
Bio-Inspired and Smart Nanoparticles for Triple Negative Breast Cancer Microenvironment


Abstract
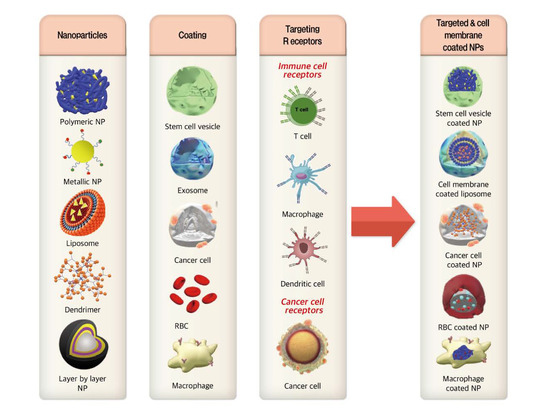
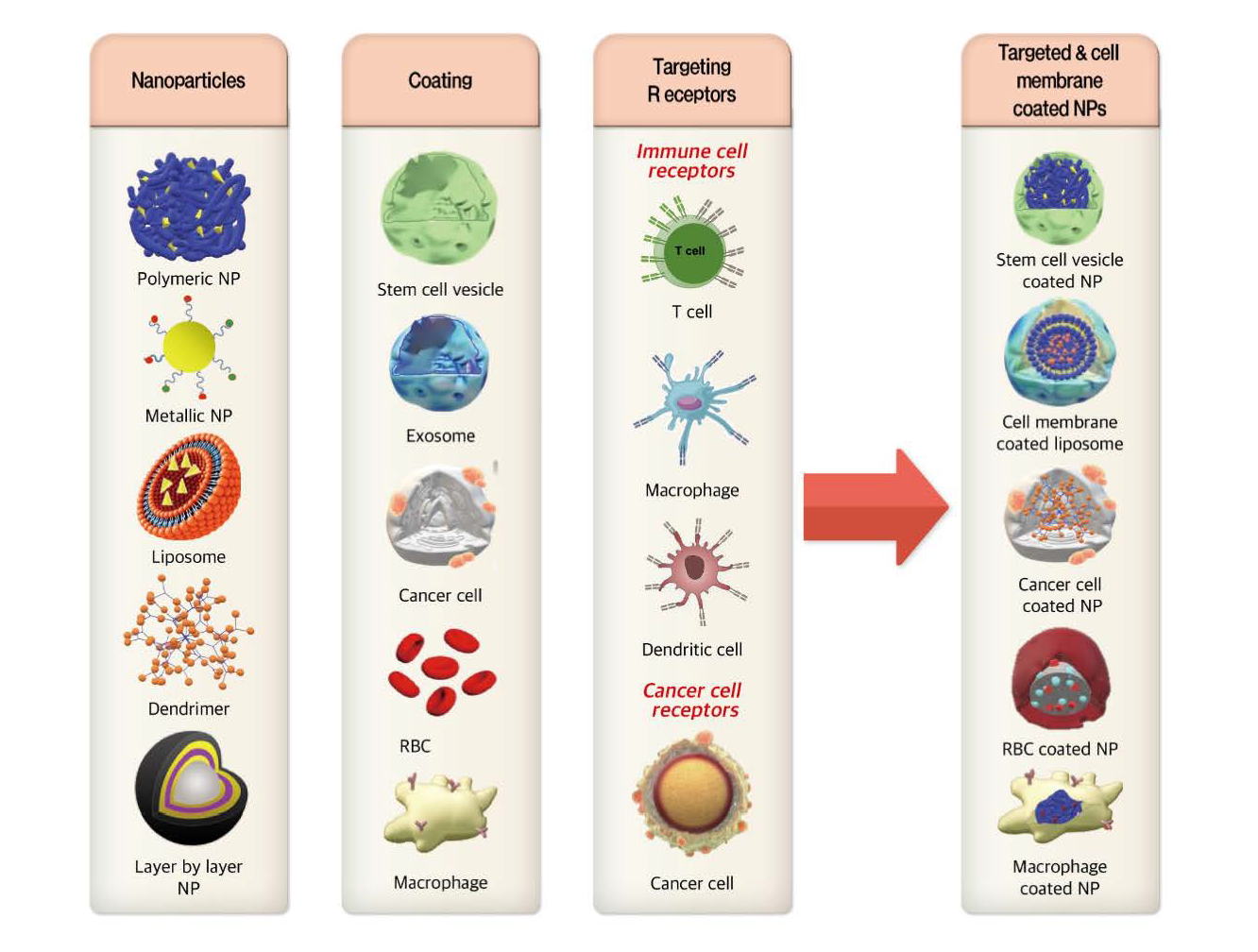
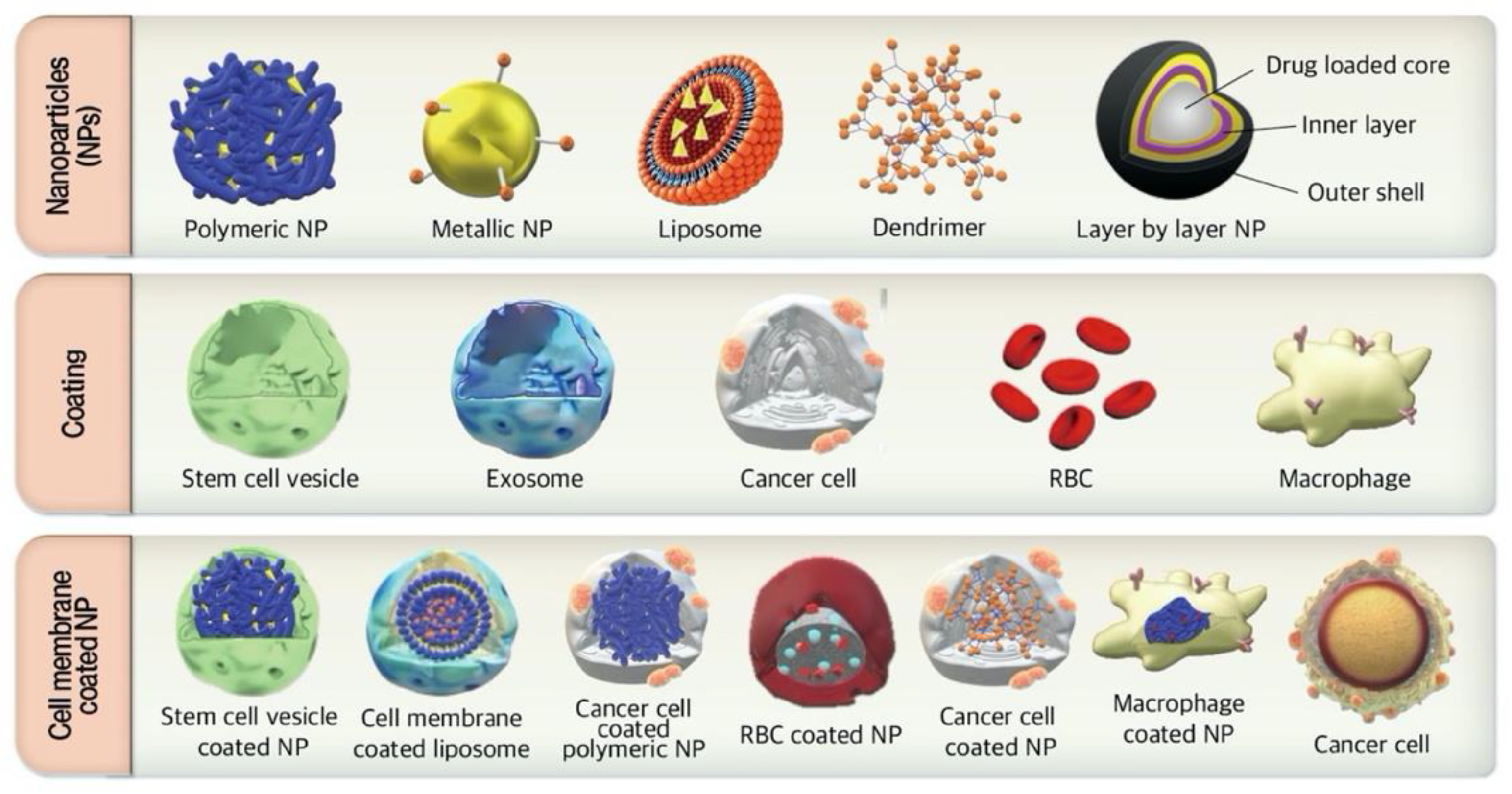
:
1. Introduction
2. Current Strategies for TNBC Treatment
3. Bio-Inspired Tumor-Homing Nanosystems for TNBC Treatment
3.1. Cell Membrane-Coated NPs
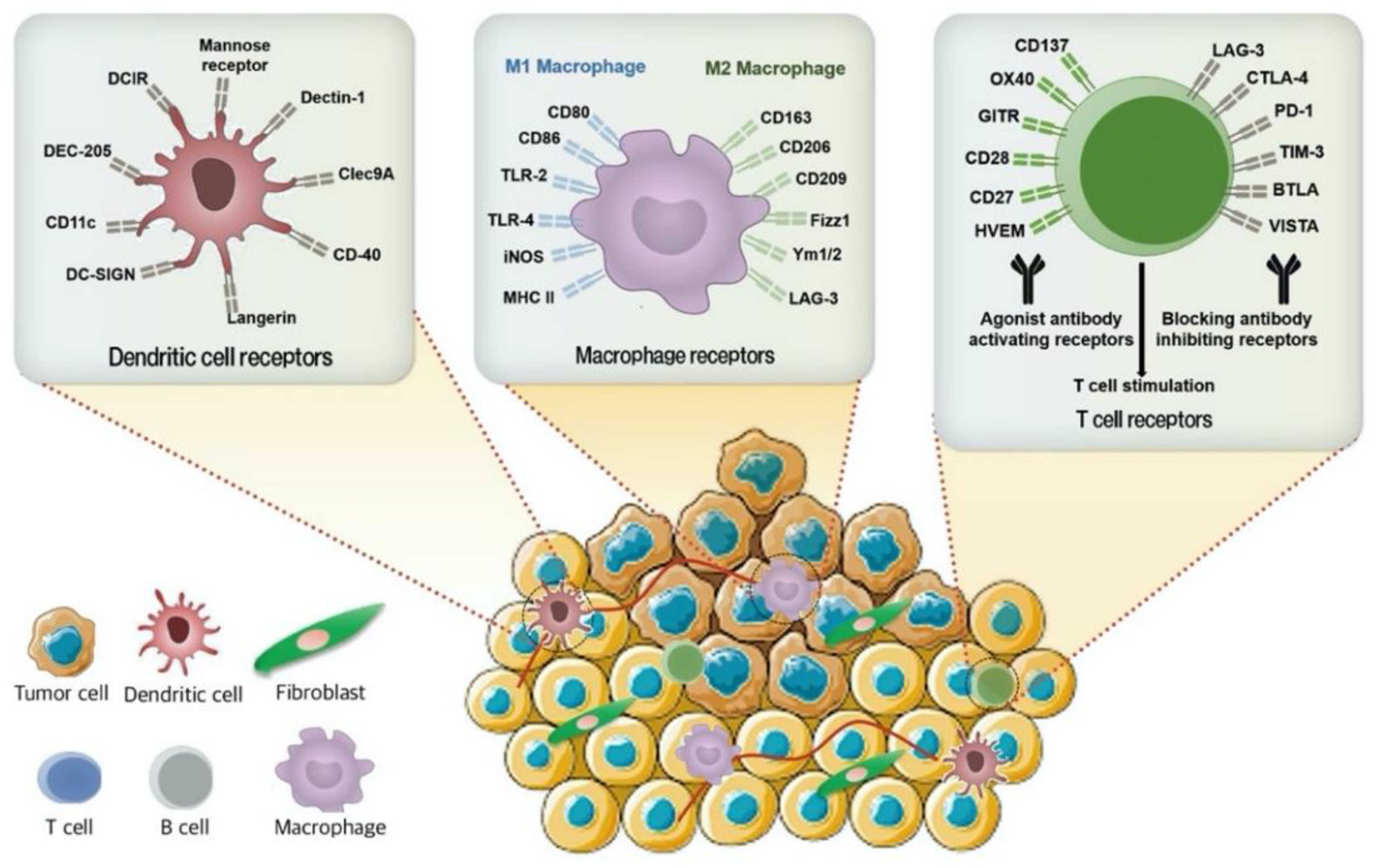
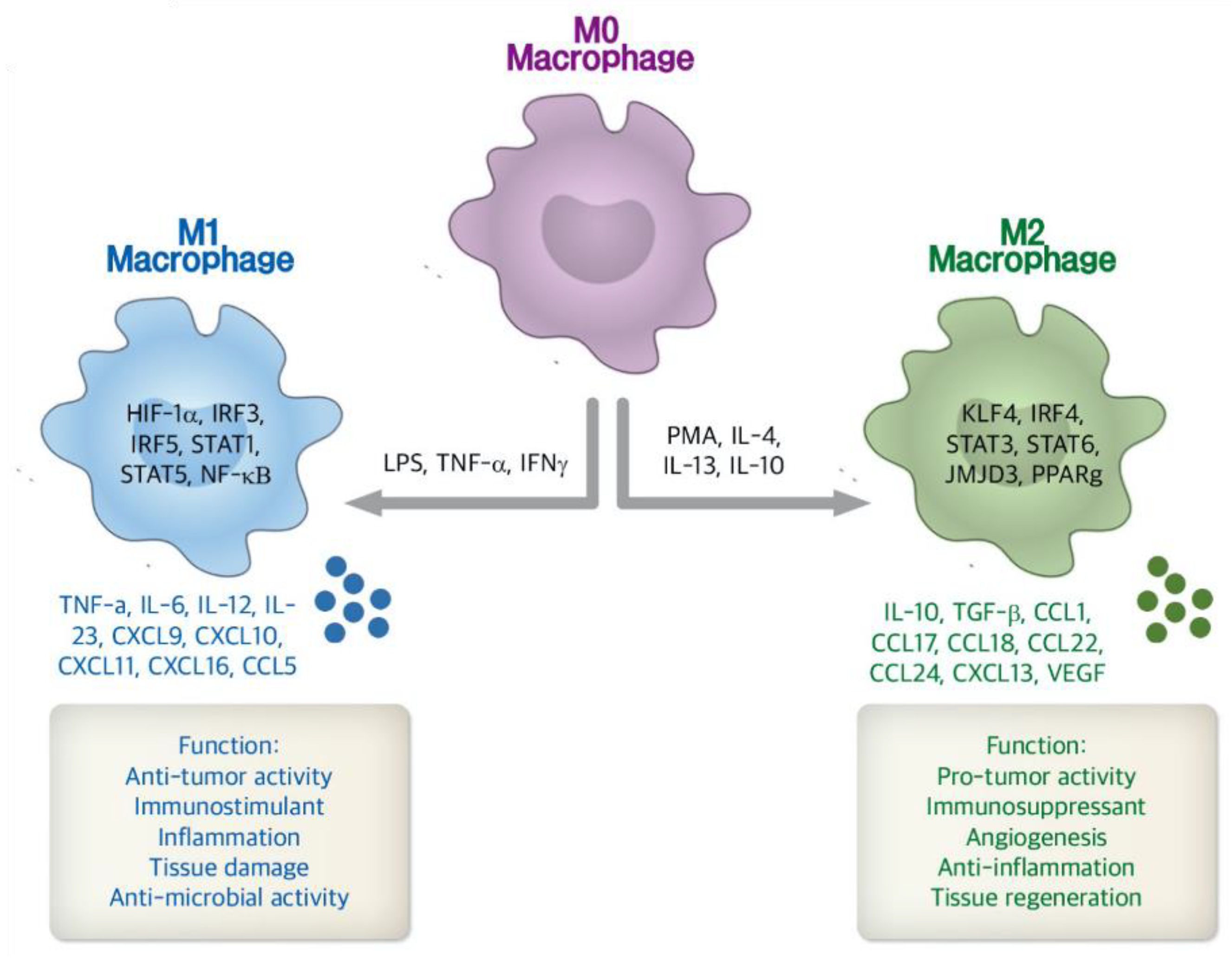
3.2. Immune Cell Targeted Nanosystems
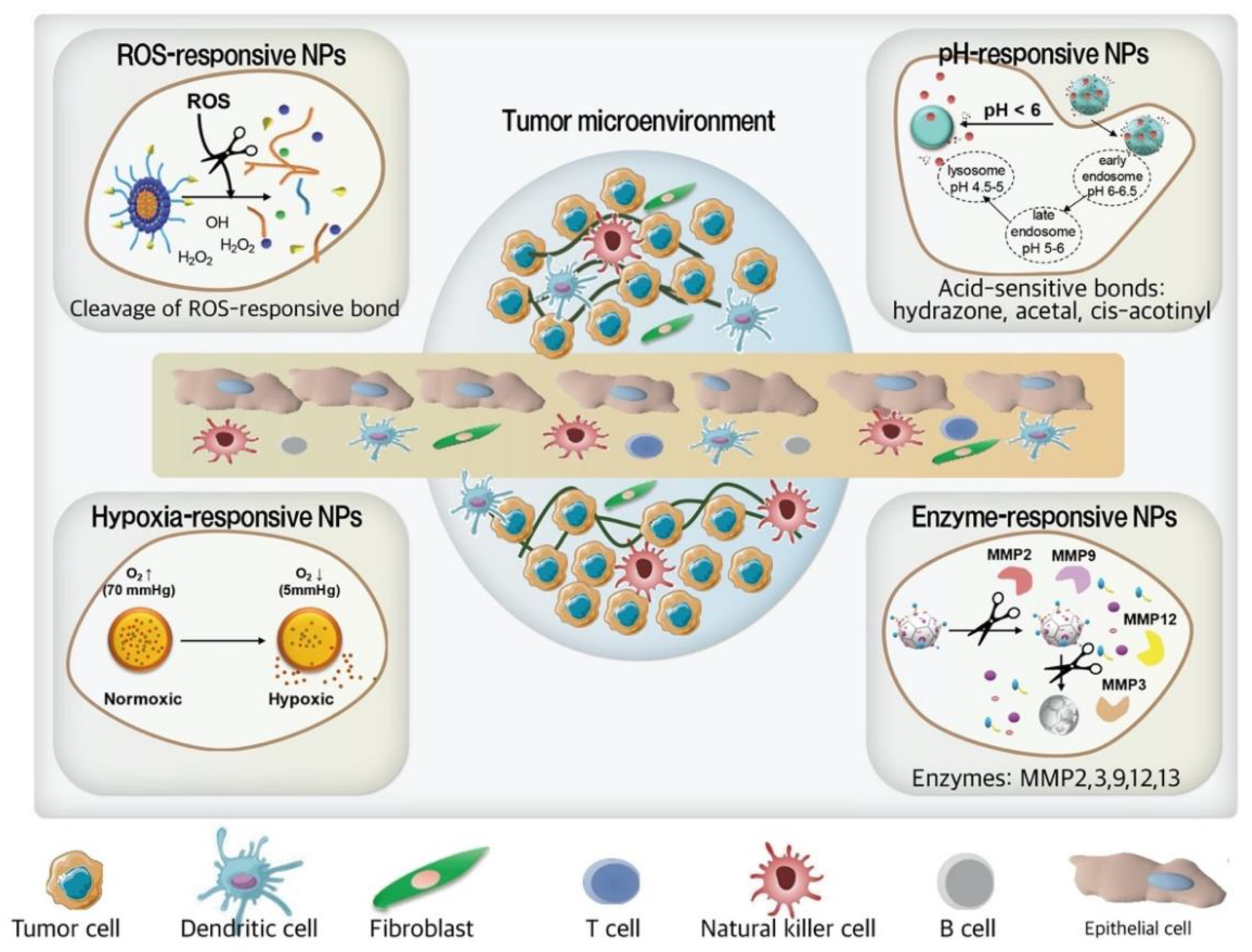
3.3. Smart NPs for TNBC Treatment
3.3.1. ROS-Responsive NPs
3.3.2. pH-Responsive NPs
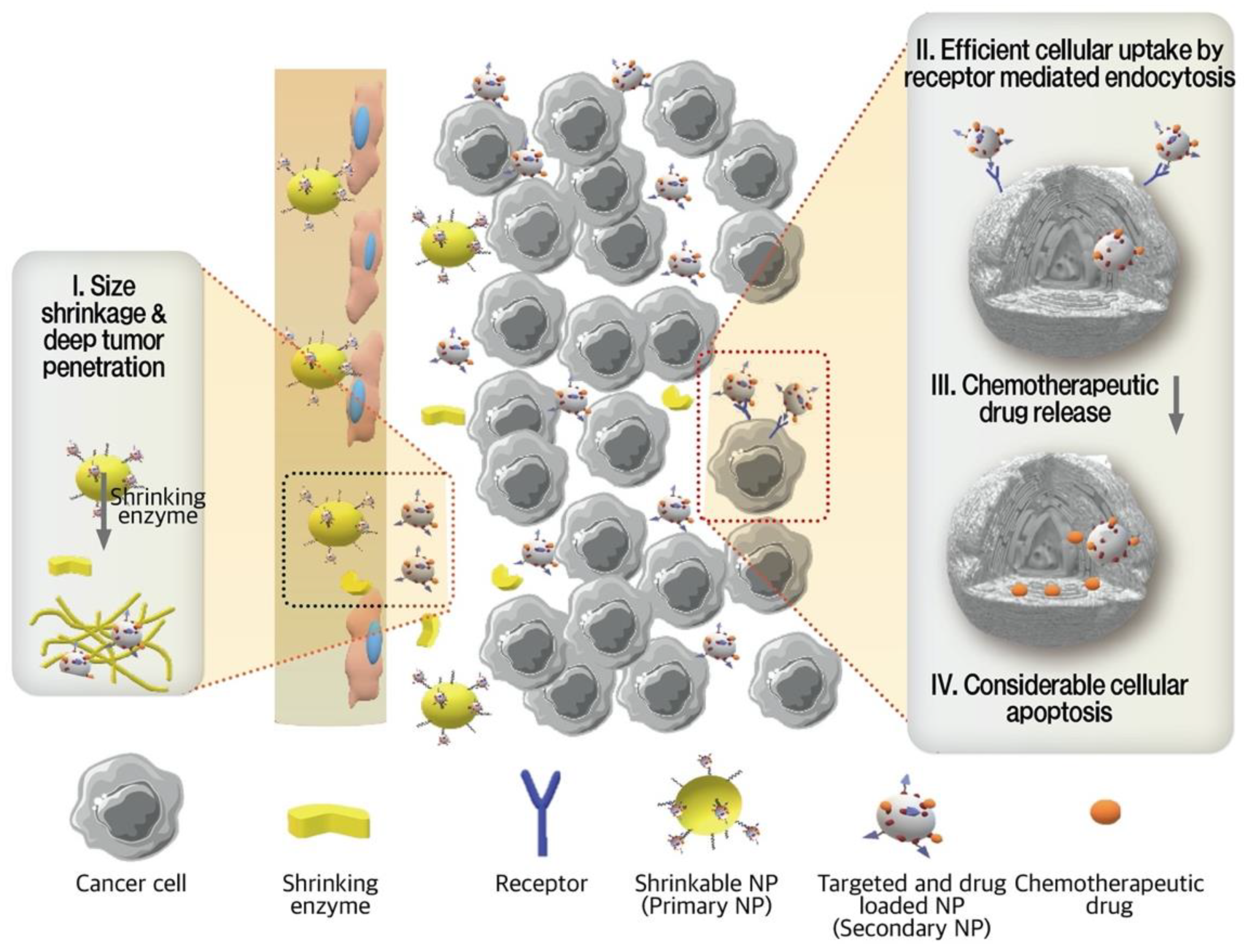
3.3.3. Enzyme-Responsive NPs
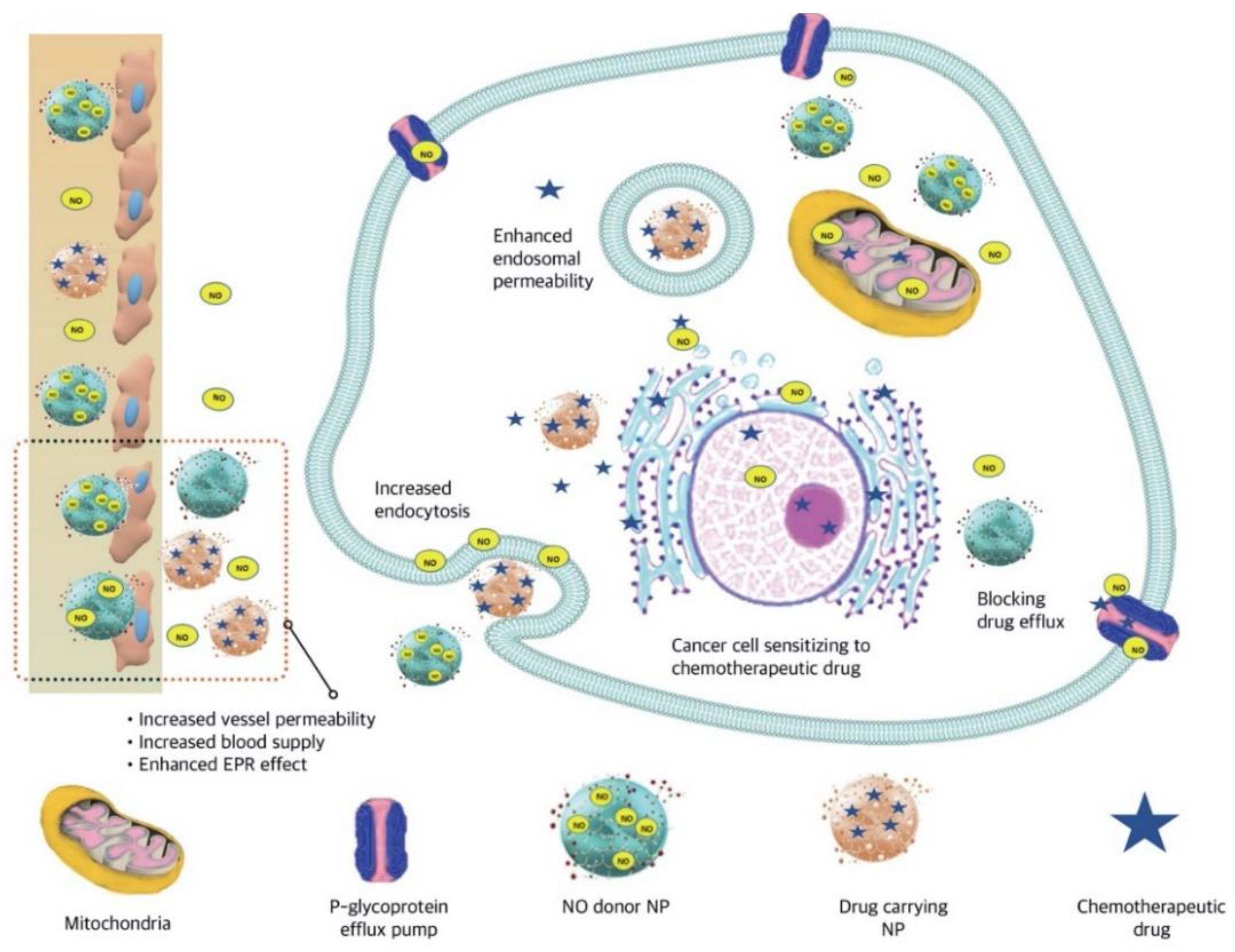
3.3.4. Nitric Oxide (NO)-Responsive NPs
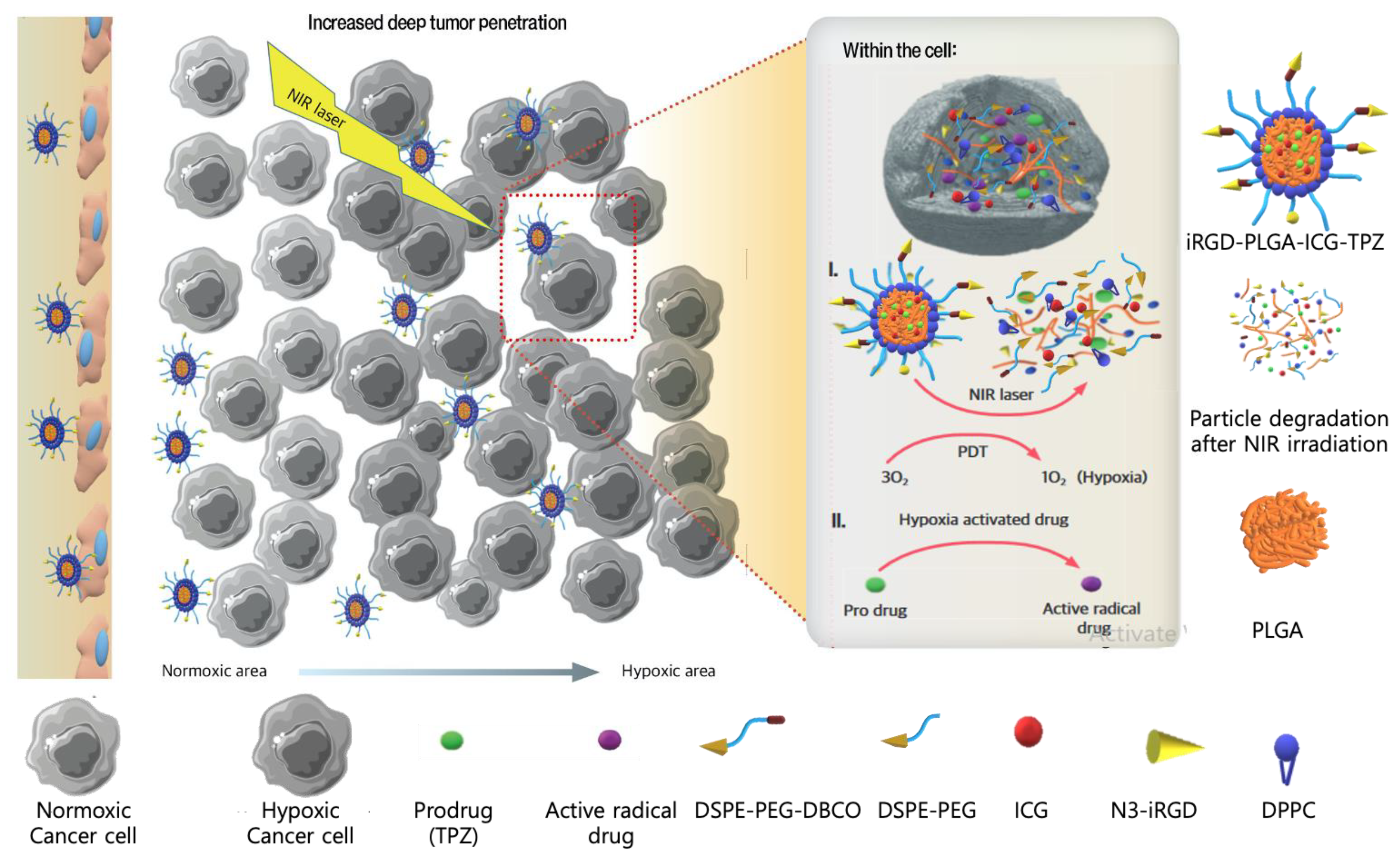
3.3.5. Hypoxia-Activated NPs
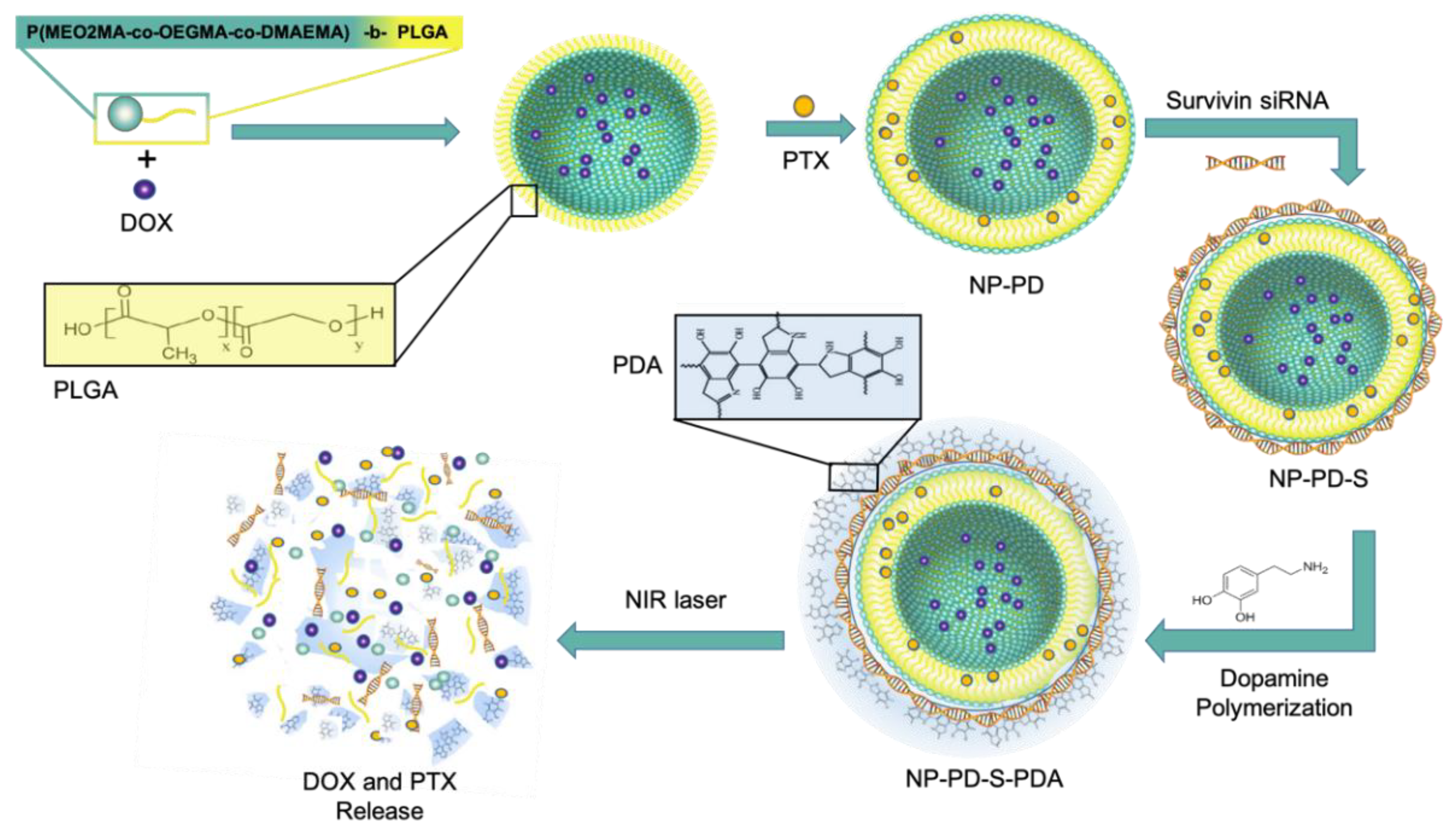
3.3.6. Thermo-Responsive NPs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.; Lee, S.-E.; Kim, D.-H.; Pyo, Y.-C.; Park, J.-S. Recent advances of nanotechnology for the delivery of anticancer drugs for breast cancer treatment. J. Pharm. Investig. 2020, 50, 261–270. [Google Scholar] [CrossRef]
- Katz, H.; Alsharedi, M.J.M.O. Immunotherapy in triple-negative breast cancer. Med. Oncol. 2018, 35, 13. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Sabnis, N.; McConathy, W.J.; Lacko, A.G.J.P. The potential role of nanotechnology in therapeutic approaches for triple negative breast cancer. Pharmaceutics 2013, 5, 353–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami, F.; Pathak, S.; Nguyen, T.T.; Shrestha, P.; Maharjan, S.; Kim, J.O.; Jeong, J.-H.; Yook, S. Photoimmunotherapy with cetuximab-conjugated gold nanorods reduces drug resistance in triple negative breast cancer spheroids with enhanced infiltration of tumor-associated macrophages. J. Control. Release 2020. [Google Scholar] [CrossRef]
