
Topical Treatments and Their Molecular/Cellular Mechanisms in Patients with Peripheral Neuropathic Pain—Narrative Review

 ,
,  ,
,  , ,
, ,
Abstract
:
1. Introduction
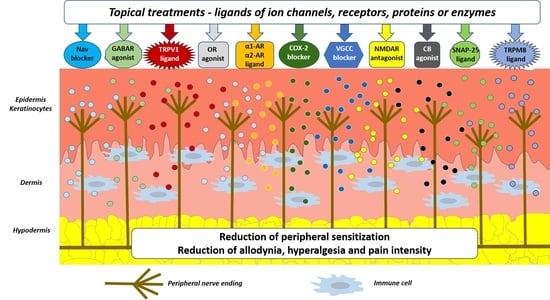
2. Topical Treatments in Patients with Neuropathic Pain
- Input from hyperexcitable peripheral neurons is crucial for development, modulation, and maintenance of NP [11].
- Upon physiological nociception, peripheral neurons exert complex interactions with immunocompetent cells and keratinocytes via neuropeptides, neurotransmitters, cytokines, and other signaling molecules acting on corresponding ion channels or receptors (Figure 1) [12]. Once pathological conditions (i.e., nerve injury, inflammation) occur, these interactions result in overactivation and disturbed functioning of neuronal and non-neuronal cells, finally contributing to neuronal hyperexcitability, peripheral sensitization, and pain [11].
3. Molecular/Cellular Mechanisms of Topical Treatments in Patients with Localized Neuropathic Pain
3.1. Treatments Acting on Voltage-Gated Sodium Channels
3.1.1. Lidocaine
- Blockade of muscarinic acetylcholine receptors (mAChR) at concentrations 1000-fold lower than needed for Nav blockade [54];
- Inhibition of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels at concentrations which block Nav1.8 [58];
- Inhibition of Toll-like receptor 4 (TLR4) at concentrations which block Nav1.8 [59];
- Inhibition of acid-sensing ion channels (ASIC), but at doses 100-fold higher than needed for Nav blockade [68];
- Inhibition of P2X purinoceptors receptor 7 (P2X7) subunits, expressed in microglia, but the exact mechanism of interaction between lidocaine and the purine receptor remains unclear [69];
- Suppression of NGF/tropomyosin receptor kinase A (TrkA) signaling due to the structural similarity of Nav and TrkA [70];
- Anti-inflammatory properties—reduction in neuroinflammation, probably via G protein-coupled receptors (GPCR), inhibition of granulocytes migration and microglial activation, reduced release of inflammatory cytokines TNFα, IL-6, and IL-1β from microglia and macrophages, reduced sensitization of peripheral nerve endings; moreover, prolonged (hours) exposure of cells to lidocaine enhances its effects on GPCR signaling [71,72,73,74,75];
3.1.2. Phenytoin
- Blockade of L-type VGCCs, observed in smooth muscle preparations as the inhibition of their spontaneous activity [86];
- Potentiation of gamma-aminobutyric acid (GABA)-induced currents through modulation of the gamma-aminobutyric acid A receptor (GABAAR) in cultured rat cortical neurons [87];
- Anti-inflammatory properties—reduction in tissue edema, decrease in inflammatory cell infiltration, and increase in epidermal growth factor, vascular endothelial growth factor, and transforming growth factor-β (TGFβ) in a rat model of wound healing [88];
- Antinociceptive effect in inflammatory pain models [89].
3.1.3. Ambroxol
3.1.4. Amitriptyline
- Inhibition of neuronal reuptake of noradrenaline and serotonin in the spinal cord [44];
- Increase in dopamine concentration in the spinal cord [44];
- Activation of the locus coeruleus in the posterior brainstem and activation of the descending noradrenergic endogenous antinociceptive system [44];
- Blockade of NMDAR in cultured rat brain neurons [109];
- Activation of Kv channels in vivo [110];
- Indirect involvement of opioid system, probably through endogenous opioid release [113];
- Activation of TRPA1 channels and subsequent probable desensitization contributing to the analgesic effect [108];
- Down-regulation of α1 adrenergic receptor (α1-AR) in the rat brain [114];
- Inhibition of the neuronal uptake of adenosine [117];
- Inhibition of the production of nitric oxide and PGE2 in synovial tissue cultures [118].
3.1.5. Doxepin
3.1.6. Funapide
3.1.7. Other Drugs—Nonsteroidal Anti-Inflammatory Drugs, Opioids, Ketamine, Menthol, Cannabidiol
3.2. Treatments Acting on Transient Receptor Potential Channels
3.2.1. Treatments Acting on Transient Receptor Potential Vanilloid 1
Capsaicin
Other Drugs—NSAIDs, Cannabinoids
3.2.2. Treatments Acting on Transient Receptor Potential Melastatin 8
Menthol
3.3. Treatments Acting on Voltage-Gated Calcium Channels
3.3.1. Gabapentin
3.3.2. Other Drugs
3.4. Treatments Acting on N-methyl-D-Aspartic Acid Receptors
3.4.1. Ketamine
- Ketamine (1.5%), baclofen (0.8%), amitriptyline (3%) [215];
- Ketamine (10%), baclofen (2%), gabapentin (6%), amitriptyline (4%), bupivacaine (2%), nifedipine (2%), clonidine (0.2%) [195];
- Ketamine, pentoxifylline, clonidine, dimethyl sulfoxide [216];
- Ketamine (5%), clonidine (0.5%), gabapentin (6%) [196].
3.4.2. Other Drugs
3.5. Treatments Acting on Cyclooxygenase-2
Nonsteroidal Anti-Inflammatory Drugs—Diclofenac
3.6. Treatments Acting on Gamma-Aminobutyric Acid Receptors
3.6.1. Baclofen
- Baclofen (0.8%), amitriptyline (3%), ketamine (1.5%) [209];
- Baclofen (2%), ketamine (10%), gabapentin (6%), amitriptyline (4%), bupivacaine (2%), nifedipine (2%), clonidine (0.2%) [195];
- Baclofen (5%), palmitoylethanolamide (1%) [246];
- Baclofen, diclofenac, ibuprofen, cyclobenzaprine, bupivacaine, gabapentin, pentoxifylline [234].
3.6.2. Other Drugs
- Antidepressants (amitriptyline, fluoxetine)—their antinociceptive effect has been observed after intraperitoneal administration in rats [111];
- Ketamine, acting as an agonist at GABAAR, revealed in an anesthetic mouse model [206];
- Phenytoin, potentiating GABA-induced currents in cultured rat cortical neurons through modulation of GABAAR [87];
- Menthol, increasing GABA-induced currents by activation of human recombinant GABAAR expressed in Xenopus oocytes [174].
3.7. Treatments Acting on α Adrenergic Receptors
3.7.1. Clonidine
3.7.2. Prazosin
3.7.3. Other Drugs
3.8. Treatments Acting on SNAP-25 and 23
Botulinum Toxin A
3.9. Treatments Acting on Peripheral Opioid Receptors
3.9.1. Loperamide
3.9.2. Morphine
3.9.3. Other Drugs Modulating Opioid System
3.10. Treatments Acting on Peripheral Cannabinoid Receptors
Cannabidiol
4. Topical Treatments in Patients with Neuropathic Pain—Summary of Possible Mechanisms of Antinociception and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.; Treede, R.-D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Attal, N.; Lanteri-Minet, M.; Laurent, B.; Fermanian, J.; Bouhassira, D. The specific disease burden of neuropathic pain: Results of a French nationwide survey. Pain 2011, 152, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Haroutounian, S.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpaa, M.; Jensen, T.S.; Kamerman, P.R.; McNicol, E.; Moore, A.; et al. Neuropathic pain clinical trials: Factors associated with decreases in estimated drug efficacy. Pain 2018, 159, 2339–2346. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Moisset, X.; Bouhassira, D.; Couturier, J.A.; Alchaar, H.; Conradi, S.; Delmotte, M.; Lanteri-Minet, M.; Lefaucheur, J.; Mick, G.; Piano, V.; et al. Pharmacological and non-pharmacological treatments for neuropathic pain: Systematic review and French recommendations. Rev. Neurol. 2020, 176, 325–352. [Google Scholar] [CrossRef]
- Pickering, G.; Lucchini, C. Topical Treatment of Localized Neuropathic Pain in the Elderly. Drugs Aging 2020, 37, 83–89. [Google Scholar] [CrossRef]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Colleoni, M.; Sacerdote, P. Murine models of human neuropathic pain. Biochim. Biophys. Acta 2010, 1802, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Kocot-Kępska, M.; Zajączkowska, R.; Mika, J.; Wordliczek, J.; Dobrogowski, J.; Przeklasa-Muszyńska, A. Peripheral Mechanisms of Neuropathic Pain—The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain. Pharmaceuticals 2021, 14, 77. [Google Scholar] [CrossRef]
- Raja, S.N.; Ringkamp, M.; Guan, Y.; Campbell, J.N.; John, J. Bonica Award Lecture: Peripheral neuronal hyperexcitability: The “low-hanging” target for safe therapeutic strategies in neuropathic pain. Pain 2020, 161, S14–S26. [Google Scholar] [CrossRef]
- Yucha, S.E.V.; Tamamoto, K.A.; Kaplan, D.L. The importance of the neuro-immuno-cutaneous system on human skin equivalent design. Cell Prolif. 2019, 52, e12677. [Google Scholar] [CrossRef] [Green Version]
- Haroutounian, S.; Nikolajsen, L.; Bendtsen, T.F.; Finnerup, N.B.; Kristensen, A.D.; Hasselstrøm, J.B.; Jensen, T.S. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 2014, 155, 1272–1279. [Google Scholar] [CrossRef]
- Haroutounian, S.; Ford, A.L.; Frey, K.; Nikolajsen, L.; Finnerup, N.B.; Neiner, A.; Kharasch, E.D.; Karlsson, P.; Bottros, M.M. How central is central poststroke pain? The role of afferent input in poststroke neuropathic pain: A prospective, open-label pilot study. Pain 2018, 159, 1317–1324. [Google Scholar] [CrossRef]
- Casale, R.; Mattia, C. Building a diagnostic algorithm on localized neuropathic pain (LNP) and targeted topical treatment: Focus on 5% lidocaine-medicated plaster. Ther. Clin. Risk Manag. 2014, 10, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Mick, G.; Baron, R.; Finnerup, N.B.; Hans, G.; Kern, K.-U.; Brett, B.; Dworkin, R.H. What is localized neuropathic pain? A first proposal to characterize and define a widely used term. Pain Manag. 2012, 2, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Casale, R.; Symeonidou, Z.; Bartolo, M. Topical Treatments for Localized Neuropathic Pain. Curr. Pain Headache Rep. 2017, 21, 15. [Google Scholar] [CrossRef] [Green Version]
- Hesselink, J.M.K.; Kopsky, D.J.; Bhaskar, A.K. Skin matters! The role of keratinocytes in nociception: A rational argument for the development of topical analgesics. J. Pain Res. 2016, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shipton, E.A. Skin Matters: Identifying Pain Mechanisms and Predicting Treatment Outcomes. Neurol. Res. Int. 2013, 2013, 329364. [Google Scholar] [CrossRef] [Green Version]
- Sawynok, J. Topical and Peripherally Acting Analgesics. Pharmacol. Rev. 2003, 55, 1–20. [Google Scholar] [CrossRef] [Green Version]
- De Leon-Casasola, O.A. Multimodal Approaches to the Management of Neuropathic Pain: The Role of Topical Analgesia. J. Pain Symptom Manag. 2007, 33, 356–364. [Google Scholar] [CrossRef]
