
Dexibuprofen Therapeutic Advances: Prodrugs and Nanotechnological Formulations


Abstract
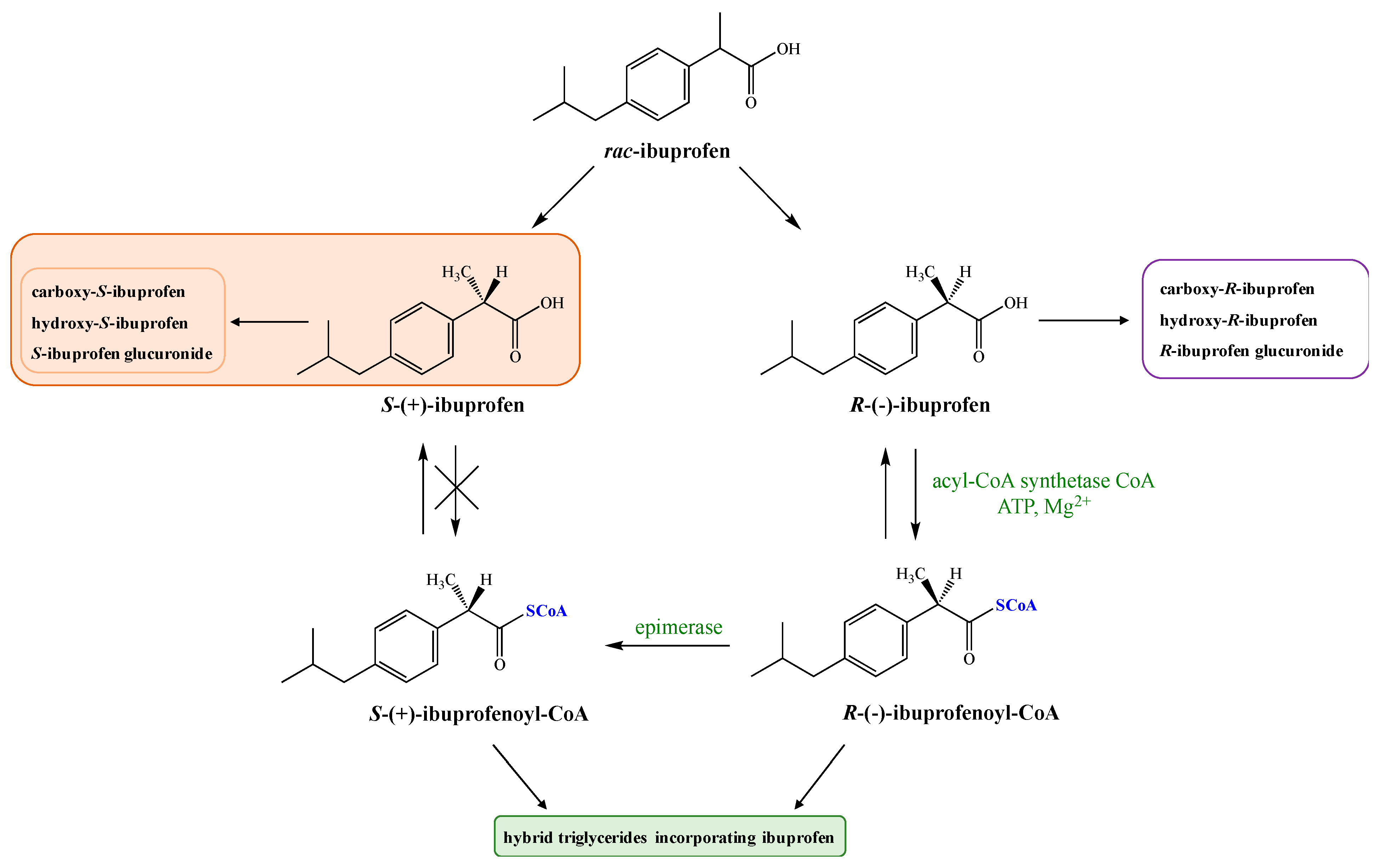
:1. Introduction
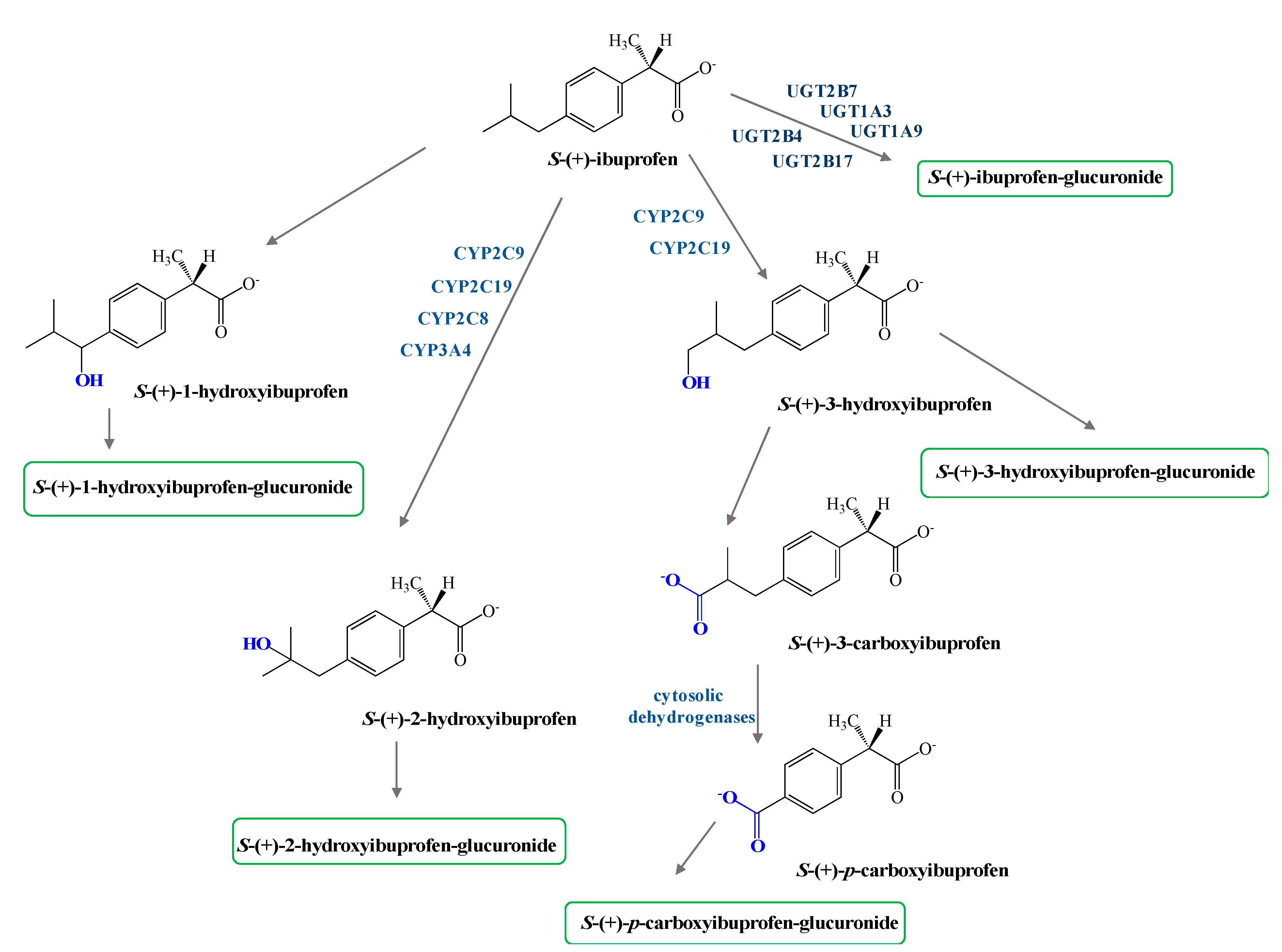
2. Dexibuprofen Main Pharmacological Properties
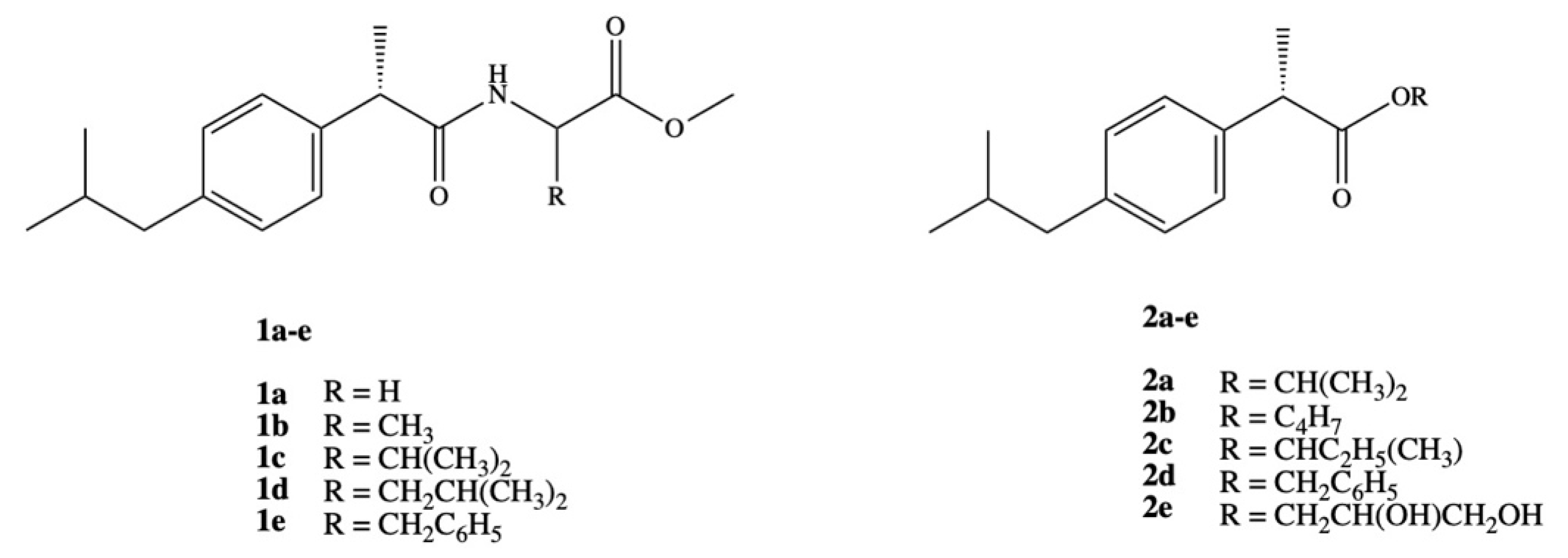
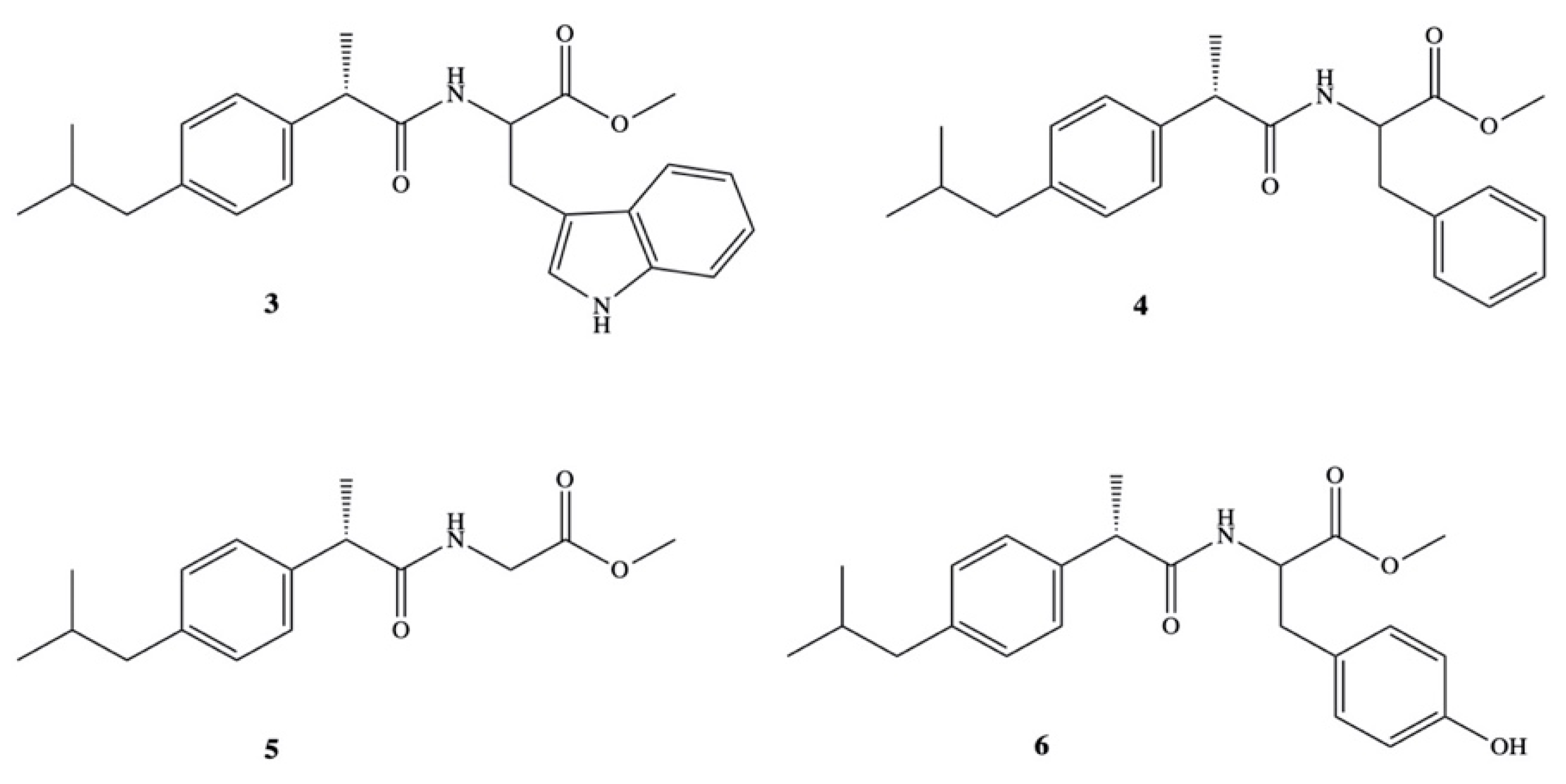
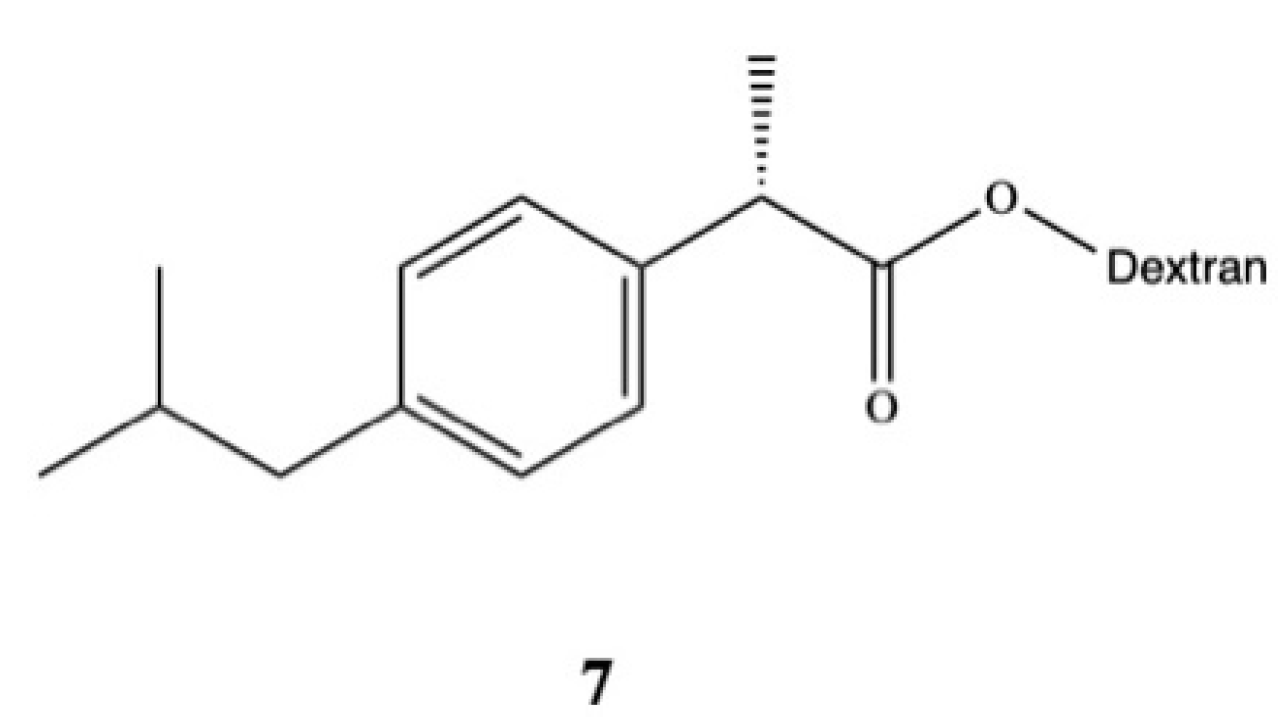
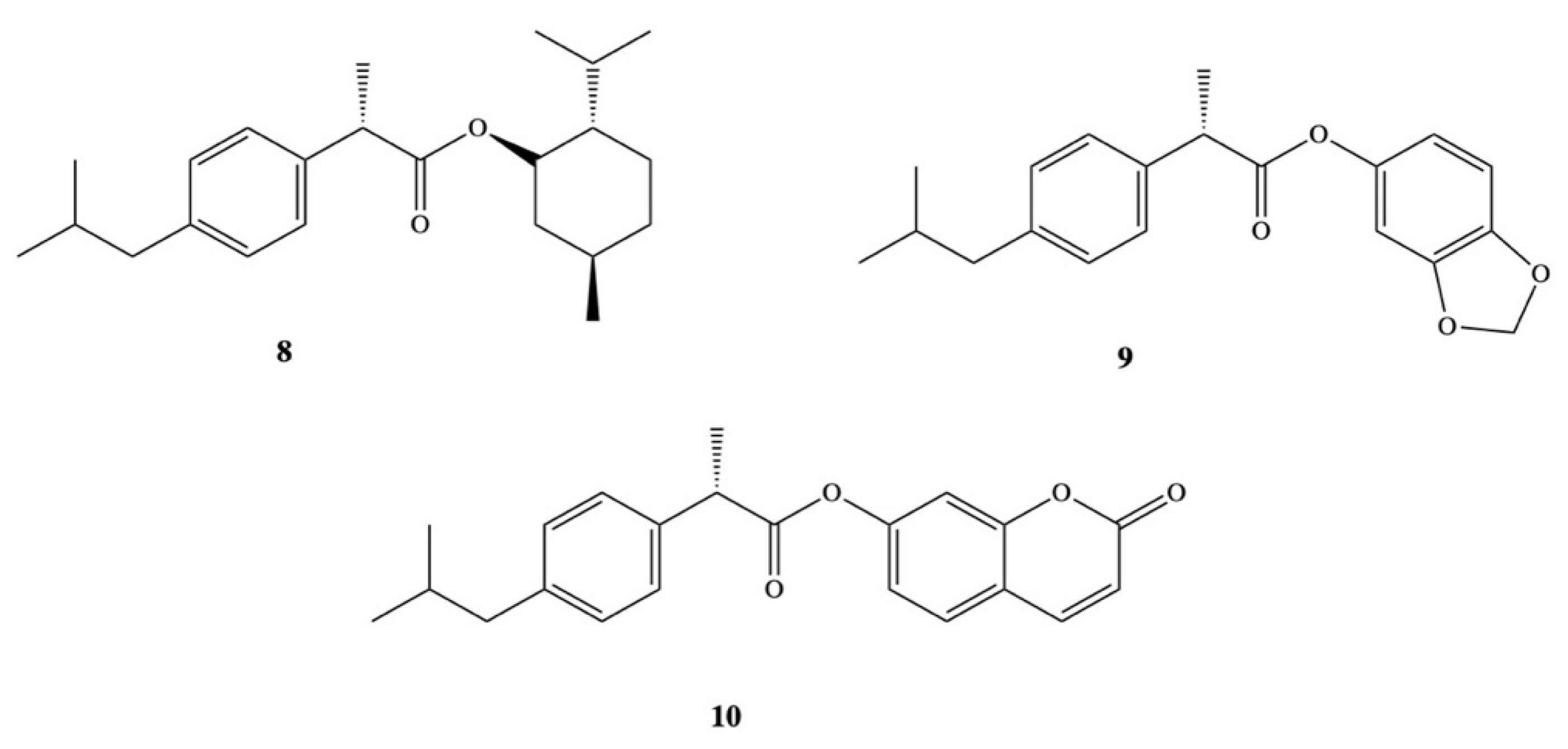
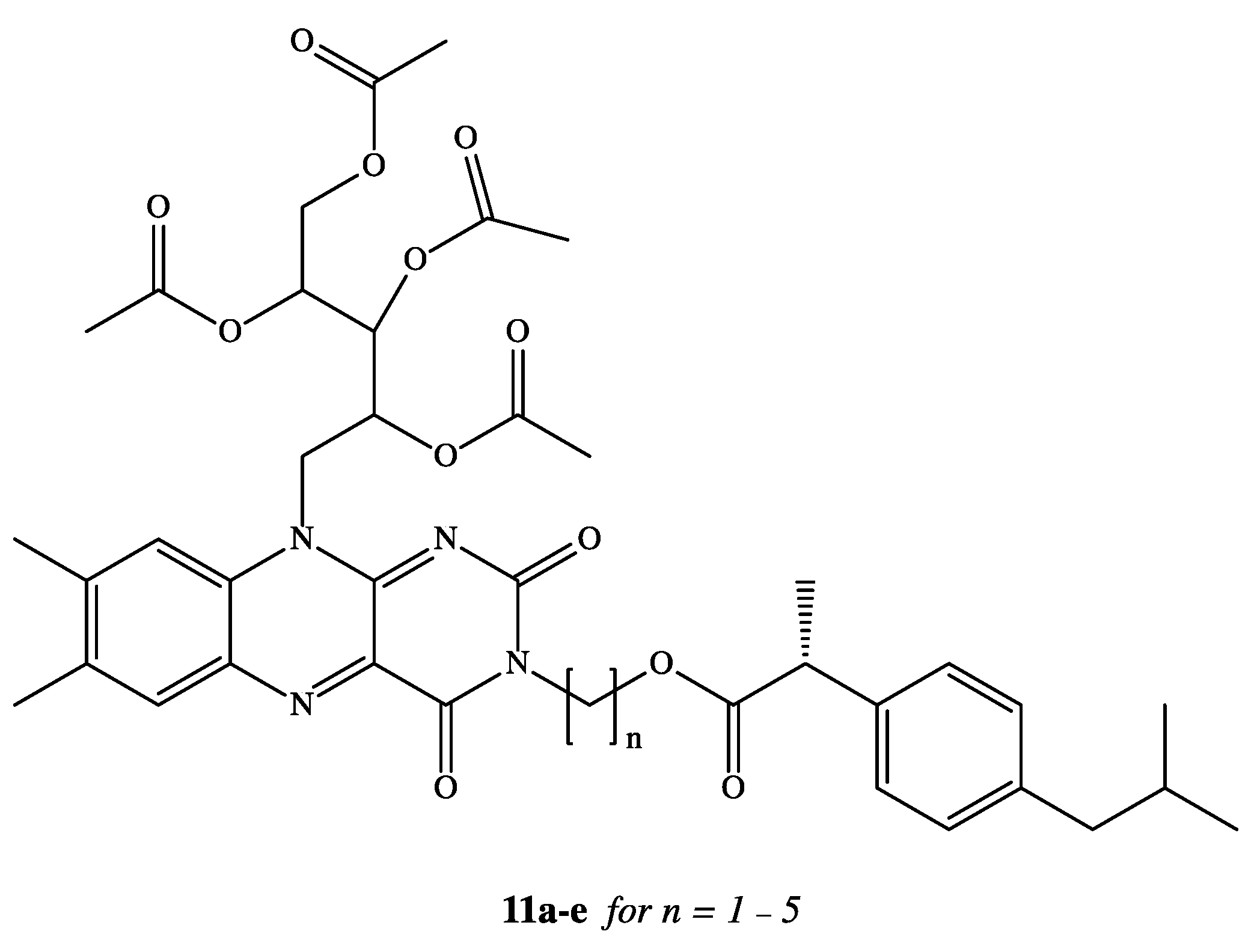
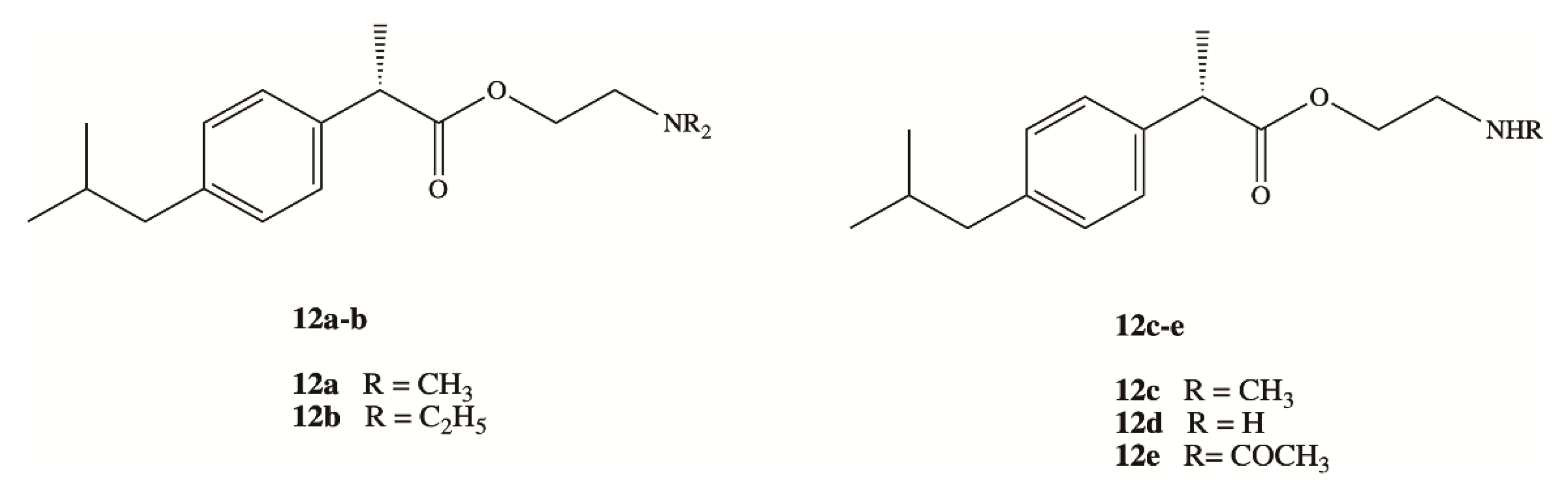
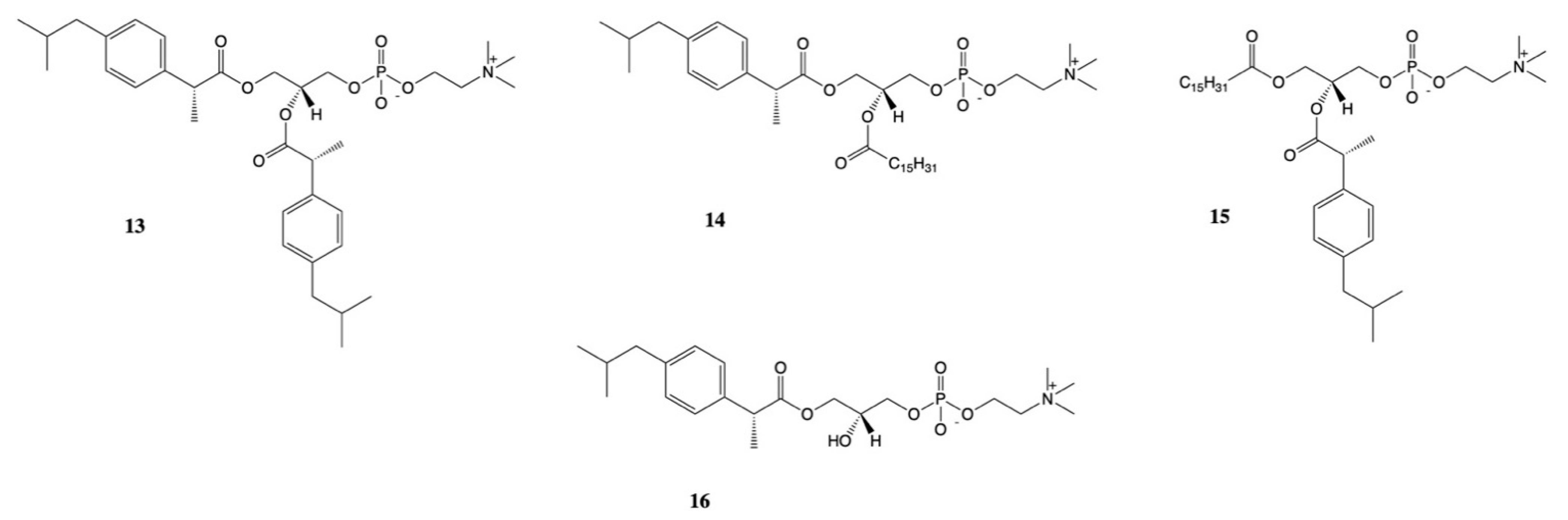
3. Dexibuprofen Prodrugs
4. Novel Strategies for Dexibuprofen Drug Delivery
4.1. Novel Strategies for Ocular Dexibuprofen Delivery
4.2. Novel Strategies for Skin Dexibuprofen Delivery
4.3. Novel Strategies for Oral Dexibuprofen Delivery
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| BBB | blood brain barrier |
| BRB | blood retinal barrier |
| CNS CoA | central nervous system coenzyme A |
| COX | cyclooxygenase |
| CSF | synovial fluid |
| DXI | dexibuprofen |
| HEC | hydroxyethyl cellulose |
| HPC | hydroxypropyl cellulose |
| HPβCD | hydroxypropyl-β- cyclodextrin |
| HPMC | hydroxypropylmethyl cellulose |
| MDD | maximum daily dose (MDD) |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| PAA | polyacrylic acid |
| PVA | polyvinyl alcohol |
| RGCs | retinal ganglion cells |
| RPE | Retinal pigment epithelium |
| SB | stratum basale |
| SC | stratum corneum |
| SG | stratum granulosum |
| SS | stratum spinosum |
| NaCMC | sodium carboxymethylcellulose |
References