- Ruan, S.; Zhang, L.; Chen, J.; Cao, T.; Yang, Y.; Liu, Y.; He, Q.; Gao, F.; Gao, H. Targeting delivery and deep penetration using multistage nanoparticles for triple-negative breast cancer. RSC Adv. 2015, 5, 64303–64317. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Trivers, K.F.; Lund, M.J.; Porter, P.L.; Liff, J.M.; Flagg, E.W.; Coates, R.J.; Eley, J.W. The epidemiology of triple-negative breast cancer, including race. Cancer Causes Control 2009, 20, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [Green Version]
- Khosravi-Shahi, P.; Cabezon-Gutierrez, L.; Custodio-Cabello, S. Metastatic triple negative breast cancer: Optimizing treatment options, new and emerging targeted therapies. Asia Pac. J. Clin. Oncol. 2018, 14, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Djamgoz, M.B.A. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat. Rev. 2018, 62, 110–122. [Google Scholar] [CrossRef]
- Sharma, P. Biology and Management of Patients with Triple-Negative Breast Cancer. Oncologist 2016, 21, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, L.N.; Wilkinson, K.H.; Kong, A. Triple-Negative Breast Cancer: Who Should Receive Neoadjuvant Chemotherapy? Surg. Oncol. Clin. N. Am. 2018, 27, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Andreopoulou, E.; Schweber, S.J.; Sparano, J.A.; McDaid, H.M. Therapies for triple negative breast cancer. Expert Opin. Pharmacother. 