsky, J. Overview of immunosuppression in liver transplantation. World J. Gastroenterol. 2009, 15, 4225–4233. [Google Scholar] [CrossRef] [PubMed]
- Metselaar, J.M.; Wauben, M.H.M.; Wagenaar-Hilbers, J.P.A.; Boerman, O.C.; Storm, G. Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Rheum. 2003, 48, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Pope, J.E. Novel treatment strategies in rheumatoid arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef]
- Zampeli, E.; Vlachoyiannopoulos, P.G.; Tzioufas, A.G. Treatment of rheumatoid arthritis: Unraveling the conundrum. J. Autoimmun. 2015, 65, 1–18. [Google Scholar] [CrossRef]
- Angelini, J.; Talotta, R.; Roncato, R.; Fornasier, G.; Barbiero, G.; Dal Cin, L.; Brancati, S.; Scaglione, F. JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future. Biomolecules 2020, 10, 1002. [Google Scholar] [CrossRef]
- Mian, A.; Ibrahim, F.; Scott, D.L. A systematic review of guidelines for managing rheumatoid arthritis. BMC Rheumatol. 2019, 3, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffing, V. Rheumatoid arthritis. In Scientific Basis of Healthcare: Arthritis, 1st ed.; Martin, C.R., Preedy, V.R., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 1–26. [Google Scholar]
- Lau, C.S.; Chia, F.; Dans, L.; Harrison, A.; Hsieh, T.Y.; Jain, R.; Jung, S.M.; Kishimoto, M.; Kumar, A.; Leong, K.P.; et al. 2018 update of the APLAR recommendations for treatment of rheumatoid arthritis. Int. J. Rheum. Dis. 2019, 22, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Van den Hoven, J.M.; Van Tomme, S.R.; Metselaar, J.M.; Nuijen, B.; Beijnen, J.H.; Storm, G. Liposomal drug formulations in the treatment of rheumatoid arthritis. Mol. Pharm. 2011, 8, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Kraus, V.B.; Setton, L.A. Progress in intra-articular therapy. Nat. Rev. Rheumatol. 2014, 10, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Aalbers, C.J.; Bevaart, L.; Loiler, S.; de Cortie, K.; Wright, J.F.; Mingozzi, F.; Tak, P.P.; Vervoordeldonk, M.J. Preclinical Potency and Biodistribution Studies of an AAV 5 Vector Expressing Human Interferon-β (ART-I02) for Local Treatment of Patients with Rheumatoid Arthritis. PLoS ONE 2015, 10, e0130612. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, T.D.; Mikuls, T.R. Recent advances in the treatment of rheumatoid arthritis. Curr. Opin. Rheumatol. 2018, 30, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, E.; Gomes, A.C.; Preto, A.; Cavaco-Paulo, A. Folate-targeted nanoparticles for rheumatoid arthritis therapy. Nanomedicine 2016, 12, 1113–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadat, E.; Shakor, N.; Gholami, M.; Dorkoosh, F.A. Hyaluronic acid based micelle for articular delivery of triamcinolone, preparation, in vitro and in vivo evaluation. Int. J. Pharm. 2015, 489, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S. Starving the synovium: Angiogenesis and inflammation in rheumatoid arthritis. J. Clin. Investig. 1999, 103, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Al-Soudi, A.; Kaaij, M.H.; Tas, S.W. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun. Rev. 2017, 16, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Maruotti, N.; Annese, T.; Cantatore, F.P.; Ribatti, D. Macrophages and angiogenesis in rheumatic diseases. Vasc. Cell 2013, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Lowin, T.; Straub, R.H. Integrins and their ligands in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 244. [Google Scholar] [CrossRef] [Green Version]
- Kiselyov, A.; Balakin, K.V.; Tkachenko, S.E. VEGF/VEGFR signalling as a target for inhibiting angiogenesis. Expert Opin. Investig. Drugs 2007, 16, 83–107. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Angiogenic and cell survival functions of Vascular Endothelial Growth Factor (VEGF). J. Cell. Mol. Med. 2005, 9, 777–794. [Google Scholar] [CrossRef]
- Azizi, G.; Boghozian, R.; Mirshafiey, A. The potential role of angiogenic factors in rheumatoid arthritis. Int. J. Rheum. Dis. 2014, 17, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, C.; Kneuer, C.; Bakowsky, U. Selectins—An emerging target for drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 527–549. [Google Scholar] [CrossRef]
- Etzerodt, A.; Maniecki, M.B.; Graversen, J.H.; Moller, H.J.; Torchilin, V.P.; Moestrup, S.K. Efficient intracellular drug-targeting of macrophages using stealth liposomes directed to the hemoglobin scavenger receptor CD163. J. Control. Release 2012, 160, 72–80. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, A.; Zhang, Y.; Zuo, Z.-Q.; Cao, Z.-T.; Zhang, H.-B.; Xu, C.-F.; Wang, J. Nanoparticle-delivered siRNA targeting Bruton’s tyrosine kinase for rheumatoid arthritis therapy. Biomater. Sci. 2019, 7, 4698–4707. [Google Scholar] [CrossRef]