- Wu, D.; Bai, X.; Lee, P.; Yang, Y.; Windsor, J.; Qian, J. A systematic review of NSAIDs treatment for acute pancreatitis in animal studies and clinical trials. Clin. Res. Hepatol. Gastroenterol. 2020, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fosslien, E. Adverse effects of nonsteroidal anti-inflammatory drugs on the gastrointestinal system. Ann. Clin. Lab. Sci. 1998, 28, 67–81. [Google Scholar] [PubMed]
- Brater, D.C. Clinical Pharmacology of NSAIDs. Clin. Pharmacol. 1988, 128, 1121–1132. [Google Scholar] [CrossRef]
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, S.T.; Phleps, W.; Hesse, E. Dexibuprofen: Pharmacology, therapeutic uses and safety. Inflammopharmacology 2003, 11, 371–383. [Google Scholar] [CrossRef]
- Chantaburanan, T.; Teeranachaideekul, V.; Chantasart, D.; Jintapattanakit, A.; Junyaprasert, V.B. Effect of binary solid lipid matrix of wax and triglyceride on lipid crystallinity, drug-lipid interaction and drug release of ibuprofen-loaded solid lipid nanoparticles (SLN) for dermal delivery. J. Colloid Interface Sci. 2017, 504, 247–256. [Google Scholar] [CrossRef]
- Hanif, A.M.; Sial, A.A.; Ali, H.; Zafar, F.; Baig, M.T.; Bushra, R.; Khan, M.A.; Nawab, A.; Mustapha, O.; Shafique, S. Dexibuprofen: Statistical assessment of drug release kinetics and investigative quality perspective. Pak. J. Pharm. Sci. 2018, 31, 2157–2162. [Google Scholar]
- Phleps, W. Overview on clinical data of dexibuprofen. Clin. Rheumatol. 2001, 20, S15–S21. [Google Scholar] [CrossRef]
- Khalid, Q.; Ahmad, M.; Usman, M.; Batool, F.; Shamshad, N.; Rehman, M. Novel β -cyclodextrin nanosponges by chain growth condensation for solubility enhancement of dexibuprofen: Characterization and acute oral toxicity studies. J. Drug Deliv. Sci. Technol. 2021, 61, 102089. [Google Scholar] [CrossRef]