- Müller-Schwefe, G.; Morlion, B.; Ahlbeck, K.; Alon, E.; Coaccioli, S.; Coluzzi, F.; Huygen, F.; Jaksch, W.; Kalso, E.; Kocot-Kępska, M.; et al. Treatment for chronic low back pain: The focus should change to multimodal management that reflects the underlying pain mechanisms. Curr. Med. Res. Opin. 2017, 33, 1199–1210. [Google Scholar] [CrossRef]
- Bos, J.D.; Meinardi, M.M. The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Exp. Dermatol. 2000, 9, 165–169. [Google Scholar] [CrossRef]
- Fialho, M.F.P.; Brum, E.D.S.; Pegoraro, N.S.; Couto, A.C.G.; Trevisan, G.; Cruz, L.; Oliveira, S.M. Topical transient receptor potential ankyrin 1 antagonist treatment attenuates nociception and inflammation in an ultraviolet B radiation-induced burn model in mice. J. Dermatol. Sci. 2020, 97, 135–142. [Google Scholar] [CrossRef]
- Ann, J.; Kim, H.S.; Thorat, S.A.; Kim, H.; Ha, H.-J.; Choi, K.; Kim, Y.H.; Kim, M.; Hwang, S.W.; Pearce, L.V.; et al. Discovery of Nonpungent Transient Receptor Potential Vanilloid 1 (TRPV1) Agonist as Strong Topical Analgesic. J. Med. Chem. 2019, 63, 418–424. [Google Scholar] [CrossRef]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef]
- Cardoso, F.C.; Lewis, R.J. Sodium channels and pain: From toxins to therapies. Br. J. Pharmacol. 2018, 175, 2138–2157. [Google Scholar] [CrossRef] [Green Version]
- Hameed, S. Nav1.7 and Nav1.8: Role in the pathophysiology of pain. Mol. Pain 2019, 15, 1744806919858801. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.L.H.; Woods, C.G. Painful and painless channelopathies. Lancet Neurol. 2014, 13, 587–599. [Google Scholar] [CrossRef]
- Tanaka, B.S.; Zhao, P.; Dib-Hajj, F.B.; Morisset, V.; Tate, S.; Waxman, S.G.; Dib-Hajj, S.D. A Gain-of-Function Mutation in Nav1.6 in a Case of Trigeminal Neuralgia. Mol. Med. 2016, 22, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Reimann, F.; Cox, J.J.; Belfer, I.; Diatchenko, L.; Zaykin, D.V.; McHale, D.P.; Drenth, J.P.H.; Dai, F.; Wheeler, J.; Sanders, F.; et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc. Natl. Acad. Sci. USA 2010, 107, 5148–5153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.D.; Nassar, M.A. Painful and painless mutations of SCN9A and SCN11A voltage-gated sodium channels. Pflüger’s Arch. 2020, 472, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Chahine, M.; O’Leary, M.E. Regulation/Modulation of Sensory Neuron Sodium Channels. Handb. Exp. Pharmacol. 2014, 221, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, T.; Happel, L.T.; England, J.D.; Nguyen, D.H.; Tiel, R.L.; Beuerman, R.W.; Kline, D.G. Accumulation of PN1 and PN3 Sodium Channels in Painful Human Neuroma-Evidence from Immunocytochemistry. Acta Neurochir. 2002, 144, 803–810. [Google Scholar] [CrossRef]
- Coward, K.; Plumpton, C.; Facer, P.; Birch, R.; Carlstedt, T.; Tate, S.; Bountra, C.; Anand, P. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain 2000, 85, 41–50. [Google Scholar] [CrossRef]
- Liu, M.; Wood, J.N. The Roles of Sodium Channels in Nociception: Implications for Mechanisms of Neuropathic Pain. Pain Med. 2011, 12, S93–S99. [Google Scholar] [CrossRef] [Green Version]
- McEntire, D.M.; Kirkpatrick, D.R.; Dueck, N.P.; Kerfeld, M.J.; Smith, T.A.; Nelson, T.J.; Reisbig, M.D.; Agrawal, D.K. Pain transduction: A pharmacologic perspective. Expert Rev. Clin. Pharmacol. 2016, 9, 1069–1080. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Barr, T.P.; Hou, Q.; Dib-Hajj, S.D.; Black, J.A.; Albrecht, P.J.; Petersen, K.; Eisenberg, E.; Wymer, J.P.; Rice, F.L.; et al. Voltage-gated sodium channel expression in rat and human epidermal keratinocytes: Evidence for a role in pain. Pain 2008, 30, 90–105. [Google Scholar] [CrossRef]
- Kodaira, M.; Inui, K.; Kakigi, R. Evaluation of nociceptive Aδ- and C-fiber dysfunction with lidocaine using intraepidermal electrical stimulation. Clin. Neurophysiol. 2014, 125, 1870–1877. [Google Scholar] [CrossRef]
- Thorn, C.F.; Whirl-Carrillo, M.; Leeder, J.S.; Klein, T.E.; Altman, R.B. PharmGKB summary: Phenytoin pathway. Pharmacogenet. Genom. 2012, 22, 466–470. [Google Scholar] [CrossRef]
- Zhu, W.; Li, T.; Silva, J.R.; Chen, J. Conservation and divergence in NaChBac and NaV1.7 pharmacology reveals novel drug interaction mechanisms. Sci. Rep. 2020, 10, 10730. [Google Scholar] [CrossRef]
- Kern, K.-U.; Weiser, T. Topical ambroxol for the treatment of neuropathic pain. An initial clinical observation. Schmerz 2015, 29, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Pancrazio, J.J.; Kamatchi, G.L.; Roscoe, A.K.; Lynch, C., III. Inhibition of neuronal Na+ channels by antidepressant drugs. J. Pharmacol. Exp. Ther. 1998, 284, 208–214. [Google Scholar]
- Obata, H. Analgesic Mechanisms of Antidepressants for Neuropathic Pain. Int. J. Mol. Sci. 2017, 18, 2483. [Google Scholar] [CrossRef] [Green Version]
- Sheets, P.L.; Jarecki, B.W.; Cummins, T.R. Lidocaine reduces the transition to slow inactivation in Na v 1.7 voltage-gated sodium channels. Br. J. Pharmacol. 2011, 164, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Price, N.; Namdari, R.; Neville, J.; Proctor, K.J.; Kaber, S.; Vest, J.; Fetell, M.; Malamut, R.; Sherrington, R.P.; Pimstone, S.N.; et al. Safety and Efficacy of a Topical Sodium Channel Inhibitor (TV-45070) in Patients With Postherpetic Neuralgia (PHN): A Randomized, Controlled, Proof-of-Concept, Crossover Study, With a Subgroup Analysis of the Nav1.7 R1150W Genotype. Clin. J. Pain 2017, 33, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Cummins, T.R. Setting up for the block: The mechanism underlying lidocaine’s use-dependent inhibition of sodium channels. J. Physiol. 2007, 582, 11. [Google Scholar] [CrossRef]
- Hermanns, H.; Hollmann, M.W.; Stevens, M.F.; Lirk, P.; Brandenburger, T.; Piegeler, T.; Werdehausen, R. Molecular mechanisms of action of systemic lidocaine in acute and chronic pain: A narrative review. Br. J. Anaesth. 2019, 123, 335–349. [Google Scholar] [CrossRef]
- Chevrier, P.; Vijayaragavan, K.; Chahine, M. Differential modulation of Nav 1.7 and Nav 1.8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br. J. Pharmacol. 2004, 142, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Leffler, A.; Reiprich, A.; Mohapatra, D.P.; Nau, C. Use-Dependent Block by Lidocaine but Not Amitriptyline Is More Pronounced in Tetrodotoxin (TTX)-Resistant Nav1.8 Than in TTX-Sensitive Na+ Channels. J. Pharmacol. Exp. Ther. 2006, 320, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Kirillova, I.; Teliban, A.; Gorodetskaya, N.; Grossmann, L.; Bartsch, F.; Rausch, V.H.; Struck, M.; Tode, J.; Baron, R.; Jänig, W. Effect of local and intravenous lidocaine on ongoing activity in injured afferent nerve fibers. Pain 2011, 152, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Sagen, J.; Castellanos, D.A.; Hama, A.T. Antinociceptive effects of topical mepivacaine in a rat model of HIV-associated peripheral neuropathic pain. J. Pain Res. 2016, 9, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.-T.; Donaldson, L.F.; Lumb, B.M. Differential contributions of A- and C-nociceptors to primary and secondary inflammatory hypersensitivity in the rat. Pain 2015, 156, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Picardi, S.; Stevens, M.F.; Hahnenkamp, K.; Lirk, P.; Hollmann, M.W.; Durieux, M.E. Time-dependent modulation of muscarinic m1/m3 receptor signalling by local anaesthetics. Br. J. Anaesth. 2014, 112, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.; Uchida, I.; Mashimo, T. Local anaesthetics have different mechanisms and sites of action at the recombinant N-methyl-D-aspartate (NMDA) receptors. Br. J. Pharmacol. 2003, 138, 876–882. [Google Scholar] [CrossRef] [Green Version]
- Hahnenkamp, K.; Durieux, M.E.; Schauerte, S.K.; Hoenemann, C.W.; Vegh, V.; Theilmeier, G.; Hollmann, M.W. Local anaesthetics inhibit signalling of human NMDA receptors recombinantly expressed in Xenopus laevis oocytes: Role of protein kinase C. Br. J. Anaesth. 2006, 96, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-Y.; Chung, C.-Y.; Lu, C.-W.; Huang, S.-K.; Shieh, J.-S.; Wang, S.-J. Local anesthetics inhibit glutamate release from rat cerebral cortex synaptosomes. Synapse 2013, 67, 568–579. [Google Scholar] [CrossRef]
- Hu, T.; Liu, N.; Lv, M.; Ma, L.; Peng, H.; Peng, S.; Liu, T. Lidocaine Inhibits HCN Currents in Rat Spinal Substantia Gelatinosa Neurons. Anesth. Analg. 2016, 122, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.-Y.; Tsai, P.-S.; Huang, Y.-H.; Huang, C.-J. Inhibition of toll-like receptor-4, nuclear factor-kappaB and mitogen-activated protein kinase by lignocaine may involve voltage-sensitive sodium channels. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1052–1058. [Google Scholar] [CrossRef]
- Oyama, Y.; Sadoshima, J.-I.; Tokutomi, N.; Akaike, N. Some properties of inhibitory action of lidocaine on the Ca2þ current of single isolated frog sensory neurons. Brain Res. 1988, 442, 223–228. [Google Scholar] [CrossRef]
- Lingamaneni, R.; Hemmings, H.C., Jr. Differential interaction of anaesthetics and antiepileptic drugs with neuronal Naþ channels, Ca2þ channels, and GABAA receptors. Br. J. Anaesth. 2003, 90, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Kindler, C.H.; Yost, S.C. Two-Pore Domain Potassium Channels: New Sites of Local Anesthetic Action and Toxicity. Reg. Anesth. Pain Med. 2005, 30, 260–274. [Google Scholar] [CrossRef]
- Wolff, M.; Schnöbel-Ehehalt, R.; Mühling, J.; Weigand, M.A.; Olschewski, A. Mechanisms of Lidocaine’s Action on Subtypes of Spinal Dorsal Horn Neurons Subject to the Diverse Roles of Na+ and K+ Channels in Action Potential Generation. Anesth. Analg. 2014, 119, 463–470. [Google Scholar] [CrossRef]
- Nakahira, K.; Oshita, K.; Itoh, M.; Takano, M.; Sakaguchi, Y.; Ishihara, K. Clinical concentrations of local anesthetics bupi-vacaine and lidocaine differentially inhibit human Kir2.x inward rectifier Kþ channels. Anesth. Analg. 2016, 122, 1038–1047. [Google Scholar] [CrossRef]
- Docherty, R.J.; Ginsberg, L.; Jadoon, S.; Orrell, R.W.; Bhattacharjee, A. TRPA1 insensitivity of human sural nerve axons after exposure to lidocaine. Pain 2013, 154, 1569–1577. [Google Scholar] [CrossRef]
- Leffler, A.; Fischer, M.J.; Rehner, D.; Kienel, S.; Kistner, K.; Sauer, S.K.; Gavva, N.R.; Reeh, P.W.; Nau, C. The vanilloid receptor TRPV1 is activated and sensitized by local anesthetics in rodent sensory neurons. J. Clin. Investig. 2008, 118, 763–776. [Google Scholar] [CrossRef]