- Pirmardvand Chegini, S.; Varshosaz, J.; Taymouri, S. Recent approaches for targeted drug delivery in rheumatoid arthritis diagnosis and treatment. Artif. Cells Nanomed. Biotechnol. 2018, 46, 502–514. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [Green Version]
- Vilar, G.; Tulla-Puche, J.; Albericio, F. Polymers and drug delivery systems. Curr. Drug Deliv. 2012, 9, 367–394. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar] [CrossRef]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.K.O.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.J.; Neumann, V.C.; Hill, J.; Astbury, C.; Le Gallez, P.; Dixon, J.S. 5-Aminosalicylic acid or sulphapyridine. Which is the active moiety of sulphasalazine in rheumatoid arthritis? Drugs 1986, 32 (Suppl. S1), 27–34. [Google Scholar] [CrossRef]
- Kapoor, B.; Gulati, M.; Singh, S.K.; Khatik, G.L.; Gupta, R.; Kumar, R.; Kumar, R.; Gowthamarajan, K.; Mahajan, S.; Gupta, S. Fail-safe nano-formulation of prodrug of sulfapyridine: Preparation and evaluation for treatment of rheumatoid arthritis. Mater. Sci. Eng. C. Mater. Biol. Appl. 2021, 118, 111332. [Google Scholar] [CrossRef] [PubMed]
- Durymanov, M.; Kamaletdinova, T.; Lehmann, S.E.; Reineke, J. Exploiting passive nanomedicine accumulation at sites of enhanced vascular permeability for non-cancerous applications. J. Control. Release 2017, 261, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Zhang, X.; Qi, J.; Shu, G.; Du, Y.; Ying, X. Sinomenine hydrochloride loaded thermosensitive liposomes combined with microwave hyperthermia for the treatment of rheumatoid arthritis. Int. J. Pharm. 2020, 576, 119001. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Hao, B.; Ju, D.; Liu, M.; Zhao, H.; Du, Z.; Xia, J. Pharmacokinetic and pharmacodynamic study of triptolide-loaded liposome hydrogel patch under microneedles on rats with collagen-induced arthritis. Acta Pharm. Sin. B 2015, 5, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, I.M.; Gonçalves, C.; Reis, R.L.; Oliveira, J.M. Engineering nanoparticles for targeting rheumatoid arthritis: Past, present, and future trends. Nano Res. 2018, 11, 4489–4506. [Google Scholar] [CrossRef] [Green Version]
- Dolati, S.; Sadreddini, S.; Rostamzadeh, D.; Ahmadi, M.; Jadidi-Niaragh, F.; Yousefi, M. Utilization of nanoparticle technology in rheumatoid arthritis treatment. Biomed. Pharmacother. 2016, 80, 30–41. [Google Scholar] [CrossRef]
- Maity, S.; Misra, A.; Wairkar, S. Novel injectable carrier based corticosteroid therapy for treatment of rheumatoid arthritis and osteoarthritis. J. Drug Deliv. Sci. Technol. 2021, 61, 102309. [Google Scholar] [CrossRef]
- Vanniasinghe, A.S.; Bender, V.; Manolios, N. The potential of liposomal drug delivery for the treatment of inflammatory arthritis. Semin. Arthritis Rheum. 2009, 39, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Katare, O.P.; Vyas, S.P.; Dixit, V.K. Enhanced in vivo performance of liposomal indomethacin derived from effervescent granule based proliposomes. J. Microencapsul. 1995, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Srinath, P.; Vyas, S.P.; Diwan, P.V. Preparation and Pharmacodynamic Evaluation of Liposomes of Indomethacin. Drug Dev. Ind. Pharm. 2000, 26, 313–321. [Google Scholar] [CrossRef]
- Dave, V.; Gupta, A.; Singh, P.; Tak, K.; Sharma, S. PEGylated Lipova E120 liposomes loaded with celecoxib: In-vitro characterization and enhanced in-vivo anti-inflammatory effects in rat models. J. Biosci. 2019, 44. [Google Scholar] [CrossRef]
- Türker, S.; Erdoğan, S.; Ozer, Y.A.; Bilgili, H.; Deveci, S. Enhanced efficacy of diclofenac sodium-loaded lipogelosome formulation in intra-articular treatment of rheumatoid arthritis. J. Drug Target. 2008, 16, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Singh, R.; Asati, R.K. Liposomally encapsulated diclofenac for sonophoresis induced systemic delivery. J. Microencapsul. 1995, 12, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Hofkens, W.; Grevers, L.C.; Walgreen, B.; de Vries, T.J.; Leenen, P.J.M.; Everts, V.; Storm, G.; van den Berg, W.B.; van Lent, P.L. Intravenously delivered glucocorticoid liposomes inhibit osteoclast activity and bone erosion in murine antigen-induced arthritis. J. Control. Release 2011, 152, 363–369. [Google Scholar] [CrossRef]
- Van der Geest, T.; Metselaar, J.M.; Gerrits, D.; van Lent, P.L.; Storm, G.; Laverman, P.; Boerman, O.C. [18]F FDG PET/CT imaging to monitor the therapeutic effect of liposome-encapsulated prednisolone in experimental rheumatoid arthritis. J. Control. Release 2015, 209, 20–26. [Google Scholar] [CrossRef]