2015, 16, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Lee, S.J.; Ohtani, S.; Im, Y.H.; Lee, E.S.; Yokota, I.; Kuroi, K.; Im, S.A.; Park, B.W.; Kim, S.B.; et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N. Engl. J. Med. 2017, 376, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Syed, Y.Y. Atezolizumab (in Combination with Nab-Paclitaxel): A Review in Advanced Triple-Negative Breast Cancer. Drugs 2020, 80, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Giuliano, M.; De Placido, S.; Arpino, G. Nab-paclitaxel for the treatment of triple-negative breast cancer: Rationale, clinical data and future perspectives. Cancer Treat. Rev. 2016, 50, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.; Glode, A.; Messersmith, W.A.; Diamond, J. Sacituzumab govitecan: Breakthrough targeted therapy for triple-negative breast cancer. Expert Rev. Anticancer Ther. 2019, 19, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Prabhu, P. Nanosoldiers: A promising strategy to combat triple negative breast cancer. Biomed. Pharmacother. 2019, 110, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Ghosh, B.; Biswas, S. Nanocarriers for cancer-targeted drug delivery. J. Drug Target. 2016, 24, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Avvakumova, S.; Pandolfi, L.; Soprano, E.; Moretto, L.; Bellini, M.; Galbiati, E.; Rizzuto, M.; Colombo, M.; Allevi, R.; Corsi, F.J.N.A. Does conjugation strategy matter? Cetuximab-conjugated gold nanocages for targeting triple-negative breast cancer cells. Nanoscale Adv. 2019, 1, 3626–3638. [Google Scholar] [CrossRef] [Green Version]
- Bose, R.J.; Paulmurugan, R.; Moon, J.; Lee, S.-H.; Park, H. Cell membrane-coated nanocarriers: The emerging targeted delivery system for cancer theranostics. Drug Discov. Today 2018, 23, 891–899. [Google Scholar] [CrossRef]