- Piao, L.-H.; Fujita, T.; Yu, T.; Kumamoto, E. Presynaptic facilitation by tetracaine of glutamatergic spontaneous excitatory transmission in the rat spinal substantia gelatinosa—Involvement of TRPA1 channels. Brain Res. 2017, 1657, 245–252. [Google Scholar] [CrossRef]
- Lin, J.; Chu, X.; Maysami, S.; Li, M.; Si, H.; Cottrell, J.E.; Simon, R.P.; Xiong, Z. Inhibition of Acid Sensing Ion Channel Currents by Lidocaine in Cultured Mouse Cortical Neurons. Anesth. Analg. 2011, 112, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Okura, D.; Horishita, T.; Ueno, S.; Yanagihara, N.; Sudo, Y.; Uezono, Y.; Minami, T.; Kawasaki, T.; Sata, T. Lidocaine Preferentially Inhibits the Function of Purinergic P2X7 Receptors Expressed in Xenopus Oocytes. Anesth. Analg. 2015, 120, 597–605. [Google Scholar] [CrossRef]
- Hirose, M.; Kuroda, Y.; Murata, E. NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pract. 2016, 16, 175–182. [Google Scholar] [CrossRef]
- Hollmann, M.W.; Durieux, M.E.; Fisher, D.M. Local anesthetics and the inflammatory response: A new therapeutic indication? Anesthesiology 2000, 93, 858–875. [Google Scholar] [CrossRef]
- Lahav, M.; Levite, M.; Bassani, L.; Lang, A.; Fidder, H.; Tal, R.; Bar-Meir, S.; Mayer, L.; Chowers, Y. Lidocaine inhibits secretion of IL-8 and IL-1β and stimulates secretion of IL-1 receptor antagonist by epithelial cells. Clin. Exp. Immunol. 2002, 127, 226–233. [Google Scholar] [CrossRef]
- Hollmann, M.W. Ca-signaling G-protein-coupled receptors: A new site of local anesthetic action? Reg. Anesth. Pain Med. 2001, 26, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Van Der Wal, S.; Van Den Heuvel, S.; Radema, S.; Van Berkum, B.; Vaneker, M.; Steegers, M.; Scheffer, G.; Vissers, K. Thein vitromechanisms andin vivoefficacy of intravenous lidocaine on the neuroinflammatory response in acute and chronic pain. Eur. J. Pain 2016, 20, 655–674. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Hou, X.; Yang, S. Lidocaine Potentiates SOCS3 to Attenuate Inflammation in Microglia and Suppress Neuropathic Pain. Cell. Mol. Neurobiol. 2019, 39, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Werdehausen, R.; Kremer, D.; Brandenburger, T.; Schlösser, L.; Jadasz, J.; Küry, P.; Bauer, I.; Aragón, C.; Eulenburg, V.; Hermanns, H. Lidocaine metabolites inhibit glycine transporter 1: A novel mechanism for the analgesic action of systemic lidocaine? Anesthesiology 2012, 116, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Werdehausen, R.; Mittnacht, S.; McLaughlin, L.A.; Minett, M.S.; Armbruster, A.; Bauer, I.; Wood, J.N.; Hermanns, H.; Eulenburg, V. The lidocaine metabolite N-ethylglycine has antinociceptive effects in experimental inflammatory and neuropathic pain. Pain 2015, 156, 1647–1659. [Google Scholar] [CrossRef] [Green Version]
- Mick, G.; Correa-Illanes, G. Topical pain management with the 5% lidocaine medicated plaster—A review. Curr. Med. Res. Opin. 2012, 28, 937–951. [Google Scholar] [CrossRef]
- Hans, G.; Sabatowski, R.; Binder, A.; Boesl, I.; Rogers, P.; Baron, R. Efficacy and tolerability of a 5% lidocaine medicated plaster for the topical treatment of post-herpetic neuralgia: Results of a long-term study. Curr. Med. Res. Opin. 2009, 25, 1295–1305. [Google Scholar] [CrossRef]
- Binder, A.; Bruxelle, J.; Rogers, P.; Hans, G.; Bösl, I.; Baron, R. Topical 5% lidocaine (lignocaine) medicated plaster treatment for post-herpetic neuralgia: Results of a double-blind, placebo-controlled, multinational efficacy and safety trial. Clin. Drug Investig. 2009, 29, 393–408. [Google Scholar] [CrossRef]
- Geha, P.Y.; Baliki, M.N.; Chialvo, D.R.; Harden, R.N.; Paice, J.A.; Apkarian, A.V. Brain activity for spontaneous pain of postherpetic neuralgia and its modulation by lidocaine patch therapy. Pain 2007, 128, 88–100. [Google Scholar] [CrossRef]
- Baron, R.; Allegri, M.; Correa-Illanes, G.; Hans, G.; Serpell, M.; Mick, G.; Mayoral, V. The 5% Lidocaine-Medicated Plaster: Its Inclusion in International Treatment Guidelines for Treating Localized Neuropathic Pain, and Clinical Evidence Supporting its Use. Pain Ther. 2016, 5, 149–169. [Google Scholar] [CrossRef] [Green Version]
- Derry, S.; Wiffen, P.J.; Kalso, E.A.; Bell, R.F.; Aldington, D.; Phillips, T.; Gaskell, H.; Moore, R.A. Topical analgesics for acute and chronic pain in adults—An overview of Cochrane Reviews. Cochrane Database Syst. Rev. 2017, 5, CD008609. [Google Scholar] [CrossRef]
- Derry, S.; Wiffen, P.J.; Moore, R.A.; Quinlan, J. Topical lidocaine for neuropathic pain in adults. Cochrane Database Syst. Rev. 2014, 7, CD010958. [Google Scholar] [CrossRef]
- Wang, Y.; Jones, P.J.; Batts, T.W.; Landry, V.; Patel, M.K.; Brown, M.L. Ligand-based design and synthesis of novel sodium channel blockers from a combined phenytoin–lidocaine pharmacophore. Bioorg. Med. Chem. 2009, 17, 7064–7072. [Google Scholar] [CrossRef] [Green Version]
- Patejdl, R.; Leroux, A.-C.; Noack, T. Phenytoin inhibits contractions of rat gastrointestinal and portal vein smooth muscle by inhibiting calcium entry. Neurogastroenterol. Motil. 2015, 27, 1453–1465. [Google Scholar] [CrossRef]
- Granger, P.; Biton, B.; Faure, C.; Vige, X.; Depoortere, H.; Graham, D.; Langer, S.Z.; Scatton, B.; Avenet, P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 1995, 47, 1189–1196. [Google Scholar]
- Şimşek, G.; Çiftçi, O.; Karadag, N.; Karatas, E.; Kizilay, A. Effects of topical phenytoin on nasal wound healing after mechanical trauma: An experimental study. Laryngoscope 2014, 124, E449–E454. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, R.B.; De Carvalho, F.L.; Da Fonsêca, D.V.; Barbosa-Filho, J.M.; Salgado, P.R.R.; Paulo, L.L.; De Queiroz, A.B.M.; Pordeus, L.C.D.M.; De Souza, S.A.; Souza, H.D.D.S.; et al. Antinociceptive Effect of Hydantoin 3-Phenyl-5-(4-ethylphenyl)-imidazolidine-2,4-dione in Mice. Molecules 2015, 20, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Bendtsen, L.; Zakrzewska, J.M.; Abbott, J.; Braschinsky, M.; Di Stefano, G.; Donnet, A.; Eide, P.K.; Leal, P.R.L.; Maarbjerg, S.; May, A.; et al. European Academy of Neurology guideline on trigeminal neuralgia. Eur. J. Neurol. 2019, 26, 831–849. [Google Scholar] [CrossRef] [Green Version]
- Schnell, S.; Marrodan, M.; Acosta, J.N.; Bonamico, L.; Goicochea, M.T. Trigeminal Neuralgia Crisis—Intravenous Phenytoin as Acute Rescue Treatment. Headache 2020, 27. [Google Scholar] [CrossRef]
- McCleane, G.J. Intravenous Infusion of Phenytoin Relieves Neuropathic Pain: A Randomized, Double-Blinded, Placebo-Controlled, Crossover Study. Anesth. Analg. 1999, 89, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, J.M.K. Phenytoin repositioned in wound healing: Clinical experience spanning 60 years. Drug Discov. Today 2018, 23, 402–408. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Caruso, G.I.; De Pasquale, R.; Sortino, M.A.; Merlo, S. The Treatment of Impaired Wound Healing in Diabetes: Looking among Old Drugs. Pharmaceuticals 2020, 13, 60. [Google Scholar] [CrossRef] [Green Version]
- Kopsky, D.J.; Hesselink, J.M.K. Phenytoin Cream for the Treatment for Neuropathic Pain: Case Series. Pharmaceuticals 2018, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Kopsky, D.J.; Hesselink, J.M.K. Single-Blind Placebo-Controlled Response Test with Phenytoin 10% Cream in Neuropathic Pain Patients. Pharmaceuticals 2018, 11, 122. [Google Scholar] [CrossRef] [Green Version]
- Kopsky, D.J.; Hesselink, J.M.K. Topical phenytoin for the treatment of neuropathic pain. J. Pain Res. 2017, 10, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Kopsky, D.J.; Vrancken, A.F.J.E.; Hesselink, J.M.K.; Van Eijk, R.P.A.; Notermans, N.C. Usefulness of a Double-Blind Placebo-Controlled Response Test to Demonstrate Rapid Onset Analgesia with Phenytoin 10% Cream in Polyneuropathy. J. Pain Res. 2020, 13, 877–882. [Google Scholar] [CrossRef]
- Russell, A.L.; Kopsky, D.J.; Hesselink, J.M.K. Phenytoin Cream for the Treatment of Sciatic Pain: Clinical Effects and Theoretical Considerations: Case Report. J. Pain Palliat. Care Pharmacother. 2020, 34, 99–105. [Google Scholar] [CrossRef]
- Hesselink, J.M.K.; Kopsky, D.J. Topical Phenytoin in Neuralgic Pain, Peripheral Modulation of Central Sensitization: Two Case Reports. J. Pain Relief 2017, 6, 284. [Google Scholar] [CrossRef] [Green Version]
- Keppel Hesselink, J.M.; Kopsky, D.J. Topical Phenytoin Cream in Small Fiber Neuropathic Pain: Fast Onset of Perceptible Pain Relief. Int. J. Pain Relief 2017, 1, 15–19. [Google Scholar]
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT04647877URL (accessed on 25 January 2021).
- Sunkari, S.; Thatikonda, S.; Pooladanda, V.; Challa, V.S.; Godugu, C. Protective effects of ambroxol in psoriasis like skin inflammation: Exploration of possible mechanisms. Int. Immunopharmacol. 2019, 71, 301–312. [Google Scholar] [CrossRef]
- Weiser, T. Comparison of the effects of four Na+ channel analgesics on TTX-resistant Na+ currents in rat sensory neurons and recombinant Nav 1.2 channels. Neurosci. Lett. 2006, 395, 179–184. [Google Scholar] [CrossRef]
- Kern, P.D.K.; Schwickert-Nieswandt, M.; Maihöfner, C.; Gaul, P.D.C. Topical Ambroxol 20% for the Treatment of Classical Trigeminal Neuralgia—A New Option? Initial Clinical Case Observations. Headache 2019, 59, 418–429. [Google Scholar] [CrossRef]
- Maihöfner, C.; Schneider, S.; Bialas, P.; Gockel, H.; Beer, K.-G.; Bartels, M.; Kern, K.-U. Successful treatment of complex regional pain syndrome with topical ambroxol: A case series. Pain Manag. 2018, 8, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Dick, I.E.; Brochu, R.M.; Purohit, Y.; Kaczorowski, G.J.; Martin, W.J.; Priest, B.T. Sodium Channel Blockade May Contribute to the Analgesic Efficacy of Antidepressants. J. Pain 2007, 8, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Genevois, A.-L.; Ruel, J.; Penalba, V.; Hatton, S.; Petitfils, C.; Ducrocq, M.; Principe, P.; Dietrich, G.; Greco, C.; Delmas, P. Analgesic Effects of Topical Amitriptyline in Patients With Chemotherapy-Induced Peripheral Neuropathy: Mechanistic Insights From Studies in Mice. J. Pain 2020, 20. [Google Scholar] [CrossRef]