- Metselaar, J.M.; van den Berg, W.B.; Holthuysen, A.E.M.; Wauben, M.H.M.; Storm, G.; van Lent, P.L.E.M. Liposomal targeting of glucocorticoids to synovial lining cells strongly increases therapeutic benefit in collagen type II arthritis. Ann. Rheum. Dis. 2004, 63, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Van den Hoven, J.M.; Hofkens, W.; Wauben, M.H.M.; Wagenaar-Hilbers, J.P.A.; Beijnen, J.H.; Nuijen, B.; Metselaar, J.M.; Storm, G. Optimizing the therapeutic index of liposomal glucocorticoids in experimental arthritis. Int. J. Pharm. 2011, 416, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Avnir, Y.; Ulmansky, R.; Wasserman, V.; Even-Chen, S.; Broyer, M.; Barenholz, Y.; Naparstek, Y. Amphipathic weak acid glucocorticoid prodrugs remote-loaded into sterically stabilized nanoliposomes evaluated in arthritic rats and in a Beagle dog: A novel approach to treating autoimmune arthritis. Arthritis Rheum. 2008, 58, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Ulmansky, R.; Turjeman, K.; Baru, M.; Katzavian, G.; Harel, M.; Sigal, A.; Naparstek, Y.; Barenholz, Y. Glucocorticoids in nano-liposomes administered intravenously and subcutaneously to adjuvant arthritis rats are superior to the free drugs in suppressing arthritis and inflammatory cytokines. J. Control. Release 2012, 160, 299–305. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Bruin, P.; de Boer, L.W.T.; de Vringer, T.; Snel, C.; Oussoren, C.; Wauben, M.H.M.; Crommelin, D.J.A.; Storm, G.; Hennink, W.E. A novel family of L-amino acid-based biodegradable polymer-lipid conjugates for the development of long-circulating liposomes with effective drug-targeting capacity. Bioconjug. Chem. 2003, 14, 1156–1164. [Google Scholar] [CrossRef]
- Gouveia, V.M.; Lopes-de-Araújo, J.; Costa Lima, S.A.; Nunes, C.; Reis, S. Hyaluronic acid-conjugated pH-sensitive liposomes for targeted delivery of prednisolone on rheumatoid arthritis therapy. Nanomedicine 2018, 13, 1037–1049. [Google Scholar] [CrossRef]
- Vanniasinghe, A.S.; Manolios, N.; Schibeci, S.; Lakhiani, C.; Kamali-Sarvestani, E.; Sharma, R.; Kumar, V.; Moghaddam, M.; Ali, M.; Bender, V. Targeting fibroblast-like synovial cells at sites of inflammation with peptide targeted liposomes results in inhibition of experimental arthritis. Clin. Immunol. 2014, 151, 43–54. [Google Scholar] [CrossRef]
- Rauchhaus, U.; Kinne, R.W.; Pohlers, D.; Wiegand, S.; Wölfert, A.; Gajda, M.; Bräuer, R.; Panzner, S. Targeted delivery of liposomal dexamethasone phosphate to the spleen provides a persistent therapeutic effect in rat antigen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 1933–1934. [Google Scholar] [CrossRef] [PubMed]
- Rauchhaus, U.; Schwaiger, F.; Panzner, S. Separating therapeutic efficacy from glucocorticoid side-effects in rodent arthritis using novel, liposomal delivery of dexamethasone phosphate: Long-term suppression of arthritis facilitates interval treatment. Arthritis Res. Ther. 2009, 11, R190. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Franch, A.; Castell, M.; Perez-Cano, F.J.; Bräuer, R.; Pohlers, D.; Gajda, M.; Siskos, A.P.; Katsila, T.; Tamvakopoulos, C.; et al. Liposomal encapsulation enhances and prolongs the anti-inflammatory effects of water-soluble dexamethasone phosphate in experimental adjuvant arthritis. Arthritis Res. Ther. 2010, 12, R147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Deng, C.; Luo, J.; Zhang, P.; Sun, X.; Zhang, Z.; Gong, T. A novel dexamethasone-loaded liposome alleviates rheumatoid arthritis in rats. Int. J. Pharm. 2018, 540, 57–64. [Google Scholar] [CrossRef]
- Wang, Q.; He, L.; Fan, D.; Liang, W.; Fang, J. Improving the anti-inflammatory efficacy of dexamethasone in the treatment of rheumatoid arthritis with polymerized stealth liposomes as a delivery vehicle. J. Mater. Chem. B 2020, 8, 1841–1851. [Google Scholar] [CrossRef]
- Hu, L.; Luo, X.; Zhou, S.; Zhu, J.; Xiao, M.; Li, C.; Zheng, H.; Qiu, Q.; Lai, C.; Liu, X.; et al. Neutrophil-Mediated Delivery of Dexamethasone Palmitate-Loaded Liposomes Decorated with a Sialic Acid Conjugate for Rheumatoid Arthritis Treatment. Pharm. Res. 2019, 36, 97. [Google Scholar] [CrossRef]
- Wang, S.; Yang, S.; Lai, X.; Song, Y.; Hu, L.; Li, C.; Shi, T.; Liu, X.; Deng, Y.; Chen, G. Sialic Acid Conjugate–Modified Liposomal Dexamethasone Palmitate Targeting Neutrophils for Rheumatoid Arthritis Therapy: Influence of Particle Size. AAPS Pharm. Sci. Tech. 2021, 22, 16. [Google Scholar] [CrossRef]
- Sultana, F.; Neog, M.K.; Rasool, M. Targeted delivery of morin, a dietary bioflavanol encapsulated mannosylated liposomes to the macrophages of adjuvant-induced arthritis rats inhibits inflammatory immune response and osteoclastogenesis. Eur. J. Pharm. Biopharm. 2017, 115, 229–242. [Google Scholar] [CrossRef]
