
Structural Polymorphism of Sorafenib Tosylate as a Key Factor in Its Solubility Differentiation

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and methods
2.1. Materials
2.2. Characterization of Polymorphic Forms of Sorafenib Tosylate
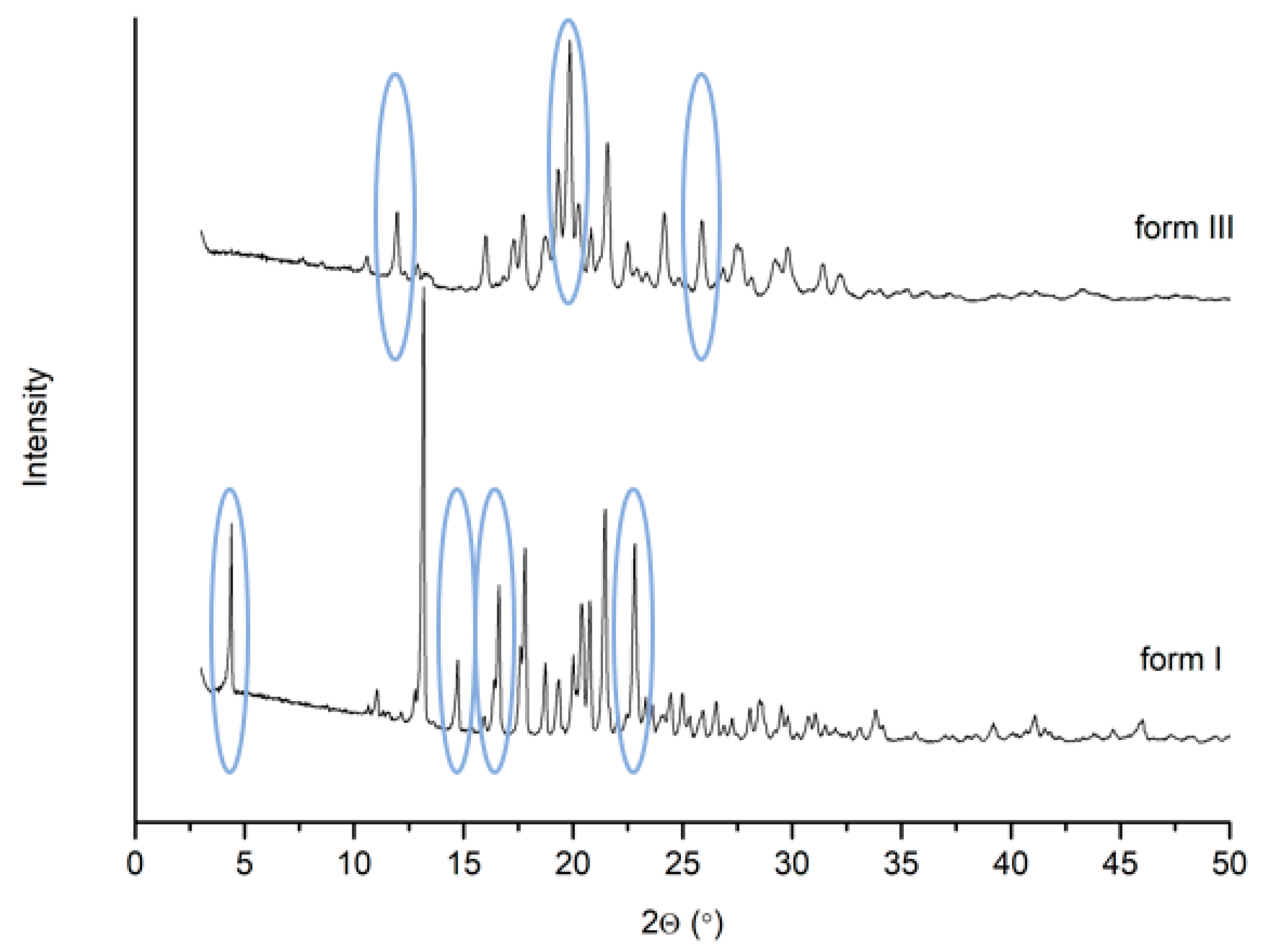
2.2.1. X-ray Powder Diffraction
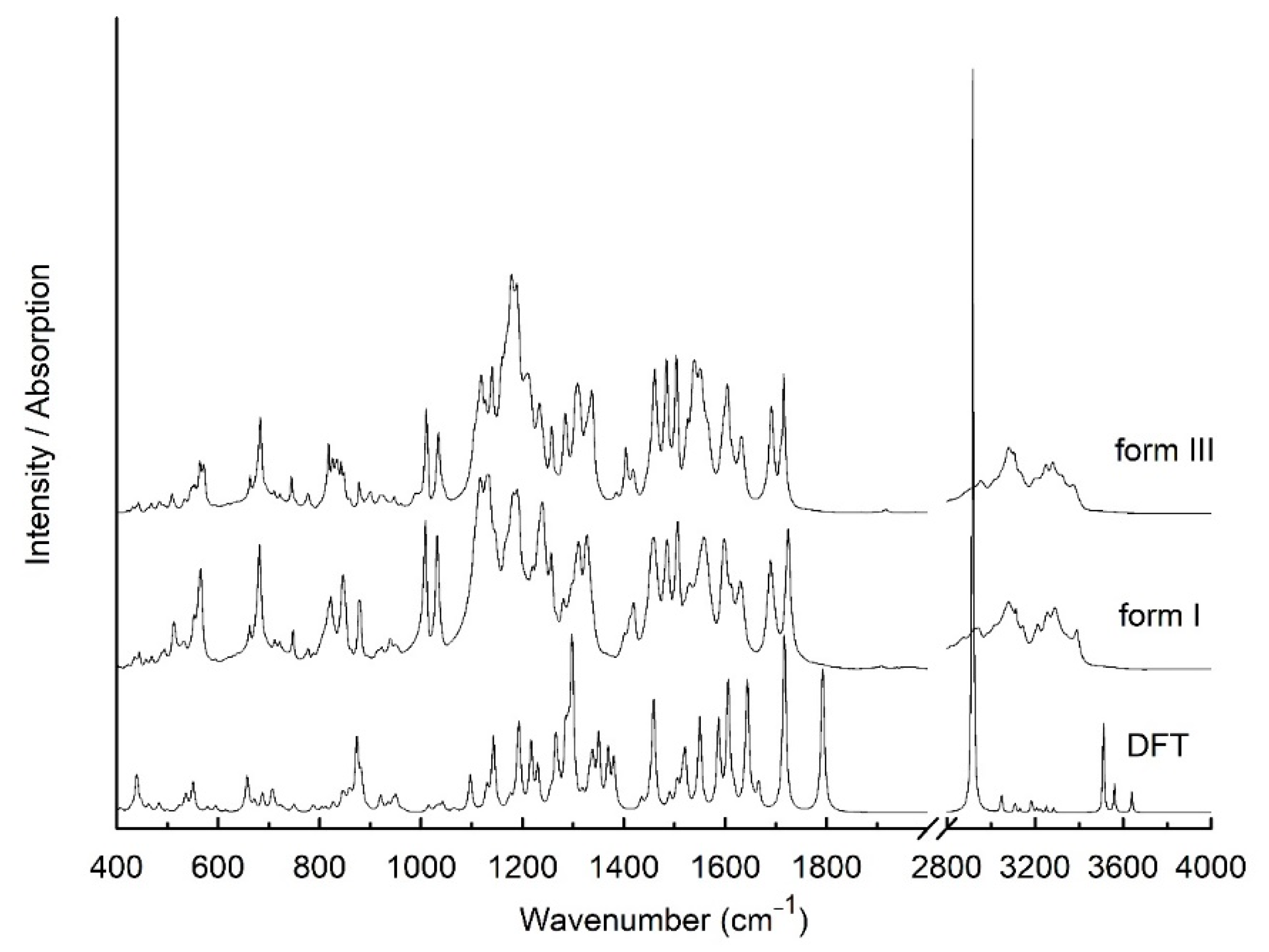
2.2.2. FT-IR Spectroscopy
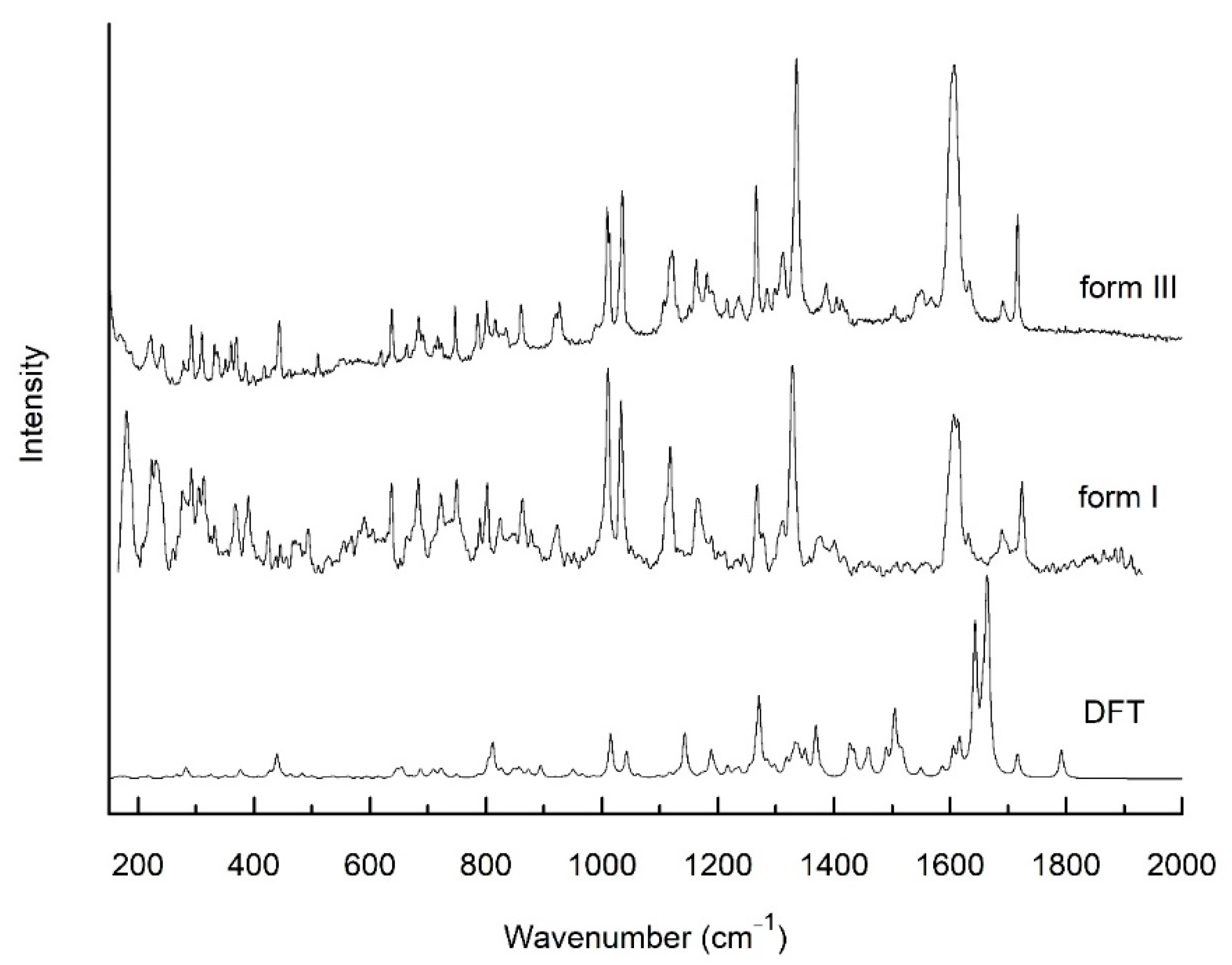
2.2.3. Raman Spectroscopy
2.3. Solubility and Permeability Studies of Polymorphic Forms of Sorafenib Tosylate
2.3.1. High-Performance Liquid Chromatography (HPLC) Method
2.3.2. Solubility Studies