- Barygin, O.I.; Nagaeva, E.I.; Tikhonov, D.B.; Belinskaya, D.A.; Vanchakova, N.P.; Shestakova, N.N. Inhibition of the NMDA and AMPA receptor channels by antidepressants and antipsychotics. Brain Res. 2017, 1660, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, N.; Ghelardini, C.; Bartolini, A. Involvement of potassium channels in amitriptyline and clomipramine analgesia. Neuropharmacology 2001, 40, 75–84. [Google Scholar] [CrossRef]
- McCarson, K.E.; Duric, V.; Reisman, S.A.; Winter, M.; Enna, S. GABAB receptor function and subunit expression in the rat spinal cord as indicators of stress and the antinociceptive response to antidepressants. Brain Res. 2006, 1068, 109–117. [Google Scholar] [CrossRef]
- Malatynska, E.; Miller, C.; Schindler, N.; Cecil, A.; Knapp, A.; Crites, G.; Rogers, H. Amitriptyline increases GABA-stimulated 36Cl− influx by recombinant (α1γ2) GABAA receptors. Brain Res. 1999, 851, 277–280. [Google Scholar] [CrossRef]
- Wattiez, A.-S.; Libert, F.; Privat, A.-M.; Loiodice, S.; Fialip, J.; Eschalier, A.; Courteix, C. Evidence for a differential opioidergic involvement in the analgesic effect of antidepressants: Prediction for efficacy in animal models of neuropathic pain? Br. J. Pharmacol. 2011, 163, 792–803. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishn, D.; Subhash, M. Effect of Amitriptyline on Adrenergic Receptor Number and Second Messenger Function in Rat Brain. Pak. J. Biol. Sci. 2012, 15, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, J.E.; Whitney, D.H.; Soltani, K. Inhibition of histamine-induced pruritus by topical tricyclic antidepressants. J. Am. Acad. Dermatol. 1981, 5, 582–585. [Google Scholar] [CrossRef]
- Lawson, K. A Brief Review of the Pharmacology of Amitriptyline and Clinical Outcomes in Treating Fibromyalgia. Biomedicines 2017, 5, 24. [Google Scholar] [CrossRef]
- Sawynok, J.; Esser, M.J.; Reid, A.R. Antidepressants as analgesics: An overview of central and peripheral mechanisms of action. J. Psychiatry Neurosci. 2001, 26, 21–29. [Google Scholar]
- Yaron, I.; Shirazi, I.; Judovich, R.; Levartovsky, D.; Caspi, D.; Yaron, M. Fluoxetine and amitriptyline inhibit nitric oxide, prostaglandin E2, and hyaluronic acid production in human synovial cells and synovial tissue cultures. Arthritis Rheum. 1999, 42, 2561–2568. [Google Scholar] [CrossRef]
- Khan, M.A.; Gerner, P.; Wang, G.K. Amitriptyline for Prolonged Cutaneous Analgesia in the Rat. Anesthesiology 2002, 96, 109–116. [Google Scholar] [CrossRef]
- Ulugol, A.; Karadag, H.C.; Tamer, M.; Firat, Z.; Aslantas, A.; Dokmeci, I. Involvement of adenosine in the anti-allodynic effect of amitriptyline in streptozotocin-induced diabetic rats. Neurosci. Lett. 2002, 328, 129–132. [Google Scholar] [CrossRef]
- Haderer, A.; Gerner, P.; Kao, G.; Srinivasa, V.; Wang, G.K. Cutaneous Analgesia After Transdermal Application of Amitriptyline Versus Lidocaine in Rats. Anesth. Analg. 2003, 96, 1707–1710. [Google Scholar] [CrossRef]
- Fridrich, P.; Eappen, S.; Jaeger, W.; Schernhammer, E.; Zizza, A.M.; Wang, G.K.; Gerner, P. Phase Ia and Ib study of ami-triptyline for ulnar nerve block in humans: Side effects and efficacy. Anesthesiology 2004, 100, 1511–1518. [Google Scholar] [CrossRef]
- Gerner, P. Topical amitriptyline in healthy volunteers. Reg. Anesth. Pain Med. 2003, 28, 289–293. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Hesselink, J.M.K. High Doses of Topical Amitriptyline in Neuropathic Pain: Two Cases and Literature Review. Pain Pract. 2012, 12, 148–153. [Google Scholar] [CrossRef]
- Lynch, M.E.; Clark, A.J.; Sawynok, J. A Pilot Study Examining Topical Amitriptyline, Ketamine, and a Combination of Both in the Treatment of Neuropathic Pain. Clin. J. Pain 2003, 19, 323–328. [Google Scholar] [CrossRef]
- Lynch, M.E.; Clark, A.J.; Sawynok, J.; Sullivan, M.J.L. Topical 2% Amitriptyline and 1% Ketamine in Neuropathic Pain Syndromes. Anaesthesiology 2005, 103, 140–146. [Google Scholar] [CrossRef]
- Ho, K.-Y.; Huh, B.K.; White, W.D.; Yeh, C.-C.; Miller, E.J. Topical Amitriptyline Versus Lidocaine in the Treatment of Neuropathic Pain. Clin. J. Pain 2008, 24, 51–55. [Google Scholar] [CrossRef]
- Lockhart, E. Topical combination of amitriptyline and ketamine for post herpetic neuralgia. J. Pain 2004, 5, 82. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Hesselink, J.M.K. Multimodal Stepped Care Approach Involving Topical Analgesics for Severe Intractable Neuropathic Pain in CRPS Type 1: A Case Report. Case Rep. Med. 2011, 2011, 319750. [Google Scholar] [CrossRef] [Green Version]
- Liebregts, R.; Kopsky, D.J.; Hesselink, J.M.K. Topical Amitriptyline in Post-Traumatic Neuropathic Pain. J. Pain Symptom Manag. 2011, 41, e6–e7. [Google Scholar] [CrossRef]
- Thompson, D.F.; Brooks, K.G. Systematic review of topical amitriptyline for the treatment of neuropathic pain. J. Clin. Pharm. Ther. 2015, 40, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.; Cozzi, B.; Liebaert, F.; Hatton, S.; Viallard, M.-L.; Hermine, O.; Greco, C. High concentration of topical amitriptyline for treating chemotherapy-induced neuropathies. Support. Care Cancer 2019, 27, 3053–3059. [Google Scholar] [CrossRef] [PubMed]
- McCleane, G. Topical application of doxepin hydrochloride, capsaicin and a combination of both produces analgesia in chronic human neuropathic pain: A randomized, double-blind, placebo-controlled study. Br. J. Clin. Pharmacol. 2000, 49, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Dworsky, Z.D.; Bennett, R.; Kim, J.M.; Kuo, D.J. Severe medication-induced peripheral neuropathy treated with topical doxepin cream in a paediatric patient with leukaemia. BMJ Case Rep. 2017, 2017, bcr2017219900. [Google Scholar] [CrossRef] [PubMed]
- Sio, T.T.; Le-Rademacher, J.G.; Leenstra, J.L.; Loprinzi, C.L.; Rine, G.; Curtis, A.; Singh, A.K.; Martenson, J.A.; Novotny, P.J.; Tan, A.D.; et al. Effect of Doxepin Mouthwash or Diphenhydramine-Lidocaine-Antacid Mouthwash vs Placebo on Radiotherapy-Related Oral Mucositis Pain: The Alliance A221304 Randomized Clinical Trial. JAMA 2019, 321, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Leenstra, J.L.; Miller, R.C.; Qin, R.; Martenson, J.A.; Dornfeld, K.J.; Bearden, J.D.; Puri, D.R.; Stella, P.J.; Mazurczak, M.A.; Klish, M.D.; et al. Doxepin Rinse Versus Placebo in the Treatment of Acute Oral Mucositis Pain in Patients Receiving Head and Neck Radiotherapy With or Without Chemotherapy: A Phase III, Randomized, Double-Blind Trial (NCCTG-N09C6 [Alliance]). J. Clin. Oncol. 2014, 32, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Sudoh, Y.; Cahoon, E.E.; Gerner, P.; Wang, G.K. Tricyclic antidepressants as long-acting local anesthetics. Pain 2003, 103, 49–55. [Google Scholar] [CrossRef]
- Kumamoto, E. Inhibition of Fast Nerve Conduction Produced by Analgesics and Analgesic Adjuvants—Possible Involvement in Pain Alleviation. Pharmaceuticals 2020, 13, 62. [Google Scholar] [CrossRef] [Green Version]
- Leffler, A.; Frank, G.; Kistner, K.; Niedermirtl, F.; Koppert, W.; Reeh, P.W.; Nau, C. Local Anesthetic-like Inhibition of Voltage-gated Na(+) Channels by the Partial μ-opioid Receptor Agonist Buprenorphine. Anesthesiology 2012, 116, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zou, B.; Liang, L.; Tao, Y.-X.; Yu, H.; Wang, X.; Li, M. Loperamide inhibits sodium channels to alleviate inflammatory hyperalgesia. Neuropharmacology 2017, 117, 282–291. [Google Scholar] [CrossRef]
- Stoetzer, C.; Martell, C.; De La Roche, J.; Leffler, A. Inhibition of Voltage-Gated Na+ Channels by Bupivacaine Is Enhanced by the Adjuvants Buprenorphine, Ketamine, and Clonidine. Reg. Anesth. Pain Med. 2017, 42, 462–468. [Google Scholar] [CrossRef]
- Gaudioso, C.; Hao, J.; Martin-Eauclaire, M.-F.; Gabriac, M.; Delmas, P. Menthol pain relief through cumulative inactivation of voltage-gated sodium channels. Pain 2012, 153, 473–484. [Google Scholar] [CrossRef]
- Ghovanloo, M.-R.; Shuart, N.G.; Mezeyova, J.; Dean, R.A.; Ruben, P.C.; Goodchild, S.J. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J. Biol. Chem. 2018, 293, 16546–16558. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.H.; Cullen, B.D.; Tang, M.; Fang, Y. The Effectiveness of Topical Cannabidiol Oil in Symptomatic Relief of Peripheral Neuropathy of the Lower Extremities. Curr. Pharm. Biotechnol. 2020, 21, 390–402. [Google Scholar] [CrossRef]
- Caterina, M.J.; Pang, Z. TRP Channels in Skin Biology and Pathophysiology. Pharmaceuticals 2016, 9, 77. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y. TRPs and pain. Semin. Immunopathol. 2016, 38, 277–291. [Google Scholar] [CrossRef]
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef] [Green Version]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and pharmacology of endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar] [CrossRef]
- Zheng, J. Molecular Mechanism of TRP Channels. Compr. Physiol. 2013, 3, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar] [CrossRef] [Green Version]
- Starkus, J.; Jansen, C.; Shimoda, L.M.N.; Stokes, A.J.; Small-Howard, A.L.; Turner, H. Diverse TRPV1 responses to cannabinoids. Channels 2019, 13, 172–191. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Shinoda, M.; Furukawa, A.; Kita, K.; Noma, N.; Iwata, K. TRPA1 contributes to capsaicin-induced facial cold hyperalgesia in rats. Eur. J. Oral Sci. 2014, 122, 391–396. [Google Scholar] [CrossRef]
- Zhang, N.; Inan, S.; Cowan, A.; Sun, R.; Wang, J.M.; Rogers, T.J.; Caterina, M.; Oppenheim, J.J. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl. Acad. Sci. USA 2005, 102, 4536–4541. [Google Scholar] [CrossRef] [Green Version]
- Obreja, O.; Rathee, P.K.; Lips, K.S.; Distler, C.; Kress, M. IL-1 potentiates heat-activated currents in rat sensory neurons: Involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002, 16, 1497–1503. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef]
- Southall, M.D.; Li, T.; Gharibova, L.S.; Pei, Y.; Nicol, G.D.; Travers, J.B. Activation of Epidermal Vanilloid Receptor-1 Induces Release of Proinflammatory Mediators in Human Keratinocytes. J. Pharmacol. Exp. Ther. 2003, 304, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Słoniecka, M.; Le Roux, S.; Boman, P.; Byström, B.; Zhou, Q.; Danielson, P. Expression Profiles of Neuropeptides, Neurotransmitters, and Their Receptors in Human Keratocytes In Vitro and In Situ. PLoS ONE 2015, 10, e0134157. [Google Scholar] [CrossRef] [Green Version]