- Sultana, F.; Neog, M.K.; Rasool, M. Withaferin-A, a steroidal lactone encapsulated mannose decorated liposomes ameliorates rheumatoid arthritis by intriguing the macrophage repolarization in adjuvant-induced arthritic rats. Colloids Surf. B. Biointerfaces 2017, 155, 349–365. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, B.; Huang, J.; Xiang, X.; Tang, Y.; Ma, L.; Yan, F.; Cheng, C.; Qiu, L. Ultrasound-targeted microbubble destruction augmented synergistic therapy of rheumatoid arthritis via targeted liposomes. J. Mater. Chem. B 2020, 8, 5245–5256. [Google Scholar] [CrossRef] [PubMed]
- Meka, R.R.; Venkatesha, S.H.; Acharya, B.; Moudgil, K.D. Peptide-targeted liposomal delivery of dexamethasone for arthritis therapy. Nanomedicine 2019, 14, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Poh, S.; Chelvam, V.; Kelderhouse, L.E.; Ayala-López, W.; Vaitilingam, B.; Putt, K.S.; Low, P.S. Folate-conjugated liposomes target and deliver therapeutics to immune cells in a rat model of rheumatoid arthritis. Nanomedicine 2017, 12, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- López-García, F.; Vázquez-Autón, J.M.; Gil, F.; Latoore, R.; Moreno, F.; Villalaín, J.; Gómez-Fernández, J.C. Intra-articular therapy of experimental arthritis with a derivative of triamcinolone acetonide incorporated in liposomes. J. Pharm. Pharmacol. 1993, 45, 576–578. [Google Scholar] [CrossRef]
- Foong, W.C.; Green, K.L. Retention and Distribution of Liposome-entrapped [3H]Methotrexate Injected into Normal or Arthritic Rabbit Joints. J. Pharm. Pharmacol. 1988, 40, 464–468. [Google Scholar] [CrossRef]
- Foong, W.C.; Green, K.L. Treatment of Antigen-induced Arthritis in Rabbits with Liposome-entrapped Methotrexate Injected Intra-articularly. J. Pharm. Pharmacol. 1993, 45, 204–209. [Google Scholar] [CrossRef]
- Williams, A.S.; Camilleri, J.P.; Williams, B.D. Suppression of adjuvant-induced arthritis by liposomally conjugated methotrexate in the rat. Br. J. Rheumatol. 1994, 33, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, D.; Noro, J.; Loureiro, A.; Lager, F.; Renault, G.; Cavaco-Paulo, A.; Nogueira, E. Increased Encapsulation Efficiency of Methotrexate in Liposomes for Rheumatoid Arthritis Therapy. Biomedicines 2020, 8, 630. [Google Scholar] [CrossRef]
- Williams, A.S.; Jones, S.G.; Goodfellow, R.M.; Amos, N.; Williams, B.D. Interleukin-1β (IL-1β) inhibition: A possible mechanism for the anti-inflammatory potency of liposomally conjugated methotrexate formulations in arthritis. Br. J. Pharmacol. 1999, 128, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Goodfellow, R.; Topley, N.; Amos, N.; Williams, B. The suppression of rat collagen-induced arthritis and inhibition of macrophage derived mediator release by liposomal methotrexate formulations. Inflamm. Res. 2000, 49, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Camilleri, J.P.; Goodfellow, R.M.; Williams, B.D. A single intra-articular injection of liposomally conjugated methotrexate suppresses joint inflammation in rat antigen-induced arthritis. Br. J. Rheumatol. 1996, 35, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.S.; Camilleri, J.P.; Amos, N.; Williams, B.D. Differential effects of methotrexate and liposomally conjugated methotrexate in rat adjuvant-induced arthritis. Clin. Exp. Immunol. 1995, 102, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S. Amelioration of rat antigen-induced arthritis by liposomally conjugated methotrexate is accompanied by down-regulation of cytokine mRNA expression. Rheumatology 2001, 40, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, E.; Lager, F.; Le Roux, D.; Nogueira, P.; Freitas, J.; Charvet, C.; Renault, G.; Loureiro, A.; Almeida, C.R.; Ohradanova-Repic, A.; et al. Enhancing Methotrexate Tolerance with Folate Tagged Liposomes in Arthritic Mice. J. Biomed. Nanotechnol. 2015, 11, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Neog, M.K.; Rasool, M. Targeted delivery of p-coumaric acid encapsulated mannosylated liposomes to the synovial macrophages inhibits osteoclast formation and bone resorption in the rheumatoid arthritis animal model. Eur. J. Pharm. Biopharm. 2018, 133, 162–175. [Google Scholar] [CrossRef]
- Wu, H.; He, Y.; Wu, H.; Zhou, M.; Xu, Z.; Xiong, R.; Yan, F.; Liu, H. Near-infrared fluorescence imaging-guided focused ultrasound-mediated therapy against Rheumatoid Arthritis by MTX-ICG-loaded iRGD-modified echogenic liposomes. Theranostics 2020, 10, 10092–10105. [Google Scholar] [CrossRef]
- Chen, M.; Amerigos, K.D.; Su, Z.; Guissi, N.E.I.; Xiao, Y.; Zong, L.; Ping, Q. Folate Receptor-Targeting and Reactive Oxygen Species-Responsive Liposomal Formulation of Methotrexate for Treatment of Rheumatoid Arthritis. Pharmaceutics 2019, 11, 582. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Shu, H.; Xu, X.; Shu, G.; Du, Y.; Ying, X. Tofacitinib citrate-based liposomes for effective treatment of rheumatoid arthritis. Pharmazie 2020, 75, 131–135. [Google Scholar] [CrossRef]