- Evans, A.M.; Nation, R.L.; Sansom, L.N.; Bochner, F.; Somogyi, A.A. Effect of racemic ibuprofen dose on the magnitude and duration of platelet cyclo-oxygenase inhibition: Relationship between inhibition of thromboxane production and the plasma unbound concentration of S(+)-ibuprofen. Br. J. Clin. Pharmacol. 1991, 31, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Wsól, V.; Skálová, L.; Szotáková, B. Chiral inversion of drugs: Coincidence or principle? Curr. Drug Metab. 2004, 5, 517–533. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.S.; Jeong, D.C.; Oh, J.W.; Lee, K.Y.; Lee, H.S.; Koh, Y.Y.; Kim, J.T.; Kang, J.H.; Lee, J.S. The effects and safety of dexibuprofen compared with ibuprofen in febrile children caused by upper respiratory tract infection. Br. J. Clin. Pharmacol. 2008, 66, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Laska, E.M.; Sunshine, A.; Marrero, I.; Olson, N.; Siegel, C.; McCormick, N. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin. Pharmacol. Ther. 1986, 40, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rudy, A.C.; Knight, M.P.; Craig Brater, D.; Hall, S.D. Stereoselective administration metabolism of Ibuprofen in humans: Administration of R-, S- and Racemic lbuprofen. J. Pharmacol. Exp. Ther. 1991, 259, 1133–1139. [Google Scholar]
- Brocks, D.; Jamali, F. The pharmacokinetics of ibuprofen in humans and animals. In Ibuprofen. A Critical Bibliopgraphic Review; Rainsford, K.D., Ed.; Taylor & Francis: London, UK, 1999; pp. 89–142. [Google Scholar]