- Wilder-Smith, E.P.; Ong, W.-Y.; Guo, Y.; Chow, A.W.-L. Epidermal transient receptor potential vanilloid 1 in idiopathic small nerve fibre disease, diabetic neuropathy and healthy human subjects. Histopathology 2007, 51, 674–680. [Google Scholar] [CrossRef]
- Biggs, J.E.; Yates, J.M.; Loescher, A.R.; Clayton, N.M.; Boissonade, F.M.; Robinson, P.P. Changes in vanilloid receptor 1 (TRPV1) expression following lingual nerve injury. Eur. J. Pain 2007, 11, 192–201. [Google Scholar] [CrossRef]
- Hossain, M.; Mostafeezur, R.M.; Suzuki, A.; Hitomi, S.; Suzuki, I.; Maeda, T.; Seo, K.; Yamada, Y.; Yamamura, K.; Lev, S.; et al. Expression of TRPV1 Channels after Nerve Injury Provides an Essential Delivery Tool for Neuropathic Pain Attenuation. PLoS ONE 2012, 7, e44023. [Google Scholar] [CrossRef]
- Facer, P.; Casula, M.A.; Smith, G.D.; Benham, C.D.; Chessell, I.P.; Bountra, C.; Sinisi, M.; Birch, R.; Anand, P. Differential expression of the capsaicin receptor TRPV1 and related novel receptors TRPV3, TRPV4 and TRPM8 in normal human tissues and changes in traumatic and diabetic neuropathy. BMC Neurol. 2007, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, T.K.; Basso, L.; Iftinca, M.C.; Flynn, R.; Chapman, K.; Dietrich, G.; Vergnolle, N.; Altier, C. TRPV1 sensitization mediates post inflammatory visceral pain following acute colitis. Am. J. Physical. Gastr. Liver Physical. 2015, 309, 87–99. [Google Scholar]
- Honda, K.; Shinoda, M.; Kondo, M.; Shimizu, K.; Yonemoto, H.; Otsuki, K.; Akasaka, R.; Furukawa, A.; Iwata, K. Sensitization of TRPV1 and TRPA1 via peripheral mGluR5 signaling contributes to thermal and mechanical hypersensitivity. Pain 2017, 158, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Pajak, A.; Kolosowska, N.; Kucharczyk, M.; Starowicz, K. The importance of TRPV1-sensitisation factors for the development of neuropathic pain. Mol. Cell. Neurosci. 2015, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Vij, A.S.; Sharma, M. Mechanisms and clinical uses of capsaicin. Eur. J. Pharmacol. 2013, 720, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Rice, A.S.; Cole, P.; Tan, T.; Moore, R.A. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 2017, 1, CD007393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, G.A.; Backonja, M.M.; Dunteman, E.; Blonsky, E.R.; Vanhove, G.F.; Lu, S.P.; Tobias, J.; NGX-4010 C117 Study Group. A Multicenter, Randomized, Double-Blind, Controlled Study of NGX-4010, a High-Concentration Capsaicin Patch, for the Treatment of Postherpetic Neuralgia. Pain Med. 2011, 12, 99–109. [Google Scholar] [CrossRef]
- Wagner, T.; Poole, C.; Roth-Daniek, A. The Capsaicin 8% Patch for Neuropathic Pain in Clinical Practice: A Retrospective Analysis. Pain Med. 2013, 14, 1202–1211. [Google Scholar] [CrossRef] [Green Version]
- Nozadze, I.; Tsiklauri, N.; Gurtskaia, G.; Tsagareli, M.G. NSAIDs attenuate hyperalgesia induced by TRP channel activation. Data Brief 2016, 6, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.-K.; Caterina, M.J. TRP Channel Knockout Mice Lose Their Cool. Neuron 2007, 54, 345–347. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Fan, L.; Balakrishna, S.; Sui, A.; Morris, J.B.; Jordt, S.-E. TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain 2013, 154, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, M.; Fujita, F.; Uchida, K.; Yamamoto, S.; Shimizu, M.S.; Uotsu, C.H.; Shimizu, M.; Tominaga, M. 1,8-Cineole, a TRPM8 Agonist, is a Novel Natural Antagonist of Human TRPA1. Mol. Pain 2012, 8, 86. [Google Scholar] [CrossRef] [Green Version]
- Sidell, N.; Verity, M.A.; Nord, E.P. Menthol blocks dihydropyridine-insensitive Ca2+ channels and induces neurite outgrowth in human neuroblastoma cells. J. Cell. Physiol. 1990, 142, 410–419. [Google Scholar] [CrossRef]
- Watt, E.E.; Betts, B.A.; Kotey, F.O.; Humbert, D.J.; Griffith, T.N.; Kelly, E.W.; Veneskey, K.C.; Gill, N.; Rowan, K.C.; Jenkins, A.; et al. Menthol shares general anesthetic activity and sites of action on the GABA(A) receptor with the intravenous agent, propofol. Eur. J. Pharmacol. 2008, 590, 120–126. [Google Scholar] [CrossRef]
- Hans, M.; Wilhelm, M.; Swandulla, D. Menthol Suppresses Nicotinic Acetylcholine Receptor Functioning in Sensory Neurons via Allosteric Modulation. Chem. Senses 2012, 37, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Wasner, G.; Naleschinski, D.; Binder, A.; Schattschneider, J.; McLachlan, E.M.; Baron, R. The Effect of Menthol on Cold Allodynia in Patients with Neuropathic Pain. Pain Med. 2008, 9, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.J.; Harding, L.M.; Baranowski, A.P. A Novel Treatment of Postherpetic Neuralgia Using Peppermint Oil. Clin. J. Pain 2002, 18, 200–202. [Google Scholar] [CrossRef]
- Wright, A. Oil of peppermint as a local anaesthetic. Lancet 1870, 2464, 726. [Google Scholar] [CrossRef] [Green Version]
- Hatem, S.; Attal, N.; Willer, J.-C.; Bouhassira, D. Psychophysical study of the effects of topical application of menthol in healthy volunteers. Pain 2006, 122, 190–196. [Google Scholar] [CrossRef]
- Kumamoto, J.; Goto, M.; Denda, S.; Nakatani, M.; Takasugi, Y.; Tsuchiya, K.; Shimizu, Y.; Takatsuru, Y.; Denda, M. External negative electric potential accelerates exocytosis of lamellar bodies in human skinex vivo. Exp. Dermatol. 2013, 22, 421–423. [Google Scholar] [CrossRef]
- Denda, M.; Fujiwara, S.; Hibino, T. Expression of voltage-gated calcium channel subunit alpha1C in epidermal keratinocytes and effects of agonist and antagonists of the channel on skin barrier homeostasis. Exp. Dermatol. 2006, 15, 455–460. [Google Scholar] [CrossRef]
- Evans, A.R.; Nicol, G.D.; Vasko, M.R. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 1996, 712, 265–273. [Google Scholar] [CrossRef]
- Kawabata, A. Targeting Cav3.2 T-type calcium channels as a therapeutic strategy for chemotherapy-induced neuropathic pain. Nihon Yakurigaku Zasshi 2013, 141, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Targeting of CaV3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflüger’s Arch. 2014, 466, 701–706. [Google Scholar] [CrossRef] [PubMed]
- DuBreuil, D.M.; Lopez Soto, E.J.; Li, D.; Lipscombe, D. Peripheral voltage-gated calcium channels in skin are essential for tran-sient neurogenic thermal hyperalgesia in mice. bioRxiv 2020, 225615. [Google Scholar] [CrossRef]
- Bannister, K.; Qu, C.; Navratilova, E.; Oyarzo, J.; Xie, J.Y.; King, T.; Dickenson, A.H.; Porreca, F. Multiple sites and actions of gabapentin-induced relief of ongoing experimental neuropathic pain. Pain 2017, 158, 2386–2395. [Google Scholar] [CrossRef]
- Hara, K.; Sata, T. Inhibitory effect of gabapentin on N-methyl-d-aspartate receptors expressed in Xenopus oocytes. Acta Anaesthesiol. Scand. 2007, 51, 122–128. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Olivieri, M.; Fidilio, A.; Lupo, G.; Rusciano, D.; Pezzino, S.; Gagliano, C.; Drago, F.; Bucolo, C. Gabapentin Attenuates Ocular Inflammation: In vitro and In vivo Studies. Front. Pharmacol. 2017, 8, 173. [Google Scholar] [CrossRef]
- Manville, R.W.; Abbott, G.W. Gabapentin Is a Potent Activator of KCNQ3 and KCNQ5 Potassium Channels. Mol. Pharmacol. 2018, 94, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Shahid, M.; Subhan, F.; Ahmad, N.; Ali, G.; Akbar, S.; Fawad, K.; Sewell, R. Topical gabapentin gel alleviates allodynia and hyperalgesia in the chronic sciatic nerve constriction injury neuropathic pain model. Eur. J. Pain 2017, 21, 668–680. [Google Scholar] [CrossRef]
- Shahid, M.; Subhan, F.; Ahmad, N.; Sewell, R.D.E. Efficacy of a topical gabapentin gel in a cisplatin paradigm of chemotherapy-induced peripheral neuropathy. BMC Pharmacol. Toxicol. 2019, 28, 51. [Google Scholar] [CrossRef]
- Heustess, A.; Spigener, S.; Sweitzer, S.; Romero-Sandoval, A.; Asbill, S. Analgesic Efficacy and Transdermal Penetration of Topical Gabapentin Creams: Finding an Optimal Dose and Pre-treatment Time. Int. J. Pharm. Compd. 2015, 19, 167–173. [Google Scholar]
- Hiom, S.; Patel, G.; Newcombe, R.; Khot, S.; Martin, C. Severe postherpetic neuralgia and other neuropathic pain syndromes alleviated by topical gabapentin. Br. J. Dermatol. 2015, 173, 300–302. [Google Scholar] [CrossRef]
- Boardman, L.A.; Cooper, A.S.; Blais, L.R.; Raker, C.A. Topical Gabapentin in the Treatment of Localized and Generalized Vulvodynia. Obstet. Gynecol. 2008, 112, 579–585. [Google Scholar] [CrossRef]
- Somberg, J.C.; Molnar, J. Retrospective Study on the Analgesic Activity of a Topical (TT-CTAC) Cream in Patients With Diabetic Neuropathy and Other Chronic Pain Conditions. Am. J. Ther. 2015, 22, 214–221. [Google Scholar] [CrossRef]
- Prommer, E.E. Topical Analgesic Combinations for Bortezomib Neuropathy. J. Pain Symptom Manag. 2009, 37, e3–e5. [Google Scholar] [CrossRef]
- Woolf, C.J.; Thompson, S.W. The induction and maintenance of central sensitization is dependent on N -methyl-d-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 1991, 44, 293–299. [Google Scholar] [CrossRef]
- Coggeshall, R.E.; Carlton, S.M. Ultrastructural analysis of NMDA, AMPA, and kainate receptors on unmyelinated and mye-linated axons in the periphery. J. Comp. Neurol. 1998, 391, 78–86. [Google Scholar] [CrossRef]
- Bennett, G.J. Update on the neurophysiology of pain transmission and modulation: Focus on the NMDA-receptor. J. Pain Symptom Manag. 2000, 9, S2–S6. [Google Scholar] [CrossRef]
- Richardson, J.D.; Vasko, M.R. Cellular Mechanisms of Neurogenic Inflammation. J. Pharmacol. Exp. Ther. 2002, 302, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Nam, T.S.; Jun, J.; Jung, S.J.; Kim, D.-W.; Leem, J.W. Peripheral NMDA Receptors Mediate Antidromic Nerve Stimulation-Induced Tactile Hypersensitivity in the Rat. Mediat. Inflamm. 2015, 2015, 793624. [Google Scholar] [CrossRef] [Green Version]
- Warncke, T.; Jørum, E.; Stubhaug, A. Local treatment with the N-methyl-d-aspartate receptor antagonist ketamine, inhibit development of secondary hyperalgesia in man by a peripheral action. Neurosci. Lett. 1997, 227, 1–4. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Hesselink, J.M.K.; Bhaskar, A.; Hariton, G.; Romanenko, V.; Casale, R. Analgesic effects of topical ketamine. Minerva Anestesiol. 2015, 81, 440–449. [Google Scholar] [PubMed]