- Corvo, M.L.; Boerman, O.C.; Oyen, W.J.; Jorge, J.C.; Cruz, M.E.; Crommelin, D.J.; Storm, G. Subcutaneous administration of superoxide dismutase entrapped in long circulating liposomes: In vivo fate and therapeutic activity in an inflammation model. Pharm. Res. 2000, 17, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.M.; Boerman, O.C.; Laverman, P.; Corvo, M.L.; Storm, G.; Cruz, M.E.M. Enzymosomes with surface-exposed superoxide dismutase: In vivo behaviour and therapeutic activity in a model of adjuvant arthritis. J. Control. Release 2007, 117, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Corvo, M.L.; Boerman, O.C.; Oyen, W.J.; Van Bloois, L.; Cruz, M.E.; Crommelin, D.J.; Storm, G. Intravenous administration of superoxide dismutase entrapped in long circulating liposomes. II. In vivo fate in a rat model of adjuvant arthritis. Biochim. Biophys. Acta 1999, 1419, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Corvo, M.L.; Marinho, H.S.; Marcelino, P.; Lopes, R.M.; Vale, C.A.; Marques, C.R.; Martins, L.C.D.; Laverman, P.; Storm, G.; Martins, M.B.A.F. Superoxide dismutase enzymosomes: Carrier capacity optimization, in vivo behaviour and therapeutic activity. Pharm. Res. 2015, 32, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Niwa, Y.; Somiya, K.; Michelson, A.M.; Puget, K. Effect of liposomal-encapsulated superoxide dismutase on active oxygen-related human disorders. A preliminary study. Free Radic. Res. Commun. 1985, 1, 137–153. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; García-Alvarez, F.; Basáñez, G.; Alegre-Aguarón, E.; Desportes, P.; Larrad, L.; Naval, J.; Martínez-Lorenzo, M.J.; Anel, A. Liposome-bound APO2L/TRAIL is an effective treatment in a rabbit model of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.; Louis-Plence, P.; Escriou, V.; Noel, D.; Largeau, C.; Cantos, C.; Scherman, D.; Jorgensen, C.; Apparailly, F. Efficient new cationic liposome formulation for systemic delivery of small interfering RNA silencing tumor necrosis factor α in experimental arthritis. Arthritis Rheum. 2006, 54, 1867–1877. [Google Scholar] [CrossRef]
- Khoury, M.; Escriou, V.; Courties, G.; Galy, A.; Yao, R.; Largeau, C.; Scherman, D.; Jorgensen, C.; Apparailly, F. Efficient suppression of murine arthritis by combined anticytokine small interfering RNA lipoplexes. Arthritis Rheum. 2008, 58, 2356–2367. [Google Scholar] [CrossRef]
- Sujitha, S.; Dinesh, P.; Rasool, M. Berberine encapsulated PEG-coated liposomes attenuate Wnt1/β-catenin signaling in rheumatoid arthritis via miR-23a activation. Eur. J. Pharm. Biopharm. 2020, 149, 170–191. [Google Scholar] [CrossRef] [PubMed]
- Trif, M.; Roseanu, A.; Brock, J.H.; Brewer, J.M. Designing lipid nanostructures for local delivery of biologically active macromolecules. J. Liposome Res. 2007, 17, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.C.; Campos, C.F.; Cunha, C.; Carvalho, A.; Reis, R.L.; Ferreira, H.; Neves, N.M. Biofunctionalized Liposomes to Monitor Rheumatoid Arthritis Regression Stimulated by Interleukin-23 Neutralization. Adv. Healthc. Mater. 2021, 10, 2001570. [Google Scholar] [CrossRef]
- Meka, R.R.; Venkatesha, S.H.; Moudgil, K.D. Peptide-directed liposomal delivery improves the therapeutic index of an immunomodulatory cytokine in controlling autoimmune arthritis. J. Control. Release 2018, 286, 279–288. [Google Scholar] [CrossRef]
- Verma, A.; Jain, A.; Tiwari, A.; Saraf, S.; Panda, P.K.; Agrawal, G.P.; Jain, S.K. Folate Conjugated Double Liposomes Bearing Prednisolone and Methotrexate for Targeting Rheumatoid Arthritis. Pharm. Res. 2019, 36, 123. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Li, H. Combination of NF-kB targeted siRNA and methotrexate in a hybrid nanocarrier towards the effective treatment in rheumatoid arthritis. J. Nanobiotechnol. 2018, 16, 58. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wang, D.; Zhang, X.; Xu, S.; Zhang, N. Targeted and triple therapy-based liposomes for enhanced treatment of rheumatoid arthritis. Int. J. Pharm. 2020, 586, 119642. [Google Scholar] [CrossRef]
- Sun, Z.; Wei, T.; Zhou, X. Liposomes encapsulated dimethyl curcumin regulates dipeptidyl peptidase I activity, gelatinase release and cell cycle of spleen lymphocytes in-vivo to attenuate collagen induced arthritis in rats. Int. Immunopharmacol. 2018, 65, 511–521. [Google Scholar] [CrossRef]
- Oelzner, P.; Bräuer, R.; Henzgen, S.; Thoss, K.; Wünsche, B.; Hersmann, G.; Abendroth, K.; Kinne, R.W. Periarticular Bone Alterations in Chronic Antigen-Induced Arthritis: Free and Liposome-Encapsulated Clodronate Prevent Loss of Bone Mass in the Secondary Spongiosa. Clin. Immunol. 1999, 90, 79–88. [Google Scholar] [CrossRef]