- Jeffrey, P.; Tucker, G.T.; Bye, A.; Crewe, H.K.; Wright, P.A. The Site of Inversion of R (–) -Ibuprofen: Studies Using Rat In-situ Isolated Perfused Intestine/liver Preparations. J. Pharm. Pharmacol 1991, 43, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Jamali, F.; Mehvar, R.; Russell, A.S.; Sattari, S.; Yakimets, W.W.; Koo, J. Human Pharmacokinetics of Ibuprofen Enantiomers following Different Doses and Formulations: Intestinal Chiral Inversion. Am. Pharm. Assoc. J. 1992, 81, 221–225. [Google Scholar] [CrossRef]
- Rudy, A.C.; Bradley, J.D.; Ryan, S.I.; Kalasinski, L.A.; Xiaotao, Q.; Hall, S.D. Variability in the disposition of ibuprofen enantiomers in osteoarthritis patients. Ther. Drug Monit. 1992, 14, 464–470. [Google Scholar] [CrossRef]
- Jamali, F.; Kunz-dober, C.M. Pain-mediated altered absorption and metabolism of ibuprofen: An explanation for decreased serum enantiomer concentration after dental surgery. Br. J. Clin. Pharmacol 1999, 47, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodman, T.J.; Wood, P.J.; Thompson, A.S.; Hutchings, T.J.; Steel, G.R.; Jiao, P.; Threadgill, M.D.; Lloyd, M.D. Chiral inversion of 2-arylpropionyl-CoA esters by human a-methylacyl-CoA racemase 1A (P504S)—A potential mechanism for the anti-cancer effects of ibuprofen. Chem. Commun. 2011, 47, 7332–7334. [Google Scholar] [CrossRef] [Green Version]
- Davies, N.M. Clinical Pharmacokinetics of Ibuprofen. The First 30 Years. Clin. Pharmacokinet. 1998, 34, 101–154. [Google Scholar] [CrossRef]