- Gupta, A.; Devi, L.A.; Gomes, I. Potentiation of μ-opioid receptor-mediated signaling by ketamine. J. Neurochem. 2011, 119, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.-C.; Ma, L.; Fan, G.-H.; Zhao, J.; Jiang, L.-Z.; Pei, G. Activation of N-Methyl-d-Aspartate Receptor Attenuates Acute Responsiveness of δ-Opioid Receptors. Mol. Pharmacol. 1997, 51, 583–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irifune, M.; Sato, T.; Kamata, Y.; Nishikawa, T.; Dohi, T.; Kawahara, M. Evidence for GABAA Receptor Agonistic Properties of Ketamine: Convulsive and Anesthetic Behavioral Models in Mice. Anesth. Analg. 2000, 91, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Yamakage, M.; Hirshman, C.A.; Croxton, T.L. Inhibitory effects of thiopental, ketamine, and propofol on voltage-dependent Ca2þ channels in porcine tracheal smooth muscle cells. Anesthesiology 1995, 83, 1274–1282. [Google Scholar] [CrossRef]
- Sleigh, J.; Harvey, M.; Voss, L.; Denny, B. Ketamine—More mechanisms of action than just NMDA blockade. Trends Anaesth. Crit. Care 2014, 4, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Yamakura, T.; Chavez-Noriega, L.E.; Harris, R.A. Subunit-dependent Inhibition of Human Neuronal Nicotinic Acetylcholine Receptors and Other Ligand-gated Ion Channels by Dissociative Anesthetics Ketamine and Dizocilpine. Anesthesiology 2000, 92, 1144–1153. [Google Scholar] [CrossRef]
- Wess, J.; Duttaroy, A.; Gomeza, J.; Zhang, W.; Yamada, M.; Felder, C.C.; Bernardini, N.; Reeh, P.W. Muscarinic receptor subtypes mediating central and peripheral antinociception studied with muscarinic receptor knockout mice: A review. Life Sci. 2003, 72, 2047–2054. [Google Scholar] [CrossRef]
- Liu, F.-L.; Chen, T.-L.; Chen, R.-M. Mechanisms of ketamine-induced immunosuppression. Acta Anaesthesiol. Taiwanica 2012, 50, 172–177. [Google Scholar] [CrossRef]
- Everton, D.; Bhagwat, D.; Damask, M. A multicenter, double blind, randomized, placebo controlled study of the efficacy/safety of two doses of amitriptyline/ketamine topical cream in treating post-herpetic neuralgia (PHN). J. Pain 2007, 8, S47. [Google Scholar] [CrossRef]
- Gewandter, J.S.; Mohile, S.G.; Heckler, C.E.; Ryan, J.L.; Kirshner, J.J.; Flynn, P.J.; Hopkins, J.O.; Morrow, G.R. A phase III randomized, placebo-controlled study of topical amitriptyline and ketamine for chemotherapy-induced peripheral neuropathy (CIPN): A University of Rochester CCOP study of 462 cancer survivors. Support. Care Cancer 2014, 22, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Sawynok, J.; Zinger, C. Topical amitriptyline and ketamine for post-herpetic neuralgia and other forms of neuropathic pain. Expert Opin. Pharmacother. 2016, 17, 601–609. [Google Scholar] [CrossRef]
- Barton, D.L.; Wos, E.J.; Qin, R.; Mattar, B.I.; Green, N.B.; Lanier, K.S.; Bearden, J.D.; Kugler, J.W.; Hoff, K.L.; Reddy, P.S.; et al. A double-blind, placebo-controlled trial of a topical treatment for chemotherapy-induced peripheral neuropathy: NCCTG trial N06CA. Support. Care Cancer 2011, 19, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.A.; Santarelli, D.M. A Novel Compound Analgesic Cream (Ketamine, Pentoxifylline, Clonidine, DMSO) for Complex Regional Pain Syndrome Patients. Pain Pract. 2016, 16, E14–E20. [Google Scholar] [CrossRef]
- Mahoney, J.M.; Vardaxis, V.; Moore, J.L.; Hall, A.M.; Haffner, K.E.; Peterson, M.C. Topical ketamine cream in the treatment of painful diabetic neuropathy: A randomized, placebo-controlled, double blind initial study. J. Am. Podiatr. Med. Assoc. 2012, 102, 178–183. [Google Scholar] [CrossRef]
- Barros, G.A.; Miot, H.A.; Braz, A.M.; Ramos, F.; Borges, M.A. Topical (S)-ketamine for pain management of postherpetic neuralgia. An. Bras. Dermatol. 2012, 87, 504–505. [Google Scholar] [CrossRef]
- Quan, D.; Wellish, M.; Gilden, N.H. Topical ketamine treatment of postherpetic neuralgia. Neurology 2003, 60, 1391–1392. [Google Scholar] [CrossRef]
- Gammaitoni, A.; Gallagher, R.M.; Welz-Bosna, M. Topical Ketamine Gel: Possible Role in Treating Neuropathic Pain. Pain Med. 2000, 1, 97–100. [Google Scholar] [CrossRef]
- Finch, P.M.; Knudsen, L.; Drummond, P.D. Reduction of allodynia in patients with complex regional pain syndrome: A double-blind placebo-controlled trial of topical ketamine. Pain 2009, 146, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Crowley, K.L.; Flores, J.A.; Hughes, C.N.; Iacono, R.P. Clinical application of ketamine ointment in the treatment of sympathetically mediated pain. J. Pharm. Compd. 1998, 2, 122–127. [Google Scholar]
- Ushida, T.; Tani, T.; Kanbara, T.; Zinchuk, V.S.; Kawasaki, M.; Yamamoto, H. Analgesic effects of ketamine ointment in patients with complex regional pain syndrome type 1. Reg. Anesth. Pain Med. 2002, 27, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Durham, M.J.; Mekhjian, H.S.; Goad, J.A.; Lou, M.; Ding, M.; Richeimer, S.H. Topical Ketamine in the Treatment of Complex Regional Pain Syndrome. Int. J. Pharm. Compd. 2018, 22, 172–175. [Google Scholar] [PubMed]
- Kopsky, D.J.; Hesselink, J.M.K. Treatment of chronic regional pain syndrome type 1 with palmitoylethanolamide and topical ketamine cream: Modulation of nonneuronal cells. J. Pain Res. 2013, 21, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.-D.; Svensson, P.; Cairns, B.E. The analgesic action of topical diclofenac may be mediated through peripheral NMDA receptor antagonism. Pain 2009, 147, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Chabot, J.-G.; Vercauteren, F.; Quirion, R. Injured nerve-derived COX2/PGE2 contributes to the maintenance of neuropathic pain in aged rats. Neurobiol. Aging 2010, 31, 1227–1237. [Google Scholar] [CrossRef]
- Suzuki, H.; Sasaki, E.; Nakagawa, A.; Muraki, Y.; Hatano, N.; Muraki, K. Diclofenac, a nonsteroidal anti-inflammatory drug, is an antagonist of human TRPM 3 isoforms. Pharmacol. Res. Perspect. 2016, 4, e00232. [Google Scholar] [CrossRef]
- Ortiz, M.I.; Torres-López, J.E.; Castañeda-Hernández, G.; Rosas, R.; Vidal-Cantú, G.C.; Granados-Soto, V. Pharmacological evidence for the activation of K(+) channels by diclofenac. Eur. J. Pharmacol. 2002, 438, 85–91. [Google Scholar] [CrossRef]
- Silva, L.C.R.; Castor, M.G.M.e.; Souza, T.C.; Duarte, I.D.G.; Romero, T.R.L. NSAIDs induce peripheral antinociception by interaction with the adrenergic system. Life Sci. 2015, 130, 7–11. [Google Scholar] [CrossRef]
- Silva, L.C.R.; Castor, M.G.M.E.; Navarro, L.C.; Romero, T.R.L.; Duarte, I.D.G. κ-Opioid receptor participates of NSAIDs peripheral antinociception. Neurosci. Lett. 2016, 622, 6–9. [Google Scholar] [CrossRef]
- Yarishkin, O.V.; Hwang, E.M.; Kim, D.; Yoo, J.C.; Kang, S.S.; Kim, D.R.; Shin, J.-H.-J.; Chung, H.-J.; Jeong, H.-S.; Kang, D.; et al. Diclofenac, a Non-steroidal Anti-inflammatory Drug, Inhibits L-type Ca2+ Channels in Neonatal Rat Ventricular Cardiomyocytes. Korean J. Physiol. Pharmacol. 2009, 13, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.U.; Zhang, Y.; Chen, L.; Cohen, A.; Hillary, K.S.; Vo, T.; Houghton, M.; Mao, J. Effect of 1.5% Topical Diclofenac on Clinical Neuropathic Pain. Anesthesiology 2015, 123, 191–198. [Google Scholar] [CrossRef]
- Safaeian, P.; Mattie, R.; Hahn, M.; Plastaras, C.T.; McCormick, Z.L. Novel Treatment of Radicular Pain with a Multi-Mechanistic Combination Topical Agent: A Case Series and Literature Review. Anesthesiol. Pain Med. 2016, 6, e33322. [Google Scholar] [CrossRef]
- Ngo, D.-H.; Vo, T.S. An Updated Review on Pharmaceutical Properties of Gamma-Aminobutyric Acid. Molecules 2019, 24, 2678. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Regal, M.P.; Bayón-Cordero, L.; Ordaz, R.P.; Garay, E.; Limon, A.; Arellano, R.O.; Matute, C.; Sánchez-Gómez, M.V. Expression and Function of GABA Receptors in Myelinating Cells. Front. Cell. Neurosci. 2020, 14, 256. [Google Scholar] [CrossRef]
- Magnaghi, V.; Ballabio, M.; Consoli, A.; Lambert, J.J.; Roglio, I.; Melcangi, R.C. GABA Receptor-Mediated Effects in the Peripheral Nervous System: A Cross-Interaction with Neuroactive Steroids. J. Mol. Neurosci. 2006, 28, 89–102. [Google Scholar] [CrossRef]
- Whitehead, R.; Puil, E.; Ries, C.; Schwarz, S.; Wall, R.; Cooke, J.; Putrenko, I.; Sallam, N.; MacLeod, B. GABAB receptor-mediated selective peripheral analgesia by the non-proteinogenic amino acid, isovaline. Neuroscience 2012, 213, 154–160. [Google Scholar] [CrossRef]
- Wu, C.; Qin, X.; Du, H.; Li, N.; Ren, W.; Peng, Y. The immunological function of GABAergic system. Front. Biosci. (Landmark Ed.) 2017, 22, 1162–1172. [Google Scholar]
- Denda, M.; Inoue, K.; Inomata, S.; Denda, S. γ-Aminobutyric Acid (A) Receptor Agonists Accelerate Cutaneous Barrier Recovery and Prevent Epidermal Hyperplasia Induced by Barrier Disruption. J. Investig. Dermatol. 2002, 119, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Hokazono, H.; Omori, T.; Ono, K. Effects of Single and Combined Administration of Fermented Barley Extract and γ-Aminobutyric Acid on the Development of Atopic Dermatitis in NC/Nga Mice. Biosci. Biotechnol. Biochem. 2010, 74, 135–139. [Google Scholar] [CrossRef]
- Andoh, T.; Sugiyama, K.; Fujita, M.; Iida, Y.; Nojima, H.; Saiki, I.; Kuraishi, Y. Pharmacological Evaluation of Morphine and Non-opioid Analgesic Adjuvants in a Mouse Model of Skin Cancer Pain. Biol. Pharm. Bull. 2008, 31, 520–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopsky, D.J.; Hesselink, J.M.K. Neuropathic Pain as a Result of Acromegaly, Treated with Topical Baclofen Cream. J. Pain Symptom Manag. 2013, 46, e4–e5. [Google Scholar] [CrossRef] [PubMed]
- Kopsky, D.J.; Hesselink, J.M.K.; Casale, R. Walking with Neuropathic Pain: Paradoxical Shift from Burden to Support? Case Rep. Med. 2015, 2015, 764950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crul, T.C.; Stolwijk-Swüste, J.M.; Kopsky, D.J.; Visser-Meily, J.M.A.; Post, M.W.M. Neuropathic pain in spinal cord injury: Topical analgesics as a possible treatment. Spinal Cord Ser. Cases 2020, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, J.M.K.; Kopsky, D.J.; Sajben, N.L. Vulvodynia and proctodynia treated with topical baclofen 5 % and palmitoylethanolamide. Arch. Gynaecol. Obstet. 2014, 290, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Drummond, P.D. Neuronal changes resulting in up-regulation of alpha-1 adrenoceptors after peripheral nerve injury. Neural Regen. Res. 2014, 15, 1337–1340. [Google Scholar] [CrossRef]