- Richards, P.J.; Williams, B.D.; Williams, A.S. Suppression of chronic streptococcal cell wall-induced arthritis in Lewis rats by liposomal clodronate. Rheumatology 2001, 40, 978–987. [Google Scholar] [CrossRef] [Green Version]
- Richards, P.J.; Williams, A.S.; Goodfellow, R.M.; Williams, B.D. Liposomal clodronate eliminates synovial macrophages, reduces inflammation and ameliorates joint destruction in antigen-induced arthritis. Rheumatology 1999, 38, 818–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, P.; Blom, A.; van Lent, P.L.; van Bloois, L.; Beijnen, J.H.; van Rooijen, N.; de Waal Malefijt, M.C.; van de Putte, L.B.; Storm, G.; van den Berg, W.B. Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Ceponis, A.; Waris, E.; Mönkkönen, J.; Laasonen, L.; Hyttinen, M.; Solovieva, S.A.; Hanemaaijer, R.; Bitsch, A.; Konttinen, Y.T. Effects of low-dose, noncytotoxic, intraarticular liposomal clodronate on development of erosions and proteoglycan loss in established antigen-induced arthritis in rabbits. Arthritis Rheum. 2001, 44, 1908–1916. [Google Scholar] [CrossRef]
- Zhang, Y.; He, W.; Du, Y.; Du, Y.; Zhao, C.; Zhang, Y.; Zhang, H.; Yin, L.; Li, X. Dimeric artesunate phospholipid-conjugated liposomes as promising anti-inflammatory therapy for rheumatoid arthritis. Int. J. Pharm. 2020, 579, 119178. [Google Scholar] [CrossRef]
- Mohanty, S.; Sahoo, A.K.; Konkimalla, V.B.; Pal, A.; Si, S.C. Naringin in Combination with Isothiocyanates as Liposomal Formulations Potentiates the Anti-inflammatory Activity in Different Acute and Chronic Animal Models of Rheumatoid Arthritis. ACS Omega 2020, 5, 28319–28332. [Google Scholar] [CrossRef]
- Jhun, J.; Moon, J.; Ryu, J.; Shin, Y.; Lee, S.; Cho, K.-H.; Kang, T.; Cho, M.-L.; Park, S.-H. Liposome/gold hybrid nanoparticle encoded with CoQ10 (LGNP-CoQ10) suppressed rheumatoid arthritis via STAT3/Th17 targeting. PLoS ONE 2020, 15, e0241080. [Google Scholar] [CrossRef] [PubMed]
- Isalomboto Nkanga, C.; Murhimalika Bapolisi, A.; Ikemefuna Okafor, N.; Werner Maçedo Krause, R. General Perception of Liposomes: Formation, Manufacturing and Applications. In Liposomes—Advances and Perspectives, 1st ed.; Catala, A., Ed.; IntechOpen: London, UK, 2019; pp. 31–54. [Google Scholar]
- Lee, Y.; Thompson, D.H. Stimuli-responsive liposomes for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1450. [Google Scholar] [CrossRef]
- Bas, D.B.; Su, J.; Wigerblad, G.; Svensson, C.I. Pain in rheumatoid arthritis: Models and mechanisms. Pain Manag. 2016, 6, 265–284. [Google Scholar] [CrossRef]
- Boland, E.W. Clinical Comparison of the Newer Anti-Inflammatory Corticosteroids. Ann. Rheum. Dis. 1962, 21, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Upponi, J.R.; Torchilin, V.P. Design of multifunctional non-viral gene vectors to overcome physiological barriers: Dilemmas and strategies. Int. J. Pharm. 2012, 427, 3–20. [Google Scholar] [CrossRef]
- Pedrosa, P.; Vinhas, R.; Fernandes, A.; Baptista, P.V. Gold Nanotheranostics: Proof-of-Concept or Clinical Tool? Nanomaterials 2015, 5, 1853–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias-Lopez, C.; Agustí, A.; Obach, M.; Vallano, A. Regulatory Framework for Advanced Therapy Medicinal Products in Europe and United States. Front. Pharmacol. 2019, 10, 921. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Lammers, T. Challenges in nanomedicine clinical translation. Drug Deliv. Transl. Res. 2020, 10, 721–725. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, R.; Jones, S.L. Cost–effectiveness of nanomedicine: Estimating the real size of nano-costs. Nanomedicine 2019, 14, 1367–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.S.; Florindo, H.F.; Silva, L.C.; Videira, M.A.; Corvo, M.L.; Martins, B.F.; Silva-Lima, B. Regulatory Aspects of Oncologicals: Nanosystems Main Challenges. In Nano-Oncologicals: New Targeting and Delivery Approaches, 1st ed.; Alonso, M.J., Garcia-Fuentes, M., Eds.; Springer: Cham, Switzerland, 2014; pp. 425–452. [Google Scholar]
- Taha, M.S.; Padmakumar, S.; Singh, A.; Amiji, M.M. Critical quality attributes in the development of therapeutic nanomedicines toward clinical translation. Drug Deliv. Transl. Res. 2020, 10, 766–790. [Google Scholar] [CrossRef]
- Barz, M.; Luxenhofer, R.; Schillmeier, M. Quo vadis nanomedicine? Nanomedicine 2015, 10, 3089–3091. [Google Scholar] [CrossRef]