2.3.3. Permeability Study
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| API | active pharmaceutical ingredient |
| BCS | Biopharmaceutics Classification System |
| c-KIT | receptor tyrosine kinase |
| DFT | density-functional theory |
| DMSO | dimethyl sulfoxide |
| FDA | Food and Drug Administration |
| FLT-3 | FMS-like tyrosine kinase 3 |
| FT-IR | Fourier-transform infrared spectroscopy |
| GIT | gastrointestinal tract |
| HCl | hydrochloric acid |
| HPLC | high-performance liquid chromatography |
| ICH | International Conference on Harmonization |
| MAPK | mitogen-activated protein kinase |
| PAMPA | parallel artificial membrane permeability assay |
| Papp | permeability coefficient |
| PDGFR | platelet-derived growth factor receptor |
| RAF | rapidly accelerated fibrosarcoma |
| RET RSD | rearranged during transfection relative standard deviation |
| SDS | sodium dodecyl sulfate |
| VEGFR | vascular endothelial growth factor receptor |
| XRPD | X-ray powder diffraction |
References
- Keating, G.M.; Santoro, A. Sorafenib. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Polymorphs of Sorafenib Acid Addition Salts. U.S. Patent No. 8604208B2. Available online: https://patents.google.com/patent/US8604208B2/en (accessed on 10 February 2021).