- Bosch, A.; Eroles, P.; Zaragoza, R.; Vina, J.R.; Lluch, A. Triple-negative breast cancer: Molecular features, pathogenesis, treatment and current lines of research. Cancer Treat. Rev. 2010, 36, 206–215. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Evaluation of IPI-549 Combined with Front-line Treatments in Pts. With Triple-Negative Breast Cancer or Renal Cell Carcinoma (MARIO-3) (MARIO-3). Available online: https://clinicaltrials.gov/ct2/show/NCT03961698?term=nanoparticle+targeted+therapy&type=Intr&cond=NCT03961698&draw=2&rank=1 (accessed on 20 December 2020).
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Banstola, A.; Poudel, K.; Emami, F.; Ku, S.K.; Jeong, J.-H.; Kim, J.O.; Yook, S. Localized therapy using anti-PD-L1 anchored and NIR-responsive hollow gold nanoshell (HGNS) loaded with doxorubicin (DOX) for the treatment of locally advanced melanoma. Nanomedicine 2020, 33, 102349. [Google Scholar] [CrossRef] [PubMed]
- Duwa, R.; Banstola, A.; Emami, F.; Jeong, J.-H.; Lee, S.; Yook, S. Cetuximab conjugated temozolomide-loaded poly (lactic-co-glycolic acid) nanoparticles for targeted nanomedicine in EGFR overexpressing cancer cells. J. Drug Deliv. Sci. Technol. 2020, 60, 101928. [Google Scholar] [CrossRef]
- Huang, Y.; Mei, C.; Tian, Y.; Nie, T.; Liu, Z.; Chen, T. Bioinspired tumor-homing nanosystem for precise cancer therapy via reprogramming of tumor-associated macrophages. NPG Asia Mater. 2018, 10, 1002–1015. [Google Scholar] [CrossRef] [Green Version]
- Souho, T.; Lamboni, L.; Xiao, L.; Yang, G. Cancer hallmarks and malignancy features: Gateway for improved targeted drug delivery. Biotechnol. Adv. 2018, 36, 1928–1945. [Google Scholar] [CrossRef]
- Tian, W.; Lu, J.; Jiao, D.J.P.f.A.T. Stem cell membrane vesicle–coated nanoparticles for efficient tumor-targeted therapy of orthotopic breast cancer. Polym. Adv. Technol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Kroll, A.V.; Fang, R.H.; Zhang, L. Biointerfacing and applications of cell membrane-coated nanoparticles. Bioconjug. Chem. 2017, 28, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, V.; Uthaman, S.; Park, I.