- Tinkle, S.; McNeil, S.E.; Mühlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y.C.; Tamarkin, L.; Desai, N. Nanomedicines: Addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Dias, T.H.; Pepperall, D.-G.; Yang, Y. Topical Loperamide-Encapsulated Liposomal Gel Increases the Severity of Inflammation and Accelerates Disease Progression in the Adjuvant-Induced Model of Experimental Rheumatoid Arthritis. Front. Pharmacol. 2017, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, L.; Zhang, Y.; Crielaard, B.J.; Dusad, A.; Lele, S.M.; Rijcken, C.J.F.; Metselaar, J.M.; Kostková, H.; Etrych, T.; Ulbrich, K.; et al. Nanomedicines for inflammatory arthritis: Head-to-head comparison of glucocorticoid-containing polymers, micelles, and liposomes. ACS Nano 2014, 8, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Lovrić, J.; Lakoš, G.P.; Pepić, I. Nanotherapeutics in the EU: An overview on current state and future directions. Int. J. Nanomed. 2014, 9, 1005–1023. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Therapeutic Agent | Drug Delivery Nanosystems Developed | Lipid Composition (Molar Ratio) | Diameter (nm) | Reference |
|---|---|---|---|---|
| Nonsteroidal Anti-inflammatory Drug | Liposomes with incorporated indomethacin | SL:Chol:SA/DCP (7:3:1) | n.r. | [65] |
| EPC:Chol:SA/PG (1:0.5:0.1/0.2) | 50 or 100 | [66] | ||
| Liposomes with incorporated celecoxib | Lipova E120:Chol: DSPE-PEG2000 (9:1:0.25) | 92 | [67] | |
| Gel formulation of liposomes with encapsulated diclofenac sodium | DMPC:Chol:DCP (7:1:2) | 235 | [68] | |
| Oil/water emulsion of liposomes with incorporated diclofenac | EPC:DCP (9:1 or 7:3) | 4430–5400 | [69] | |
| EPC:Chol (9:1 or 7:3) | 3590–4280 | |||
| Glucocorticoid | Liposomes with encapsulated prednisolone phosphate 1 | DPPC:Chol:DSPE-PEG2000 (1.85:1:0.15) | 90–110 or 450–500 | [25,70,71,72,73] |
| Liposomes with encapsulated methyl prednisolone hemisuccinate | HSPC:Chol:DSPE-PEG2000 (55:40:5 or 54:41:5) | 68–98 | [74,75] | |
| Poly-(hydroxyethyl L-asparagine) (PHEA)-liposomes with encapsulated prednisolone phosphate | DPPC:Chol:PHEA-DODASuc (1.85:1.0:0.15) | 144–148 | [76] | |
| pH-sensitive liposomes with incorporated prednisolone, targeted with hyaluronic acid | DPPE:CHEMS (6.5:3.5) | 113–119 | [77] | |
| Liposomes with encapsulated prednisolone phosphate, targeted with RGD or HAP-1 peptides | DPPC:Chol:DSPE-PEG2000: DSPE-PEG2000-Mal (1.85:1.0:0.075:0.075) | 95–105 | [78] | |
| Liposomes with encapsulated dexamethasone phosphate | DPPC:Chol:DSPE-PEG2000 (1.85:1.0:0.15) | 90–100 | [73] | |
| DPPC:DPPG:Chol (50:10:40) | 280–310 | [79,80,81] | ||
| Liposomes with incorporated dexamethasone | SPC:Solutol HS 15 (3:1) | 60 | [82] | |
| Polymerized liposomes with incorporated dexamethasone | DC8,9PC:DSPE-PEG2000 (9:1) | 112–131 | [83] | |
| Liposomes with incorporated dexamethasone palmitate, targeted with sialic acid | HSPC:Chol (55:40) | 130–138 | [84] | |
| DSPC:DSPG:Chol (8.9:2.4:1) | 71–79, 146–154 or 295–305 | [85] | ||
| Liposomes with incorporated dexamethasone palmitate, targeted with mannose | DSPC:Chol (60:35 or 60:32.5) | 142–146 or 176–190 | [86,87] | |
| Liposomes with encapsulated dexamethasone sodium phosphate, targeted with folate (FA) | DPPC:Chol:DSPE-PEG2000-FA (64:30:5) | 157–159 | [88] | |
| Liposomes with encapsulated dexamethasone, targeted with RGD peptide | DPPC:Chol:DSPE-PEG2000: DSPE-PEG2000-Mal (1.85:1:0.075:0.075) | 100 | [20] | |
| Liposomes with encapsulated dexamethasone, targeted with ART-2 lipopeptide | DOPC:DOPE:Chol: DSPE-PEG2000-NH2 (1:0.6:0.4:0.05) | 96–105 | [89] | |
| Liposomes with encapsulated betamethasone hemisuccinate | HSPC:Chol:DSPE-PEG2000 (55:40:5 or 54:41:5) | 68–98 | [74,75] | |
| Liposomes with encapsulated betamethasone, targeted with folate | DSPC:Chol:DSPE-PEG2000: DSPE-PEG3400-FA (56:40:4:0.1) | 90–110 | [90] | |
| Liposomes with encapsulated budesonide phosphate | DPPC:Chol:DSPE-PEG2000 (1.85:1:0.15) | 90–100 | [73] | |
| Liposomes with incorporated triamcinolone acetonide | DPPC:Chol:PA (8:3:1) | n.r. | [91] | |
| Disease-modifying Antirheumatic Drug | Liposomes with encapsulated methotrexate sodium salt | EPC:Chol:DCP (5:5:1) | 1070 | [92,93] |
| EL:Chol:PA (7:2:1) | 100 | [94] | ||
| DOPE/EPC:Chol:DSPE-PEG2000 (54:36:10) | 121–136/194–208 | [95] | ||
| Liposomes with incorporated methotrexate | EPC:Chol:PA (7:2:1) or DSPC:Chol:DSPE-PEG2000 (10:5:1) | 100 | [96,97] | |
| EL:Chol:PA (7:2:1) | 100 or 1200 | [98,99] | ||
| POPC:Chol:DMPA | 1200 | [100] | ||
| Liposomes with encapsulated methotrexate, targeted with folate | DOPE:Chol:DSPE-PEG2000-CA (n.r.) | 120 | [101] | |
| Liposomes with incorporated methotrexate, targeted with mannose | DSPC:Chol (60:35) | 122–127 | [102] | |
| Echogenic liposomes containing methotrexate and indocyanine green, targeted with iRGD peptide | DPPC:Chol:DSPE-PEG2000: DSPE-PEG2000-Mal (n.r.) | 109–117 | [103] | |
| Liposomes with co-encapsulated methotrexate and catalase, targeted with folate | POPC:Chol:S100-FA (13.2:1.9:0.6) | 141–150 | [104] | |
| Liposomes with encapsulated tofacitinib citrate | SPC:Chol (1:1) | 55–63 | [105] | |