- Lloyd, M.D.; Yevglevskis, M.; Lee, G.L.; Wood, P.J.; Threadgill, M.D.; Woodman, T.J. Progress in Lipid Research a -Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog. Lipid Res. 2013, 52, 220–230. [Google Scholar] [CrossRef]
- Bonabello, A.; Galmozzi, M.R.; Canaparo, R.; Isaia, G.C.; Serpe, L.; Muntoni, E.; Zara, G.P. Dexibuprofen (S(+)-isomer ibuprofen) reduces gastric damage and improves analgesic and antiinflammatory effects in rodents. Anesth. Analg. 2003, 97, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawel, R.; Klein, G.; Singer, F.; Mayrhofer, F.; Kähler, S.T. Comparison of the efficacy and tolerability of dexibuprofen and celecoxib in the treatment of osteoarthritis of the hip. Int. J. Clin. Pharmacol. Ther. 2003, 41, 153–164. [Google Scholar] [CrossRef]
- Repetti, M.R.; Benedetich, C.; Jos, C.; Briand, L.E.; Cornaglia, L.M.; Bosko, M.L. Isolation of ibuprofen enantiomers and racemic esters through electrodialysis. J. Membr. Sci. 2021, 618, 1187714. [Google Scholar]
- Leising, G.; Resel, R.; Stelzer, F.; Lanziner, A.; Hantich, G.; Tasch, S. Physical aspects of dexibuprofen and racemic ibuprofen. J. Clin. Pharmacol. 1996, 36, 2–6. [Google Scholar]
- Derry, S.; Best, J.; Moore, R.A. Single dose oral dexibuprofen [S(+)-ibuprofen] for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2013, 2017. [Google Scholar] [CrossRef] [Green Version]
- Apsen Farmaceutica S.A. Phase I Study to Evaluate the Safety of Dexibuprofen 300 mg Under Fasting and Fed Conditions. Available online: https://clinicaltrials.gov/ct2/show/NCT02956512 (accessed on 18 November 2020).