- DailyMed. Current Medication Information for NEXAVAR (Sorafenib) Tablet, Film Coated. Available online: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=b50667e4-5ebc-4968-a646-d605058dbef0 (accessed on 16 January 2021).
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving oral bioavailability of sorafenib by optimizing the “spring” and “parachute” based on molecular interaction mechanisms. Mol. Pharm. 2016, 13, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.W.J. Theory and origin of polymorphism. In Polymorphism in Pharmaceutical Solids; Brittain, H.G., Ed.; Informa: London, UK, 1999. [Google Scholar]
- Talaczynska, A.; Dzitko, J.; Cielecka-Piontek, J. Benefits and limitations of polymorphic and amorphous forms of active pharmaceutical ingredients. Curr. Pharm. Des. 2016, 22, 4975–4980. [Google Scholar] [CrossRef] [PubMed]
- Censi, R.; Di Martino, P. Polymorph impact on the bioavailability and stability of poorly soluble drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymorphs of Sorafenib Tosylate and Sorafenib Hemi-Tosylate, and Processes for Preparation Thereof. U.S. Patent No. 8217061B2. Available online: https://patents.google.com/patent/US8217061B2/en (accessed on 16 January 2021).
- Scalable Process for the Preparation of Sorafenib Tosylate Ethanol Solvate and Sorafenib Tosylate Form iii. U.S. Patent No. 20160376236A1. Available online: https://patents.google.com/patent/US20160376236A1/en (accessed on 16 January 2021).
- Yang, P.; Qin, C.; Du, S.; Jia, L.; Qin, Y.; Gong, J.; Wu, S. Crystal Structure, Stability and Desolvation of the Solvates of Sorafenib Tosylate. Crystals 2019, 9, 367. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Qin, Y.; Wu, S.; Xu, S.; Li, K.; Yang, P.; Zhao, K.; Lin, L.; Gong, J. Solubility correlation and thermodynamic analysis of sorafenib free base and sorafenib tosylate in monosolvents and binary solvent mixtures. J. Chem. Eng. Data 2017, 62, 259–267. [Google Scholar] [CrossRef]
- Sodeifian, G.; Razmimanesh, F.; Sajadian, S.A.; Hazaveie, S.M. Experimental data and thermodynamic modeling of solubility of Sorafenib tosylate, as an anti-cancer drug, in supercritical carbon dioxide: Evaluation of Wong-Sandler mixing rule. J. Chem. Thermodyn. 2020, 142, 105998. [Google Scholar] [CrossRef]
- Guo, Y.; Zhong, T.; Duan, X.-C.; Zhang, S.; Yao, X.; Yin, Y.-F.; Huang, D.; Ren, W.; Zhang, Q.; Zhang, X. Improving anti-tumor activity of sorafenib tosylate by lipid-and polymer-coated nanomatrix. Drug Deliv. 2017, 24, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Mai, Y.; Liu, Q.; Zhang, W.; Yang, J. Mechanism and Improved Dissolution of Glycyrrhetinic Acid Solid Dispersion by Alkalizers. Pharmaceutics 2020, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasiłowicz, A.; Tykarska, E.; Lewandowska, K.; Kozak, M.; Miklaszewski, A.; Kobus-Cisowska, J.; Szymanowska, D.; Plech, T.; Jenczyk, J.; Cielecka-Piontek, J. Hydroxypropyl-β-cyclodextrin as an effective carrier of curcumin–piperine nutraceutical system with improved enzyme inhibition properties. J. Enzym. Inhib. Med. Chem. 2020, 35, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision C. 01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed;(b) Y. Zhao and DG Truhlar. Theor. Chem. Acc 2008, 120, 215. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- European Pharmacopeia 9.0. 2.9.3. Dissolution Test for Solid Dosage Forms; European Directorate for the Quality of Medicines & HealthCare (EDQM): Strasbourg, France, 2016. [Google Scholar]
- Fischer, H.; Kansy, M.; Avdeef, A.; Senner, F. Permeation of permanently positive charged molecules through artificial membranes—Influence of physico-chemical properties. Eur. J. Pharm. Sci. 2007, 31, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Guidance for Industry. ANDAs: Pharmaceutical Solid Polymorphism. Chemistry, Manufacturing and Controls Information. 2007. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/andaspharmaceutical-solid-polymorphism-chemistry-manufacturing-and-controls-information (accessed on 16 January 2021).