| Liposomes with incorporated sulfapyridine or an amide prodrug of sulfapyridine | P-90G:Chol (6.3:3.1 or 5.5:4.7) | 455–470 or 762–930 | [57] | |
| Biologic Agent | Liposomes with encapsulated or covalently linked superoxide dismutase | EPC:Chol:SA (7:2:1) | 90, 110 or 210 | [106,107,108,109] |
| EPC:Chol:DSPE-PEG2000 (1.85:1:0.15) | 90–110, 200 or 450 | |||
| EPC:Chol:DSPE-PEG2000: DSPE-PEG2000-Mal (68.25:30.5:0.5:0.75) | 120 | |||
| n.r. | n.r. | [110] | ||
| Liposomes linked to tumor necrosis factor-related apoptosis-inducing ligand (Apo2L/TRAIL) | EPC:SM:Chol:DGS-NTA (7.1:3.9:2.6:0.5) | 150–200 | [111] | |
| Liposomes encapsulating siRNA for TNF-α, IL-1β, IL-6 or IL-18 | DOPE:RPR209120:carrier DNA (n.r.) | 1500–2000 | [112,113] | |
| Liposomes containing miR-23a/polyethylenimine (PEI) complex | DSPC:DSPE-PEG2000 (n.r.) | 104–109 | [114] | |
| Liposomes encapsulating human lactoferrin | DPPE:Chol:SA (5:5:1) | 200 | [115] | |
| Liposomes with anti-IL-23 antibody covalently linked to the surface, containing gold nanoparticles | EPC:Chol:DSPE-PEG2000-Mal (0.85:1:0.15) | 127–133 | [116] | |
| Liposomes with encapsulated IL-27, targeted with ART-1 lipopeptide | DOPC:DOPE:Chol: DSPEPEG2000-NH2 (1:0.5:0.5:0.01) | 92–95 | [117] | |
| Combination of Therapeutic Compounds from Different Classes | Double liposomes with encapsulated prednisolone and incorporated methotrexate, targeted with folate | inner liposomes: DSPC:Chol:SA (7.5:2.5:0.5)/outer layer: DSPC:Chol:DSPE-PEG2000-FA (n.r.) | 157–160/426–433 | [118] |
| Liposomes with co-encapsulated methotrexate and calcium phosphate nanoparticles that contained p65 siRNA, targeted with folate | DSPC:Chol:DSPE-PEG2000: DSPE-PEG2000-FA (4:1.2:0.15:0.04) | 170 | [119] | |
| Liposomes with incorporated dexamethasone and co-encapsulated nuclear factor-κB (NF-κB) decoy oligodeoxynucleotides and gold nanorods, targeted with folate | Lipoid E80:Chol:DSPE-PEG2000-FA (6.4:2.6:0.03) | 95–113 | [120] | |
| Non-conventional Compound | Liposomes with incorporated berberine | DSPC:Chol:DSPE-PEG2000 (60:35:2.5) | 157–161 | [114] |
| Liposomes with incorporated dimethyl curcumin | SPC:Chol (1.3:2.6) | <200 | [121] | |
| Liposomes with encapsulated clodronate | PEG400-S:Chol:SDS (1.8:1.8:0.45) | 858–942 | [122] | |
| EPC:Chol:DPPA (7:7:1) | 100 | [123,124] | ||
| EPC:Chol (2:1) | n.r. | |||
| EPC:Chol (n.r.) | 120–160 | [125] | ||
| DSPC:DSPG:Chol (n.r) | n.r. | [126] | ||
| Thermosensitive liposomes with encapsulated sinomenine hydrochloride | DPPC:SPC:Chol (5.1:1.6:0.7) | 111–121 | [59] | |
| Hydrogel patch containing liposomes with incorporated triptolide | EL:Chol (2.9:1.2) | 183–220 | [60] | |
| Dimeric artesunate phospholipid-conjugated liposomes | Di-ART-GPC | 70–83 | [127] | |
| Liposomes with incorporated naringin and encapsulated sulforaphane or phenethyl isothiocyanate | DPPC:Chol:DSPE-PEG2000 (15:4:1) | 147–159 | [128] | |
| Liposome/gold hybrid nanoparticles containing coenzyme Q10 | DSPC (n.r.) | n.r. | [129] | |
| Liposomes with incorporated morin, targeted with mannose | DSPC:Chol (60:35) | 127–137 | [86] | |
| Liposomes with incorporated p-coumaric acid, targeted with mannose | DSPC:Chol (60:35) | 114–124 | [102] | |
| Liposomes with incorporated withaferin-A, targeted with mannose | DSPC:Chol (60:32.5) | 150–155 | [87] | |
| Liposomes with encapsulated or incorporated core peptide, targeted with RGD or HAP-1 peptides | DPPC:Chol:DSPE-PEG2000: DSPE-PEG2000-Mal (1.85:1.0:0.075:0.075) | 95–105 | [78] |
| Database | Drug Delivery Nanosystem | Identifier |
|---|---|---|
| USA National Library of Medicine | Polyethylene glycol (PEG)-liposomes containing prednisolone | NCT00241982 (phase II) and NCT02534896 (phase III) |
| Recombinant adeno-associated virus vector | NCT00617032 (phase I); NCT00126724 (phase I/II); NCT02727764 (phase I) and NCT03445715 (phase I) | |
| European Union Clinical Trials Register | PEG-liposomes containing prednisolone sodium phosphate (Nanocort®) | 2015-002924-17 (phase III) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira-Silva, M.; Faria-Silva, C.; Viana Baptista, P.; Fernandes, E.; Ramos Fernandes, A.; Corvo, M.L. Liposomal Nanosystems in Rheumatoid Arthritis. Pharmaceutics 2021, 13, 454. https://doi.org/10.3390/pharmaceutics13040454
Ferreira-Silva M, Faria-Silva C, Viana Baptista P, Fernandes E, Ramos Fernandes A, Corvo ML. Liposomal Nanosystems in Rheumatoid Arthritis. Pharmaceutics. 2021; 13(4):454. https://doi.org/10.3390/pharmaceutics13040454
Chicago/Turabian StyleFerreira-Silva, Margarida, Catarina Faria-Silva, Pedro Viana Baptista, Eduarda Fernandes, Alexandra Ramos Fernandes, and Maria Luísa Corvo. 2021. "Liposomal Nanosystems in Rheumatoid Arthritis" Pharmaceutics 13, no. 4: 454. https://doi.org/10.3390/pharmaceutics13040454