- Apsen Farmaceutica S.A. Phase I Study to Evaluate the Safety of Dexibuprofen 200 mg Under Fasting and Fed Conditions. Available online: HTTPS://CLINICALTRIALS.GOV/CT2/SHOW/NCT02956525 (accessed on 14 December 2017).
- University, Inje. The Efficacy and Safety of Dexibuprofen Syrup. Available online: https://clinicaltrials.gov/ct2/show/NCT00812422 (accessed on 18 January 2010).
- Gebro Pharma GmbH. Dexibuprofen 400 mg Sachet Versus Ibuprofen 400 mg Sachet in Patients With Osteoarthritis of the Hip or Knee. Available online: https://clinicaltrials.gov/ct2/show/NCT01066676 (accessed on 10 July 2012).
- Gebro Pharma GmbH. Dexibuprofen Powder for Oral Suspension. Available online: https://www.gebro.com/en/produkte/seractil-akut-400-mg-powder-for-oral-suspension/ (accessed on 18 November 2020).
- Koziolek, M.; Grimm, M.; Schneider, F.; Jedamzik, P.; Sager, M.; Kühn, J.P.; Siegmund, W.; Weitschies, W. Navigating the human gastrointestinal tract for oral drug delivery: Uncharted waters and new frontiers. Adv. Drug Deliv. Rev. 2016, 101, 75–88. [Google Scholar] [CrossRef]
- Keep, E.R.; Sidelmann, U.G.; Hansen, S.H. Isolation and characteritztion of major phase I and II metabolites of Ibuprofen. Pharm. Res. 1997, 14, 676–680. [Google Scholar] [CrossRef]
- Hao, H.; Wang, G.; Sun, J. Enantioselective Pharmacokinetics of Ibuprofen and Involved Mechanisms. Drug Metab. Rev. 2005, 1, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.T.; Nuernberg, B.; Dietzel, K.; Brune, K.A.Y. Biliary elimination of non-steroidal anti-inflammatory drugs in patients. Br. J. Clin. Pharmacol. 1990, 29, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamman, M.A.; Thompson, G.A.; Hall, S.D. Regioselective and Stereoselective Metabolism of Ibuprofen by Human Cytochrome I? 450 2C. Biochem. Pharmacol. 1997, 54, 33–41. [Google Scholar] [CrossRef]
- Walker, J.S.; Carmody, J.J. Experimental Pain in Healthy Human Subjects: Gender Differences in Nociception and in Response to Ibuprofen. Anesth. Analg. 1998, 86, 157–1262. [Google Scholar]
- Peesa, J.P.; Yalavarthi, P.R.; Rasheed, A.; Mandava, V.B.R. A perspective review on role of novel NSAID prodrugs in the management of acute inflammation. J. Acute Dis. 2016, 5, 364–381. [Google Scholar] [CrossRef] [Green Version]
- Qandil, A.M. Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. Int. J. Mol. Sci. 2012, 13, 7244. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, Z.; Imram, M.; Amin, S. Synthesis, characteritzation and in vitro hydrolysis studies of ester and amide prodrugs of dexibuprofen. Med. Chem. Res. 2012, 21, 3361–3368. [Google Scholar] [CrossRef]
- Ashraf, Z.; Mahmood, T.; Hassan, M.; Afzal, S.; Rafique, H.; Afzal, K.; Latip, J. Dexibuprofen amide derivatives as potential anticancer agents: Synthesis, in silico docking, bioevaluation, and molecular dynamic simulation. Drug Des. Dev. Ther. 2019, 13, 1643–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, N.; Zafran, M.; Ashraf, Z.; Perveen, F. Synthesis, characterization of amide substituted dexibuprofen derivatives and their spectral, voltammetric and docking investigations for DNA binding interactions. J. Photochem. Photobiol. B Biol. 2017, 169, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Kumar, C.K.A.; Mishra, A.; Rasheed, A.; Kumar, C.K.A.; Mishra, A. Synthesis, hydrolysis studies and phamacodynamic profiles of amide prodrugs of dexibuprofen with amino acids Synthesis, hydrolysis studies and phamacodynamic profiles of amide prodrugs of dexibuprofen with amino acids. J. Enzyme Inhib. Med. Chem. 2011, 26, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; Amit Gangwal, N.S.D. Dexibuprofen-Dextran macromolecular prodrugs: Synthesis, characterization and pharmacological evaluation. Sch. Libr. Res. 2011, 2, 373–383. [Google Scholar]
- JayaPreethu, P.; Lakshmanarao, A.; Basaveswara, M.V. Design and Schematic Evaluation of Dextran Conjugated Dexibuprofen, a Gastrosparing NSAID. Am. J. Chem. 2018, 3, 30–41. [Google Scholar]
- Ashraf, Z.; Alamgeer; Rasool, R.; Hassan, M.; Ahsan, H.; Afzal, S.; Afzal, K.; Cho, H.; Kim, S.J. Synthesis, bioevaluation and molecular dynamic simulation studies of dexibuprofen-antioxidant mutual prodrugs. Int. J. Mol. Sci. 2016, 17, 2151. [Google Scholar] [CrossRef] [Green Version]
- Mazhar, D.; Ang, R.; Waxman, J. COX inhibitors and breast cancer. Br. J. Cancer 2006, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardello, F.; Offerhaus, G.J.A.; DuBois, R.N. The role of nonsteroidal anti-inflammatory drugs in colorectal cancer prevention. Eur. J. Cancer 1995, 31A, 1071. [Google Scholar] [CrossRef]
- Cai, Y.; Yousef, A.; Grandis, J.R.; Johnson, D.E. NSAID therapy for PIK3CA-Altered colorectal, breast, and head and neck cancer. Adv. Biol. Regul. 2020, 75, 1000653. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.F.; Bernstein, L.; Anton-Culver, H.; Deapen, D.; Horn-Ross, P.L.; Mohrenweiser, H.; Pell, D.; Pinder, R.; Purdie, D.M.; Reynolds, P.; et al. Nonsteroidal anti-inflammatory drug use and breast cancer by stage and hormone receptor. J. Natl. Cancer Inst. 2005, 97, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seekamp, A.; Hultquist, D.E.; Till, G.O. Protection By Vitamin B 2 Against Oxidant-Mediated Acute Lung Injury. Inflammation 1999, 23, 449–460. [Google Scholar] [CrossRef]
- Banekovich, C.; Ott, I.; Koch, T.; Matuszczak, B.; Gust, R. Synthesis and biological activities of novel dexibuprofen tetraacetylriboflavin conjugates. Bioorg. Med. Chem. Lett. 2007, 17, 683–687. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Gong, T.; Sun, X.; Zhang, Z. In vitro and in vivo investigation of dexibuprofen derivatives for CNS delivery. Acta Pharmacol. Sin. 2012, 33, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, Y.; Jiang, J.; Wang, X.; Fu, Y.; Gong, T.; Sun, X.; Zhang, Z. Mechanism of brain targeting by dexibuprofen prodrugs modified with ethanolamine-related structures. J. Cereb. Blood Flow Metab. 2015, 35, 1985–1994. [Google Scholar] [CrossRef]
- Kłobucki, M.; Urbaniak, A.; Grudniewska, A.; Kocbach, B.; Maciejewska, G.; Kiełbowicz, G.; Czesław, M.; Wawrzeńczyk, U. Syntheses and cytotoxicity of phosphatidylcholines containing ibuprofen or naproxen moieties. Sci. Rep. 2019, 9, 220. [Google Scholar] [CrossRef]
- Gliszczyńska, A.; Niezgoda, N.; Gladkowski, W.; Świtalska, M.; Wietrzyk, J. Isoprenoid-phospholipid conjugates as potential therapeutic agents: Synthesis, characterization and antiproliferative studies. PLoS ONE 2017, 12, e0172238. [Google Scholar] [CrossRef] [PubMed]