-K. Cell Membrane-Camouflaged Nanoparticles: A Promising Biomimetic Strategy for Cancer Theragnostics. Polymers 2018, 10, 983. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cai, K.; Li, C.; Guo, Q.; Chen, Q.; He, X.; Liu, L.; Zhang, Y.; Lu, Y.; Chen, X.; et al. Macrophage-membrane-coated nanoparticles for tumor-targeted chemotherapy. Nano Lett. 2018, 18, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Zhang, L. Cell membrane-camouflaged nanoparticles for drug delivery. J. Control. Release 2015, 220, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Lin, Z.; Jurado-Sanchez, B.; Lin, X.; Wu, Z.; He, Q. Stem Cell Membrane-Coated Nanogels for Highly Efficient In Vivo Tumor Targeted Drug Delivery. Small 2016, 12, 4056–4062. [Google Scholar] [CrossRef]
- Emami, F.; Yazdi, S.J.M.; Na, D.H. Poly (lactic acid)/poly (lactic-co-glycolic acid) particulate carriers for pulmonary drug delivery. J. Pharm. Investig. 2019, 49, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Ding, Y.; Zhang, Y.; Wang, B.; Zhao, X.; Cheng, K.; Huang, Y.; Taleb, M.; Zhao, J.; Dong, W.F.; et al. Surface Functionalization of Polymeric Nanoparticles with Umbilical Cord-Derived Mesenchymal Stem Cell Membrane for Tumor-Targeted Therapy. ACS Appl. Mater. Interfaces 2018, 10, 22963–22973. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Ye, X.; Wang, C.; Xing, C.; Miao, Q.; Xie, Z.; Chen, X.; Zhang, X.; Zhang, H.; Mei, L. Photothermal cancer immunotherapy by erythrocyte membrane-coated black phosphorus formulation. J. Control. Release 2019, 296, 150–161. [Google Scholar] [CrossRef]