- Thermodynamically Stable Form of Bay 43-9006 Tosylate. U.S. Patent No. 8217061B2. Available online: https://patents.google.com/patent/US8217061B2/en (accessed on 16 January 2021).
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W. Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ito, S.; Itai, S.; Yamamoto, K. Physicochemical properties and bioavailability of carbamazepine polymorphs and dihydrate. Int. J. Pharm. 2000, 193, 137–146. [Google Scholar] [CrossRef]
- Wei, Y.; Ling, Y.; Su, M.; Qin, L.; Zhang, J.; Gao, Y.; Qian, S. Characterization and stability of amorphous tadalafil and four crystalline polymorphs. Chem. Pharm. Bull. 2018, 66, 1114–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfilio, R.; Souza, M.C.O.; Leal, J.S.; Viana, O.M.M.S.; Doriguetto, A.C.; Araújo, M.B.D. Solubility and dissolution studies of tibolone polymorphs. Braz. J. Pharm. Sci. 2017, 53. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Medium | Solubility (mg/mL) | |
|---|---|---|
| Form I | Form III | |
| 0.1 N HCl | - | - |
| 0.1 N HCl + 1.0% SDS | 0.314 ± 0.006 | 1.103 ± 0.014 |
| Acetate buffer pH 4.5 | - | - |
| Acetate buffer pH 4.5 + 1.0% SDS | 2.404 ± 0.012 | 2.355 ± 0.009 |
| Phosphate buffer pH 6.8 | - | - |
| Phosphate buffer pH 6.8 + 1.0% SDS | 0.051 ± 0.005 | 1.805 ± 0.023 |
| pH | Papp (cm s−1) | |
|---|---|---|
| Form I | Form III | |
| 1.2 | 3.01 × 10−5 ± 4.14 × 10−7 | 3.15 × 10−5 ± 1.89 × 10−6 |
| 6.8 | 2.72 × 10−5 ± 1.56 × 10−6 | 2.81 × 10−5 ± 9.0 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiergowska, G.; Stasiłowicz, A.; Miklaszewski, A.; Lewandowska, K.; Cielecka-Piontek, J. Structural Polymorphism of Sorafenib Tosylate as a Key Factor in Its Solubility Differentiation. Pharmaceutics 2021, 13, 384. https://doi.org/10.3390/pharmaceutics13030384
Wiergowska G, Stasiłowicz A, Miklaszewski A, Lewandowska K, Cielecka-Piontek J. Structural Polymorphism of Sorafenib Tosylate as a Key Factor in Its Solubility Differentiation. Pharmaceutics. 2021; 13(3):384. https://doi.org/10.3390/pharmaceutics13030384
Chicago/Turabian StyleWiergowska, Gabriela, Anna Stasiłowicz, Andrzej Miklaszewski, Kornelia Lewandowska, and Judyta Cielecka-Piontek. 2021. "Structural Polymorphism of Sorafenib Tosylate as a Key Factor in Its Solubility Differentiation" Pharmaceutics 13, no. 3: 384. https://doi.org/10.3390/pharmaceutics13030384