- Gliszczyńska, A.; Niezgoda, N.; Gładkowski, W.; Czarnecka, M.; Świtalska, M.; Wietrzyk, J. Synthesis and biological evaluation of novel phosphatidylcholine analogues containing monoterpene acids as potent antiproliferative agents. PLoS ONE 2016, 11, e0157278. [Google Scholar] [CrossRef]
- Araujo, J.; Gonzalez, E.; Egea, M.A.; Garcia, M.L.; Souto, E.B. Nanomedicines for ocular NSAIDs: Safety on drug delivery. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Zhong-Kun, L.; Jin Mei, Y.W.; Heng, W.; Chong-jing, W.; Bing, L. Preparation and quality control of arginine-ibuprofen eye drops. Chin. Pharm. J. 2004, 39, 362–363. [Google Scholar]
- Vega, E.; Egea, M.A.; Garduño-Ramírez, M.L.; García, M.L.; Sánchez, E.; Espina, M.; Calpena, A.C. Flurbiprofen PLGA-PEG nanospheres: Role of hydroxy-β-cyclodextrin on ex vivo human skin permeation and in vivo topical anti-inflammatory efficacy. Colloids Surf. B Biointerfaces 2013, 110, 339–346. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Egea, M.A.; Cano, A.; Espina, M.; Calpena, A.C.; Ettcheto, M.; Camins, A.; Souto, E.B.; Silva, A.M.; García, M.L. PEGylated PLGA nanospheres optimized by design of experiments for ocular administration of dexibuprofen—In vitro, ex vivo and in vivo characterization. Colloids Surf. B Biointerfaces 2016, 145, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez López, E.; Esteruelas, G.; Ortiz, A.; Espina, M.; Prat, J.; Muñoz Juncosa, M.M.; Cano, A.; Calpena, A.C.; Ettcheto Arriola, M.; Camins, A.; et al. Dexibuprofen biodegradable nanoparticles: One step closer towards a better ocular interaction study. Nanomaterials 2020, 10, 720. [Google Scholar] [CrossRef] [Green Version]
- Vega, E.; Antònia Egea, M.; Calpena, A.C.; Espina, M.; Luisa García, M. Role of hydroxypropyl-β-cyclodextrin on freeze-dried and gamma-irradiated PLGA and PLGA-PEG diblock copolymer nanospheres for ophthalmic flurbiprofen delivery. Int. J. Nanomed. 2012, 7, 1357–1371. [Google Scholar] [CrossRef] [Green Version]
- Abrego, G.; Alvarado, H.L.; Egea, M.A.; Gonzalez-Mira, E.; Calpena, A.C.; Garcia, M.L. Design of nanosuspensions and freeze-dried PLGA nanoparticles as a novel approach for ophthalmic delivery of pranoprofen. J. Pharm. Sci. 2014, 103, 3153–3164. [Google Scholar] [CrossRef]
- Souto, E.B.; Doktorovova, S.; Gonzalez-Mira, E.; Egea, M.A.; Garcia, M.L. Feasibility of lipid nanoparticles for ocular delivery of anti-inflammatory drugs. Curr. Eye Res. 2010, 35, 537–552. [Google Scholar] [CrossRef]
- Li, X.; Nie, S.F.; Kong, J.; Li, N.; Ju, C.Y.; Pan, W.S. A controlled-release ocular delivery system for ibuprofen based on nanostructured lipid carriers. Int. J. Pharm. 2008, 363, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Rincón, M.; Calpena, A.C.; Fábrega, M.J.; Garduño-Ramírez, M.L.; Rodríguez-Lagunas, M.J.; García, M.L.; Abrego, G. Development of pranoprofen loaded nanostructured lipid carriers to improve its release and therapeutic efficacy in skin inflammatory disorders. Nanomaterials 2018, 8, 1022. [Google Scholar] [CrossRef] [Green Version]
- Akhlaq, M.; Arshad, M.S.; Mudassir, A.M.; Hussain, A.; Kucuk, I.; Haj-Ahmad, R.; Rasekh, M.; Ahmad, Z. Formulation and evaluation of anti-rheumatic dexibuprofen transdermal patches: A quality-by-design approach. J. Drug Target. 2016, 24, 603–612. [Google Scholar] [CrossRef]
- Ali, F.R.; Shoaib, M.H.; Yousuf, R.I.; Ali, S.A.; Imtiaz, M.S.; Bashir, L.; Naz, S. Design, development, and optimization of Dexibuprofen microemulsion based transdermal reservoir patches for controlled drug delivery. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiruppathi, M.; Lavakumar, V.; Natarajan, P.; Sowmiya, C. Pharmaceutical assessment and pharmacological evaluation of Dexibuprofen-Aloe vera trans emulgel. Pharma Innov. J. 2019, 8, 343–350. [Google Scholar]
- Vijay Rajaram, M.; Ganesh Dinkar, B. Formulation design, development and characterization of dexibuprofen emulgel for topical delivery: In-vitro and In-vivo evaluation. J. Drug Deliv. Ther. 2019, 9, 330–342. [Google Scholar]
- Li, T.; Zhao, L.; Zheng, Z.; Zhang, M.; Sun, Y.; Tian, Q.; Zhang, S. Design and preparation acid-activated montmorillonite sustained-release drug delivery system for dexibuprofen in vitro and in vivo evaluations. Appl. Clay Sci. 2018, 163, 178–185. [Google Scholar] [CrossRef]
- El Maghraby, G.; Essa, E. Salt and non-salt forming excipients to improve the dissolution of Dexibuprofen; formulation of chewable tablets. Eur. J. Biomed. Pharm. Sci. 2020, 7, 1–11. [Google Scholar]
- Khalid, Q.; Ahmad, M.; Minhas, M.U. Synthesis of β-cyclodextrin hydrogel nanoparticles for improving the solubility of dexibuprofen: Characterization and toxicity evaluation. Drug Dev. Ind. Pharm. 2017, 43, 1873–1884. [Google Scholar] [CrossRef]
- Khalid, Q.; Ahmad, M.; Usman Minhas, M. Hydroxypropyl-β-cyclodextrin hybrid nanogels as nano-drug delivery carriers to enhance the solubility of dexibuprofen: Characterization, in vitro release, and acute oral toxicity studies. Adv. Polym. Technol. 2018, 37, 2171–2185. [Google Scholar] [CrossRef]
- Ali, M.; Khan, N.R.; Hussain, Z.; Basit, H.M.; Mahmood, S. Novel Composite pH Controlled Drug Release Hydrogel Containing Dexibuprofen. RADS J. Pharm. Pharm. Sci. 2018, 6, 223–235. [Google Scholar]
- Xia, Y.N.; Li, X.H.; Xu, J.H.; Chen, L.Q.; Ahmed, A.M.Q.; Cao, D.; Du, H.H.; Deng, Y.; Cao, Q.R. Optimization and characterization of novel sustained release supermicro-pellet based dry suspensions that load dexibuprofen. J. Drug Deliv. Sci. Technol. 2020, 55. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Ettcheto, M.; Egea, M.A.; Espina, M.; Calpena, A.C.; Folch, J.; Camins, A.; García, M.L. New potential strategies for Alzheimer’s disease prevention: Pegylated biodegradable dexibuprofen nanospheres administration to APPswe/PS1dE9. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Senthilnathan, B.; Gopalasatheeskumar, K.; Vijayalakshmi, A.; Bhavya, E.; Jeyamani, V. Design and development of dexibuprofen loaded chitosan nanoparticles. Drug Invent. Today 2018, 10, 248–252. [Google Scholar]
- Khalid, Q.; Ahmad, M.; Minhas, M.U.; Rashid, H. Development and evaluation of eudragit based microparticles of dexibuprofen for site specific drug release. Pak. J. Pharm. Sci. 2018, 31, 1385–1392. [Google Scholar]
- Bertalero, G.; Addebito, P.; Bancario, C.C.; Cliente, C.A.L. Synthesis, Characterization and In Vivo Assessment of Dexibuprofen-Eudragit Solid Dispersion Nanoparticles with Supercritical Antisolvent Technique. Lat. Am. J. Pharm. 2013, 35, 1–2. [Google Scholar]