- Zhai, Y.; Ran, W.; Su, J.; Lang, T.; Meng, J.; Wang, G.; Zhang, P.; Li, Y. Traceable bioinspired nanoparticle for the treatment of metastatic breast cancer via NIR-trigged intracellular delivery of methylene blue and cisplatin. Adv. Mater. 2018, 30, 1802378. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yoon, H.J.; Yazdi, S.J.M.; Lee, J.H. A novel automated lumen segmentation and classification algorithm for detection of irregular protrusion after stents deployment. Int. J. Med. Robot. 2020, 16, e2033. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Su, J.; Meng, Q.; Yin, Q.; Chen, L.; Gu, W.; Zhang, Z.; Yu, H.; Zhang, P.; Wang, S.; et al. Cancer Cell Membrane-Coated Gold Nanocages with Hyperthermia-Triggered Drug Release and Homotypic Target Inhibit Growth and Metastasis of Breast Cancer. Adv. Funct. Mater. 2017, 27, 1604300. [Google Scholar] [CrossRef]
- Chu, D.; Zhao, Q.; Yu, J.; Zhang, F.; Zhang, H.; Wang, Z. Nanoparticle targeting of neutrophils for improved cancer immunotherapy. Adv. Healthc. Mater. 2016, 5, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Gu, W.; Li, J.; Chen, C.; Xu, Z.P. Silencing PD-1 and PD-L1 with nanoparticle-delivered small interfering RNA increases cytotoxicity of tumor-infiltrating lymphocytes. Nanomedicine 2019, 14, 955–967. [Google Scholar] [CrossRef] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.-V.; Choi, J.; Oh, Y.-K. Nano delivery systems and cancer immunotherapy. J. Pharm. Investig. 2018, 48, 527–539. [Google Scholar] [CrossRef]
- Wang, C.; Steinmetz, N.F. CD47 blockade and cowpea mosaic virus nanoparticle in situ vaccination triggers phagocytosis and tumor killing. Adv. Healthc. Mater. 2019, 8, 1801288. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, J.A.; Ou, Y.-C.; Faley, S.; Paul, E.P.; Hittinger, J.P.; Cutright, C.C.; Lin, E.C.; Bellan, L.M.; Bardhan, R. Theranostic gold nanoantennas for simultaneous multiplexed Raman imaging of immunomarkers and photothermal therapy. ACS Omega 2017, 2, 3583–3594. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 2016, 34, 2460. [Google Scholar] [CrossRef]
- Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying technologies for the stability and bioavailability of biopharmaceuticals. Pharmaceutics 2018, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.-S.; Fu, S.-Y.; Wang, S.-C.; Yu, C.-F.; Chen, F.-H.; Lin, C.-M.; Hong, J.-H. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012, 2, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.S.; Brown, J.M. The irradiated tumor microenvironment: Role of tumor-associated macrophages in vascular recovery. Front. Physiol. 2013, 4, 157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin Encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS ONE 2013, 8, e65896. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, L.; He, P.; Liu, Y.; Wang, W.; He, Y.; Du, Y.; Gao, F. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med. Oncol. 2015, 32, 14. [Google Scholar] [CrossRef]
- Niu, M.; Valdes, S.; Naguib, Y.W.; Hursting, S.D.; Cui, Z. Tumor-Associated Macrophage-Mediated Targeted Therapy of Triple-Negative Breast Cancer. Mol. Pharm. 2016, 13, 1833–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, X.-H.; Zhao, Y.-X.; Chen, C.; Xu, X.-Y. Cancer-associated fibroblasts correlate with tumor-associated macrophages infiltration and lymphatic metastasis in triple negative breast cancer patients. J. Cancer 2018, 9, 4635. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhang, J.; Huang, Y.; Ji, S.; Shao, G.; Feng, S.; Chen, D.; Zhao, K.; Wang, Z.; Wu, A. Cancer-associated fibroblasts autophagy enhances progression of triple-negative breast cancer cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef]