- Heijnen, C.; Rouppevandervoort, C.; Vandepol, M.; Kavelaars, A. Cytokines regulate α1-adrenergic receptor mRNA expression in human monocytic cells and endothelial cells. J. Neuroimmunol. 2002, 125, 66–72. [Google Scholar] [CrossRef]
- Finch, P.M.; Drummond, E.S.; Dawson, L.F.; Phillips, J.K.; Drummond, P.D. Up-Regulation of Cutaneous α1-Adrenoceptors in Complex Regional Pain Syndrome Type I. Pain Med. 2014, 15, 1945–1956. [Google Scholar] [CrossRef] [Green Version]
- Drummond, P.D.; Morellini, N.; Finch, P.M.; Birklein, F.; Knudsen, L.F. Complex regional pain syndrome: Intradermal injection of phenylephrine evokes pain and hyperalgesia in a subgroup of patients with upregulated α1-adrenoceptors on dermal nerves. Pain 2018, 159, 2296–2305. [Google Scholar] [CrossRef]
- Riedl, M.S.; Schnell, S.A.; Overland, A.C.; Chabot-Doré, A.-J.; Taylor, A.M.; Ribeiro-Da-Silva, A.; Elde, R.P.; Wilcox, G.L.; Stone, L.S. Coexpression of α2A-adrenergic and δ-opioid receptors in substance P-containing terminals in rat dorsal horn. J. Comp. Neurol. 2009, 513, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Buerkle, H. Peripheral anti-nociceptive action of alpha2-adrenoceptor agonists. Baillières Clin. Anaesthesiol. 2000, 2, 411–418. [Google Scholar] [CrossRef]
- Lavand’Homme, P.M.; Ma, W.; De Kock, M.; Eisenach, J.C. Perineural α2A-Adrenoceptor Activation Inhibits Spinal Cord Neuroplasticity and Tactile Allodynia after Nerve Injury. Anesthesiology 2002, 97, 972–980. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Kumamoto, E.; Furue, H.; Yoshimura, M. α2Adrenoceptor–mediated Presynaptic Inhibition of Primary Afferent Glutamatergic Transmission in Rat Substantia Gelatinosa Neurons. Anesthesiology 2003, 98, 682–689. [Google Scholar] [CrossRef]
- Dogrul, A.; Uzbay, T.I. Topical clonidine antinociception. Pain 2004, 111, 385–391. [Google Scholar] [CrossRef]
- Campbell, C.; Schmidt, W.; Brady, K.; Stouch, B.; Campbell, J. Topical clonidine gel reduces pain caused by diabetic neuropathy: Results of a multicenter, placebo-controlled clinical trial. J. Pain 2009, 4, S55. [Google Scholar] [CrossRef]
- Campbell, C.M.; Kipnes, M.S.; Stouch, B.C.; Brady, K.L.; Kelly, M.; Schmidt, W.K.; Petersen, K.L.; Rowbotham, M.C.; Campbell, J.N. Randomized control trial of topical clonidine for treatment of painful diabetic neuropathy. Pain 2012, 153, 1815–1823. [Google Scholar] [CrossRef] [Green Version]
- Kiani, J.; Sajedi, F.; Nasrollahi, S.A.; Esna-Ashari, F. A randomized clinical trial of efficacy and safety of the topical clonidine and capsaicin in the treatment of painful diabetic neuropathy. J. Res. Med. Sci. 2015, 20, 359–363. [Google Scholar]
- Wrzosek, A.; Woron, J.; Dobrogowski, J.; Jakowicka-Wordliczek, J.; Wordliczek, J. Topical clonidine for neuropathic pain. Cochrane Database Syst. Rev. 2015, 8, CD010967. [Google Scholar] [CrossRef]
- Khan, Z.P.; Ferguson, C.N.; Jones, R.M. Alpha-2 and imidazoline receptor agonistsTheir pharmacology and therapeutic role. Anaesthesia 1999, 54, 146–165. [Google Scholar] [CrossRef]
- Watanabe, K.; Hayasaka, S.; Hiraki, S.; Matsumoto, M.; Kadoi, C.; Nagaki, Y.; Hayasaka, Y. Effects of Topical Clonidine on Prostaglandin-E(2)-Induced Aqueous Flare Elevation in Pigmented Rabbits. Ophthalmic Res. 2000, 32, 210–214. [Google Scholar] [CrossRef]
- Drummond, E.S.; Maker, G.; Birklein, F.; Finch, P.M.; Drummond, P.D. Topical prazosin attenuates sensitivity to tactile stimuli in patients with complex regional pain syndrome. Eur. J. Pain 2016, 20, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Yokogawa, F.; Kiuchi, Y.; Ishikawa, Y.; Otsuka, N.; Masuda, Y.; Oguchi, K.; Hosoyamada, A. An Investigation of Monoamine Receptors Involved in Antinociceptive Effects of Antidepressants. Anesth. Analg. 2002, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, F.; Corradini, I.; Fossati, G.; Tomasoni, R.; Menna, E.; Matteoli, M. SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions. Front. Synaptic Neurosci. 2016, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Eubanks, H.B.; Lavoie, E.G.; Goree, J.; Kamykowski, J.A.; Gokden, N.; Fausther, M.; Dranoff, J.A. Reduction in SNAP-23 Alters Microfilament Organization in Myofibrobastic Hepatic Stellate Cells. Gene Expr. 2020, 20, 25–37. [Google Scholar] [CrossRef]
- Balkarli, A.; Sengül, C.; Tepeli, E.; Balkarli, H.; Cobankara, V. Synaptosomal-associated protein 25 (Snap-25) gene Polymorphism frequency in fibromyalgia syndrome and relationship with clinical symptoms. BMC Musculoskelet. Disord. 2014, 15, 191. [Google Scholar] [CrossRef] [Green Version]
- Welch, M.J.; Purkiss, J.R.; Foster, K.A. Sensitivity of embryonic rat dorsal root ganglia neurons to Clostridium botulinum neurotoxins. Toxicon 2000, 38, 245–258. [Google Scholar] [CrossRef]
- Meng, J.; Wang, J.; Lawrence, G.; Dolly, J.O.; Etournay, R.; Zwaenepoel, I.; Perfettini, I.; Legrain, P.; Petit, C.; El-Amraoui, A. Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J. Cell Sci. 2007, 120, 2864–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, M.; Khanijou, S.; Rubino, J.; Aoki, K.R. Subcutaneous administration of botulinum toxin A reduces formalin-induced pain. Pain 2004, 107, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Piotrowska, A.; Popiolek-Barczyk, K.; Mika, J. Botulinum Toxin Type A—A Modulator of Spinal Neuron–Glia Interactions under Neuropathic Pain Conditions. Toxins 2018, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zychowska, M.; Rojewska, E.; Makuch, W.; Luvisetto, S.; Pavone, F.; Marinelli, S.; Przewlocka, B.; Mika, J. Dataset of botulinum toxin A influence on interleukins under neuropathy. Data Brief 2016, 9, 1020–1023. [Google Scholar] [CrossRef] [Green Version]
- Cobianchi, S.; Jaramillo, J.; Luvisetto, S.; Pavone, F.; Navarro, X. Botulinum neurotoxin A promotes functional recovery after peripheral nerve injury by increasing regeneration of myelinated fibers. Neuroscience 2017, 359, 82–91. [Google Scholar] [CrossRef]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-Distance Retrograde Effects of Botulinum Neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef]
- Mika, J.; Rojewska, E.; Makuch, W.; Korostynski, M.; Luvisetto, S.; Marinelli, S.; Pavone, F.; Przewlocka, B.; Luvisetto, S. The effect of botulinum neurotoxin A on sciatic nerve injury-induced neuroimmunological changes in rat dorsal root ganglia and spinal cord. Neuroscience 2011, 175, 358–366. [Google Scholar] [CrossRef]
- Luvisetto, S.; Marinelli, S.; Lucchetti, F.; Marchi, F.; Cobianchi, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Botulinum neurotoxins and formalin-induced pain: Central vs. peripheral effects in mice. Brain Res. 2006, 1082, 124–131. [Google Scholar] [CrossRef]
- Park, J.; Park, H.J. Botulinum Toxin for the Treatment of Neuropathic Pain. Toxins 2017, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.-M.; Chung, M.E. Botulinum Toxin for Neuropathic Pain: A Review of the Literature. Toxins 2015, 7, 3127–3154. [Google Scholar] [CrossRef] [Green Version]
- Machelska, H.; Celik, M.Ö. Opioid Receptors in Immune and Glial Cells—Implications for Pain Control. Front. Immunol. 2020, 11, 300. [Google Scholar] [CrossRef] [Green Version]
- Obara, I.; Parkitna, J.R.; Korostynski, M.; Makuch, W.; Kaminska, D.; Przewlocka, B.; Przewlocki, R. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 2009, 141, 283–291. [Google Scholar] [CrossRef]
- Leel, C.-Y.; Perezl, F.M.; Wangl, W.; Guanl, X.; Zhaol, X.; Fisherl, J.L.; Guanl, Y.; Sweitzerl, S.M.; Rajal, S.N.; Taol, Y.-X. Dynamic temporal and spatial regulation of mu opioid receptor expression in primary afferent neurons following spinal nerve injury. Eur. J. Pain 2011, 15, 669–675. [Google Scholar] [CrossRef]
- Sehgal, N.; Smith, H.S.; Manchikanti, L. Peripherally acting opioids and clinical implications for pain control. Pain Physician 2011, 14, 249–258. [Google Scholar] [CrossRef]
- Bigliardi, P.L.; Bigliardi-Qi, M.; Buechner, S.; Rufli, T. Expression of μ-Opiate Receptor in Human Epidermis and Keratinocytes. J. Investig. Dermatol. 1998, 111, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigliardi-Qi, M.; Sumanovski, L.; Bigliardi, P.; Büchner, S.; Rufli, T. Mu-Opiate Receptor and Beta-Endorphin Expression in Nerve Endings and Keratinocytes in Human Skin. Dermatology 2004, 209, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Schröder, W.; Jostock, R.; Schwartz, M.; Rosson, G.; Polydefkis, M. Nociceptin/orphanin FQ opioid peptide-receptor expression in pachyonychia congenita. J. Peripher. Nerv. Syst. 2018, 23, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.F.; Guerrini, R.; Willets, J.M.; Thompson, J.P.; Caló, G.; Lambert, D.G. Nociceptin/Orphanin FQ (N/OFQ) conjugated to ATTO594: A novel fluorescent probe for the N/OFQ (NOP) receptor. Br. J. Pharmacol. 2018, 175, 4496–4506. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Ruth, L.J.; Preuss, C.V. Opioid Analgesics. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 17 February 2021. [Google Scholar]
- DeHaven-Hudkins, D.L.; Burgos, L.C.; Cassel, J.A.; Daubert, J.D.; DeHaven, R.N.; Mansson, E.; Nagasaka, H.; Yu, G.; Yaksh, T. Loperamide (ADL 2-1294), an opioid antihyperalgesic agent with peripheral selectivity. J. Pharmacol. Exp. Ther. 1999, 289, 494–502. [Google Scholar] [PubMed]
- Regnard, C.; Twycross, R.; Mihalyo, M.; Wilcock, A. Loperamide. J. Pain Symptom Manag. 2011, 42, 319–323. [Google Scholar] [CrossRef]
- Nozaki-Taguchi, N.; Yaksh, T.L. Characterization of the Antihyperalgesic Action of a Novel Peripheral Mu-opioid Receptor Agonist-Loperamide. Anesthesiology 1999, 90, 225–234. [Google Scholar] [CrossRef]
- Guan, Y.; Johanek, L.M.; Hartke, T.V.; Shim, B.; Tao, Y.-X.; Ringkamp, M.; Meyer, R.A.; Raja, S.N. Peripherally acting mu-opioid receptor agonist attenuates neuropathic pain in rats after L5 spinal nerve injury. Pain 2008, 138, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Anderson, M.; Yang, F.; Tiwari, V.; Zheng, Q.; He, S.Q.; Zhang, T.; Shu, B.; Chen, X.; Grenald, S.A.; et al. Peripherally acting m-opioid receptor agonists attenuate ongoing pain-associated behavior and spontaneous neuronal activity after nerve injury in rats. Anesthesiology 2018, 128, 1220–1236. [Google Scholar] [CrossRef]