- Abdelbary, G.; Makhlouf, A. Adoption of polymeric micelles to enhance the oral bioavailability of dexibuprofen: Formulation, in-vitro evaluation and in-vivo pharmacokinetic study in healthy human volunteers. Pharm. Dev. Technol. 2014, 19, 717–727. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmaceutical Form | Physicochemical Characteristics | In Vitro Outcomes | In Vivo Outcomes | Ref |
|---|---|---|---|---|
| Transdermal patches Compounds: DXI, Polymeric excipients (ethyl cellulose and polyvinylpyrrolidone, plasticizer (di-N-butyl phthalate), Permeation enhancer (almond oil) | Uniform thickness (0.44 ± 0.02 cm) Low moisture uptake (7.87 ± 1.11 w/w %), Highly drug loading (100.0 ± 0.026%) | Ex vivo skin permeation studieS show 42% of DXI released in 4 h and 91% within 24 h | New Zealand rabbit model PatcheS were non-irritant Better pharmacokinetics: longer tmax (8 h) compared with DXI oral tablets (2 h) and increased half-life (10.51 h) against DXI oral tablets (3.50 h) | [69] |
| DXI microemulsion based transdermal reservoir patchesMicroemulsion compounds: ethyl oleate, Tween 80: PG | Microemulsion properties: size 119–221 nm Polydispersity index 0.35-0.56DXI loading >97% Stable for 6 months at 4 °C | Zero-order release rate Q24h 79.13%; flow of 331.17 µg/cm2h | Model used: abino wistar rats No skin irritation Increased antiinflammatory effectivity against commercial hydrogel and ibuprofen emulsion gel | [70] |
| DXI Aloe vera trans emulgel | High DXI loading (78%) pH 7.56 Viscosity 112 CPs Flux 0.624 μg cm−2 h−1 Stable for the first 45 days | 78% of the drug is released within 150 min | No skin irritation Superior anti-inflammatory activity (60.94%) than diclofenac gel (49.6%) | [71] |
| DXI emulgel. Compounds: gelling agent (Carbapol 940), penetration enhancers (Clove oil and Mentha oil), gel base | Stable for 3 months | In vivo release show 55.91–57.21% DXI released within 150 min Ex vivo permeation show DXI release 59.45–61.68% within 150 min | Comparable analgesic and anti-inflammatory activity against diclofenac gel | [72] |
| No-alcoholic transdermal DXI hydrogel. Compounds: pH modifying agent, antioxidants, water miscible solvent, HPMC, others | Stable for three months | Data not shown | Data not shown | [73] |
| Pharmaceutical Form | Physicochemical Characteristics | In Vitro Outcomes | In Vivo Outcomes | Ref |
|---|---|---|---|---|
| Montmorillonite acid DXI composites | DXI loading of 298 mg/g | In vitro DXI released (92%) within 12 h in simulated intestinal fluid | Rat animal model. Better pharmacokinetic profile (AUC0–24 644.49 μg/h/mL and MRT0–24 7.65 ± 0.48 h) than DXI suspension (AUC0–24 439.88 μg/h/mL and MRT0–24 3.10 h). Increased bioavailability (154.11%) against commercial DXI | [73] |
| DXI chewable tablets | Preparation suing wet-co grinding of DXI adding mannitol and/or meglumine | DXI dissolution enhanced Mannitol based tablets showed prompt drug release Meglumine based tablets required crushing for fast drug release | Data not shown | [74] |
| DXI loaded β-cyclodextrin hydrogel nanoparticles | Nanoparticles size: 287 nm | DXI release higher than DXI tablets at pH 1.2 and 6.8 | Animal model: Wistar albino rats Acute toxicity studies: no modification of behavioral, physiological, biochemical or histopathologic parameters were observed | [75] |
| DXI loaded hydroxypropyl- β- cyclodextrin (HPβCD) hybrid nanogels | Solubility enhancement of DXI confirmed Particle size 310.65 ± 18.75 nm Polydispersity index: 0.21 Zeta potential:−36.49 ± 2.34 mV | Highly porous and amorphous nanogels DXI release higher than DXI tablets at pH 1.2 and 6.8 | Animal model: Wistar albino rats Toxicity studies: no modification of behavioral, physiological, biochemical or histopathologic parameters were observed | [76] |
| pH controlled DXI release hydrogel containing Dexibuprofen | Maximal gel swelling and drug release at pH 1.2. | Swelling and drug release pH-dependent Fast release at pH 1.2 | Data not shown | [77] |
| DXI supermicro-pellet based dry suspensions | Pellets preparation using spray dry fluid bed coating technique Suitable stability (sedimentation rate 0.8 Hu/H) Aqueate flowability (θ 27°). | DXI release around 8 h being pH dependent | Data not shown | [78] |
| DXI loaded PLGA PEG nanoparticles | Nanoparticles size: 195.4 nm Polydispersity index: <0.1 Negative surface charge Stable for 2 months at 25 and 4 °C | 100% nanoparticles uptaken by cells within 5 min Nanoparticles were able to cross trough and in vitro BBB model | Model: C57bl6 mice and APPswe/PS1dE9 transgenic Nanoparticles were effective for Alzheimer’s disease: inflammation and β-amyloid plaque reduction; behavioural improvement | [79] |
| DXI loaded chitosan nanoparticles | Particle size: 437.6 nm High entrapment efficiency (88.54%) | In vitro DXI release of 99.81% within 24 h | Data not shown | [80] |
| DXI Eudragit based microparticles | High entrapment efficiency (>70%) | In vitro DXI release at pH 1.2 < 21% while at pH 6.8 was high (around 60% within 8 h): gastro-resistant formulation developement | Data not shown | [81] |
| DXI Eudragit solid dispersed nanoparticles | Size: <300 nm | Improved dissolution rate | Animal model: sprague-dawley rats Improved pharmacokinetic parameters: AUC0–24 and Cmax increased 4.6 and 5.7 times respectively | [82] |
| DXI loaded polymeric micelle based tablets | Size: 28.11 nm Polydispersity index: 0.15 Zeta potential −2.88 mV | Faster DXI release from the polymeric micelle based tablets (80.1% of DXI was released within 30 min) than the commercial tablet (35.35% within 30 min) | Human studies developed Pharmacokinetic studies: DXI polymeric micelles based tablets show better pharmacokinetic profile (AUC0–24 407.45 µg mL h−1; Cmax 20.99 µg/mL; Tmax 1 h; MRT 12.79 h) than commercial tablets (AUC0–24 71.91 µg mL h−1; Cmax 12.94 µg/mL; Tmax 2.75 h; MRT 10.53 h) Relative bioavailability was 160.15% | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gliszczyńska, A.; Sánchez-López, E. Dexibuprofen Therapeutic Advances: Prodrugs and Nanotechnological Formulations. Pharmaceutics 2021, 13, 414. https://doi.org/10.3390/pharmaceutics13030414
Gliszczyńska A, Sánchez-López E. Dexibuprofen Therapeutic Advances: Prodrugs and Nanotechnological Formulations. Pharmaceutics. 2021; 13(3):414. https://doi.org/10.3390/pharmaceutics13030414
Chicago/Turabian StyleGliszczyńska, Anna, and Elena Sánchez-López. 2021. "Dexibuprofen Therapeutic Advances: Prodrugs and Nanotechnological Formulations" Pharmaceutics 13, no. 3: 414. https://doi.org/10.3390/pharmaceutics13030414