- Iwaszkiewicz, K.S.; Hua, S. Development of an effective topical liposomal formulation for localized analgesia and anti-inflammatory actions in the Complete Freund’s Adjuvant rodent model of acute inflammatory pain. Pain Physician 2014, 17, E719–E735. [Google Scholar]
- Uhelski, M.L.; Bruce, D.; Speltz, R.; Wilcox, G.L.; Simone, D.A. Topical Application of Loperamide/Oxymorphindole, Mu and Delta Opioid Receptor Agonists, Reduces Sensitization of C-fiber Nociceptors that Possess NaV1.8. Neuroscience 2020, 15, 102–112. [Google Scholar] [CrossRef]
- Bruce, B.D.J.; Peterson, P.C.D.; Kitto, K.F.; Akgün, P.E.; Lazzaroni, B.S.; Portoghese, P.P.S.; Fairbanks, P.C.A.; Wilcox, P.G.L. Combination of a δ-opioid Receptor Agonist and Loperamide Produces Peripherally-mediated Analgesic Synergy in Mice. Anesthesiology 2019, 131, 649–663. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Bhaskar, A.K.; Zonneveldt, H.J.; Hesselink, J.M.K. Topical loperamide for the treatment of localized neuropathic pain: A case report and literature review. J. Pain Res. 2019, 12, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Krajnik, M.; Zylicz, Z.; Finlay, I.; Łuczak, J.; Van Sorge, A.A. Potential uses of topical opioids in palliative care—Report of 6 cases. Pain 1999, 80, 121–125. [Google Scholar] [CrossRef]
- Ciałkowska-Rysz, A.; Dzierżanowski, T. Topical morphine for treatment of cancer-related painful mucosal and cutaneous lesions: A double-blind, placebo-controlled cross-over clinical trial. Arch. Med. Sci. 2019, 15, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.M.; Porreca, F.; Lai, J.; Albrecht, P.J.; Rice, F.L.; Khodorova, A.; Davar, G.; Makriyannis, A.; Vanderah, T.W.; Mata, H.P.; et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc. Natl. Acad. Sci. USA 2005, 22, 3093–3098. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, R.; Baños, J.E.; Cabañero, D. The endocannabinoid system and neuropathic pain. Pain 2016, 157, S23–S32. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Pacher, P.; Tegeder, I.; Amaya, F.; Constantin, C.E.; Brenner, G.J.; Rubino, T.; Michalski, C.W.; Marsicano, G.; Monory, K.; et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat. Neurosci. 2007, 10, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Lötsch, J.; Weyer-Menkhoff, I.; Tegeder, I. Current evidence of cannabinoid-based analgesia obtained in preclinical and human experimental settings. Eur. J. Pain 2018, 22, 471–484. [Google Scholar] [CrossRef]
- Nam, G.; Jeong, S.K.; Park, B.M.; Lee, S.H.; Kim, H.J.; Hong, S.-P.; Kim, B.; Kim, B.-W. Selective Cannabinoid Receptor-1 Agonists Regulate Mast Cell Activation in an Oxazolone-Induced Atopic Dermatitis Model. Ann. Dermatol. 2016, 28, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Bih, C.I.; Chen, T.; Nunn, A.V.W.; Bazelot, M.; Dallas, M.L.; Whalley, B.J. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics 2015, 12, 699–730. [Google Scholar] [CrossRef] [Green Version]
- Ward, S.J.; McAllister, S.D.; Kawamura, R.; Murase, R.; Neelakantan, H.; Walker, E.A. Cannabidiol inhibits paclitaxel-induced neuropathic pain through 5- HT(1A) receptors without diminishing nervous system function or chemotherapy efficacy. Br. J. Pharmacol. 2014, 171, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Nichol, K.; Stott, C.; Jones, N.; Gray, R.A.; Bazelot, M.; Whalley, B.J. The proposed multimodal mechanism of action of can-nabidiol (CBD) in epilepsy: Modulation of intracellular calcium and adenosine-mediated signaling (P5.5-007). Neurology 2019, 92. [Google Scholar]
- Bruni, N.; Della Pepa, C.; Oliaro-Bosso, S.; Pessione, E.; Gastaldi, D.; Dosio, F. Cannabinoid Delivery Systems for Pain and Inflammation Treatment. Molecules 2018, 23, 2478. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=Neuropathic+Pain&term=topical&cntry=&state=&city=&dist= (accessed on 5 March 2021).
- Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=topical+neuropathic (accessed on 5 March 2021).



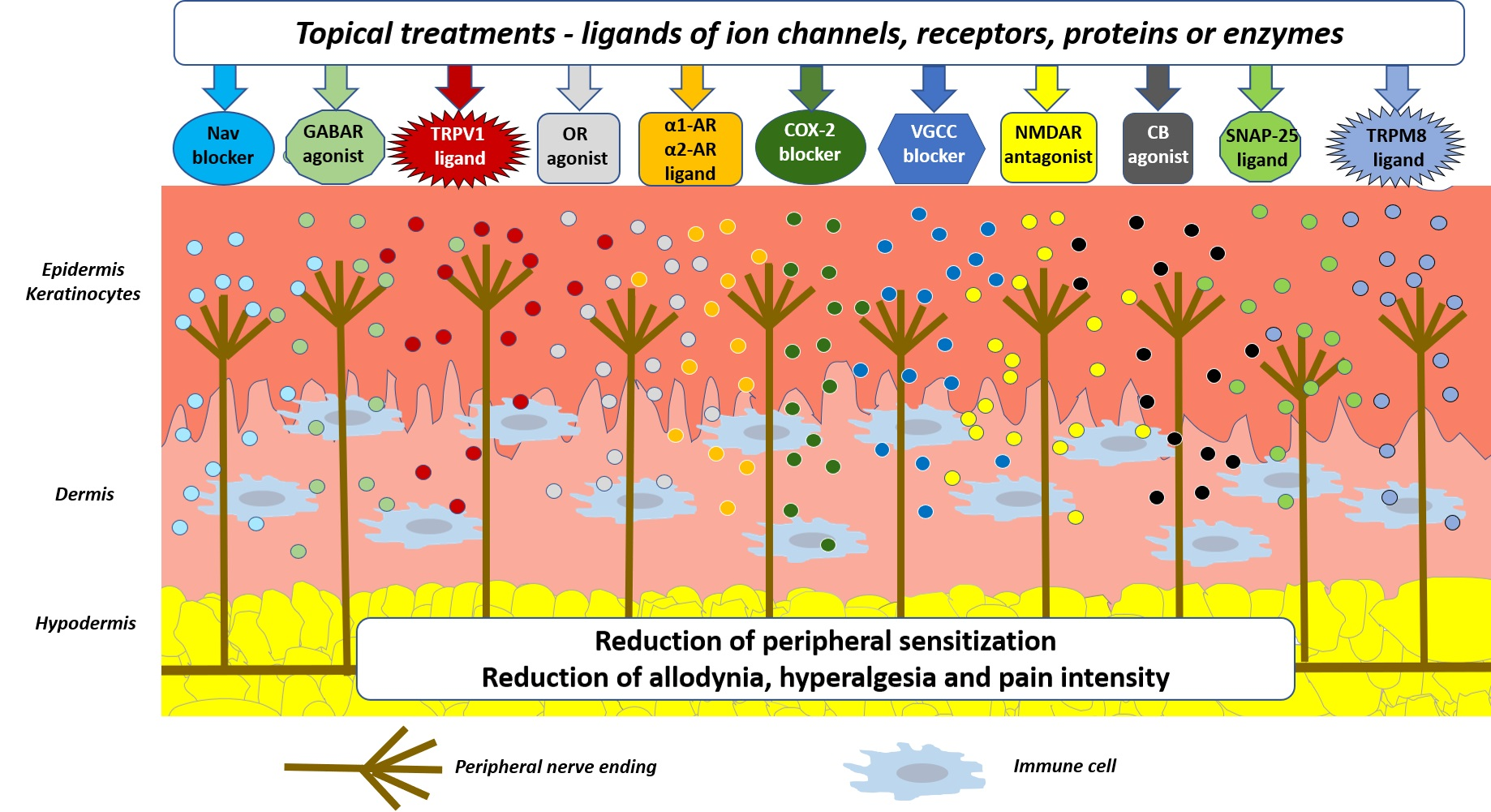{kind=link}
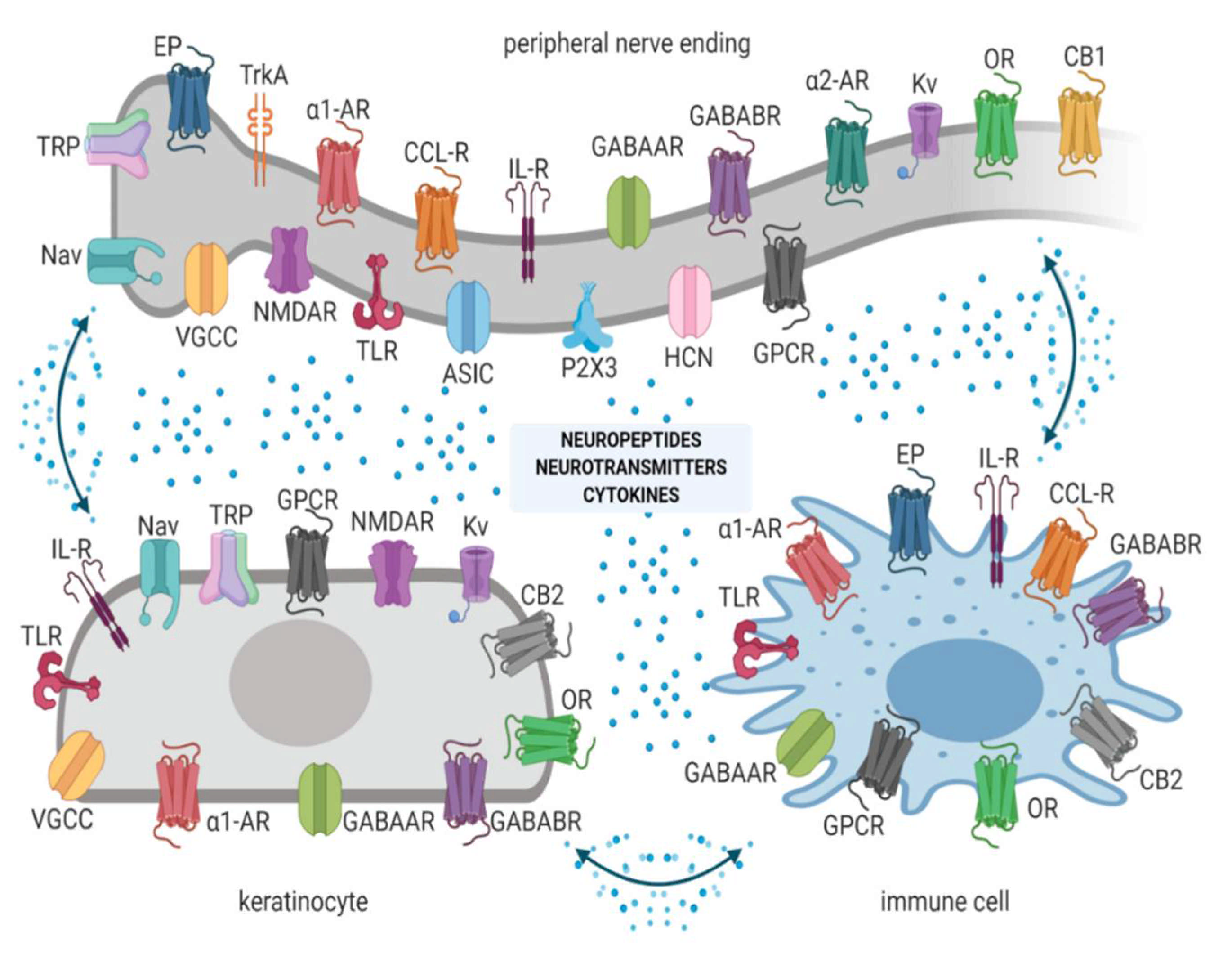{kind=link}
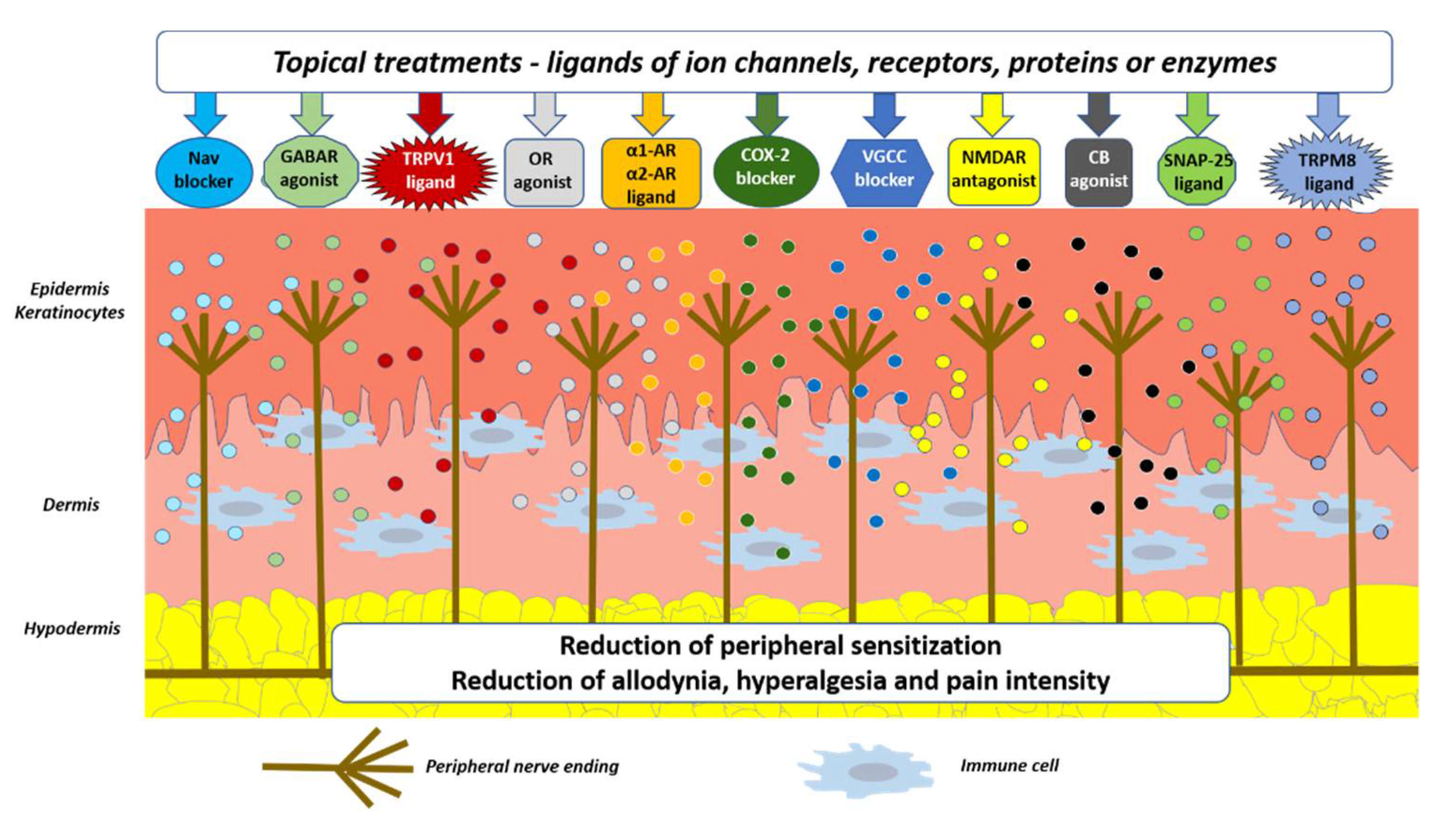{kind=link}
| Topical Agent | Direct or Indirect Mechanism of Action | Cellular Targets | Reference | Form of Drug |
|---|---|---|---|---|
| Lidocaine | Nav blockade mAChR blockade TRPA1 desensitization NMDAR antagonism ASIC blockade HCN blockade TLR4 inhibition Kv blockade VGCC blockade P2X7 inhibition NGF/TrkA modulation Gly system modulation Anti-inflammatory effect | Neurons Keratinocytes Immune cells Schwann cells | [39,45,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77] | 5% patch EMLA cream EMLA patch 2–11% cream, gel 10% spray 7% cream combined with 7% tetracaine |
| Phenytoin | Nav blockade L-VGCC blockade GABAAR modulation Anti-inflammatory effect | Neurons Keratinocytes Immune cells | [40,85,86,87,88,89] | 5–30% cream |
| Ambroxol | Nav blockade Anti-inflammatory effect | Neurons Keratinocytes Immune cells | [41,42,103,104] | 20% cream |
| Antidepressants: -Amitriptyline -Doxepin | Nav blockade VGCC blockade NMDAR antagonism Kv activation α1-AR down-regulation TRPA1 desensitization GABABR modulation 5-HT-R blockade H-R blockade mAChR blockade Reduction in NO, PGE2 NA, 5-HT, DA and adenosine reuptake inhibition Opioid system modulation | Neurons Keratinocytes | [43,44,107,108,109,110,111,112,113,114,115,116,117,118,263] | Amitriptyline: 1–10% cream Doxepin: 3.3%, 5% cream |
| Funapide | Nav1.7 blockade | Neurons Keratinocytes | [46] | hydrogel |
| Capsaicin | TRPV1 activation | Neurons Keratinocytes | [147] | 8% patch 0.025–0.1% cream |
| Menthol | TRPM8 activation GABAAR activation VGCC blockade TRPA1 inhibition nAChR blockade | Neurons Keratinocytes Immune cells | [142,171,172,173,174,175] | 2.5–16% gel 7.5%, 16% cream 16% solution 16% spray 1.25–16% patch |
| Gabapentin | VGCC blockade NMDA blockade Kv activation Anti-inflammatory effect | Neurons Keratinocytes | [186,187,188,189] | 2–6% cream |
| Ketamine | NMDA antagonism Nav blockade OR re-sensitization L-VGCC inhibition GABAA activation mAchR inhibition nAChR inhibition TLR4 inhibition Nav blockade | Neurons Keratinocytes Immune cells Schwann cells | [141,204,205,206,207,208,209,210,211] | 0.5–20% cream |
| Diclofenac | COX-2 inhibition NMDAR antagonism TRPV1, TRPA1, TRPM3 ligand Nav blockade VGCC inhibition K+ channels modulation α1-AR interaction KOR modulation | Immune cells Neurons Keratinocytes Schwann cells | [138,169,226,228,229,232] | 1–1.5% gel 140 mg patch |
| Baclofen | GABABR agonism | Neurons Keratinocytes Immune cells Schwann cells | [239] | 2%, 5% cream |
| Clonidine | α2-AR activation Nav blockade I2-R agonism Anti-inflammatory effect | Neurons Keratinocytes Immune cells | [138] [255,260,261] | 0.1%, 0.2% gel 0.1%, 0.2% cream |
| Prazosin | α1-AR blockade | Neurons Keratinocytes Immune cells | [262] | 1% cream |
| BTX-A | SNAP-25 SNAP-23 Anti-inflammatory effect | Neurons Immune cells Microglia Astroglia | [267,268,269,271] | Intradermal injections |
| Loperamide | OR agonism Nav blockade | Neurons Immune cells Keratinocytes | [138,139,140,286] | 5% loperamide cream |
| Cannabidiol | CB1 interaction Nav and Kv interaction TRPV1 desensitization 5-HT-R modulation Adenosine system modulation Opioid system modulation Synaptic GPCR interaction Anti-inflammatory effect | Neurons Immune cells Keratinocytes | [143,148,150,151,303,304,305] | ointment, cream cream: 250 mg CBD/3 fl. (around 8.3%) |
| Title of the Study | Formulations/Drugs Studied | Type of Study |
|---|---|---|
| The Effects of Topical Treatment with Clonidine + Pentoxifylline in Patients with Neuropathic Pain | Solution of clonidine (0.1%) + pentoxifylline (5%) vs. Placebo | RCT Triple blind |
| Multicentric, Open, Randomized Study Comparing Topical Treatment by Patch of Capsaicin to 8% (Qutenza) to Pregabalin Oral in the Early Treatment of Neuropathic Pain After Primary Surgery for Breast Cancer | Capsaicin 8% patch vs. Oral pregabalin | RCT Open label |
| A Phase II RCT of Topical Menthol Gel vs. Placebo in the Treatment of Chemotherapy Induced Peripheral Neuropathic Pain | Menthol gel vs. Placebo | RCT Triple blind |
| Clinical Trial Assessing the Efficacy of Capsaicin Patch (Qutenza®) in Cancer Patients with Neuropathic Pain | Capsaicin 8% patch | Open-label clinical trial |
| Intraoral Administration of Onabotulinum Toxin A for Continuous Neuropathic Pain: a Single Subject Experimental Design | BTX-A | Open-label clinical trial |
| A Multicentre, Single-Arm, Open-Label Study of the Repeated Administration of QUTENZA for the Treatment of Peripheral Neuropathic Pain | Capsaicin 8% patch | Open-label clinical trial |
| Is there a correlation between the pain relief and the A-delta- and C-fiber function after topical application of lidocaine (5%) in patients with peripheral neuropathic pain? | Lidocaine 5% patch vs. Placebo | RCT Double blind |
| Amitriptyline 10% and ketamine 10% cream in neuropathic pain: A randomised, double-blind, placebo-controlled cross-over pilot study with a three months open follow-up | Amitriptyline 10% cream vs. Ketamine 10% cream vs. Placebo | RCT Double blind |
| Enrichment randomized double-blind, placebo-controlled cross-over trial with PHEnytoin cream in patients with painful chronic idiopathic axonal polyNEuropathy | phenytoin 10% cream vs. Phenytoin 20% cream vs. Placebo | RCT Double blind |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocot-Kępska, M.; Zajączkowska, R.; Mika, J.; Kopsky, D.J.; Wordliczek, J.; Dobrogowski, J.; Przeklasa-Muszyńska, A. Topical Treatments and Their Molecular/Cellular Mechanisms in Patients with Peripheral Neuropathic Pain—Narrative Review. Pharmaceutics 2021, 13, 450. https://doi.org/10.3390/pharmaceutics13040450
Kocot-Kępska M, Zajączkowska R, Mika J, Kopsky DJ, Wordliczek J, Dobrogowski J, Przeklasa-Muszyńska A. Topical Treatments and Their Molecular/Cellular Mechanisms in Patients with Peripheral Neuropathic Pain—Narrative Review. Pharmaceutics. 2021; 13(4):450. https://doi.org/10.3390/pharmaceutics13040450
Chicago/Turabian StyleKocot-Kępska, Magdalena, Renata Zajączkowska, Joanna Mika, David J. Kopsky, Jerzy Wordliczek, Jan Dobrogowski, and Anna Przeklasa-Muszyńska. 2021. "Topical Treatments and Their Molecular/Cellular Mechanisms in Patients with Peripheral Neuropathic Pain—Narrative Review" Pharmaceutics 13, no. 4: 450. https://doi.org/10.3390/pharmaceutics13040450