- Rao, N.V.; Ko, H.; Lee, J.; Park, J.H. Recent Progress and Advances in Stimuli-Responsive Polymers for Cancer Therapy. Front. Bioeng. Biotechnol. 2018, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- Hajebi, S.; Rabiee, N.; Bagherzadeh, M.; Ahmadi, S.; Rabiee, M.; Roghani-Mamaqani, H.; Tahriri, M.; Tayebi, L.; Hamblin, M.R. Stimulus-responsive polymeric nanogels as smart drug delivery systems. Acta Biomater. 2019, 92, 1–18. [Google Scholar] [CrossRef]
- Chen, W.-L.; Li, F.; Qu, C.-X.; Liu, Y.; Wang, Y.; Wang, D.-D.; Zhang, X.-N. Programmed pH/reduction-responsive nanoparticles for efficient delivery of antitumor agents in vivo. Acta Biomater. 2018, 81, 219–230. [Google Scholar] [CrossRef]
- Du, J.; Lane, L.A.; Nie, S.J.J.o.C.R. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J. Control. Release 2015, 219, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.; Shin, S.; Rao, N.V.; Um, W.; Jeon, J.; Ko, H.; Deepagan, V.; Kwon, S.; Lee, J.Y.; Park, J.H. Anti-Trop2 antibody-conjugated bioreducible nanoparticles for targeted triple negative breast cancer therapy. Int. J. Biol. Macromol. 2018, 110, 406–415. [Google Scholar] [CrossRef]
- Yazdi, S.J.M.; Cho, K.S.; Kang, N. Characterization of the viscoelastic model of in vivo human posterior thigh skin using ramp-relaxation indentation test. Korea Aust. Rheol. 2018, 30, 293–307. [Google Scholar] [CrossRef]
- Ding, Y.; Su, S.; Zhang, R.; Shao, L.; Zhang, Y.; Wang, B.; Li, Y.; Chen, L.; Yu, Q.; Wu, Y.J.B. Precision combination therapy for triple negative breast cancer via biomimetic polydopamine polymer core-shell nanostructures. Biomaterials 2017, 113, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yu, Y.; Sun, Y.; Kong, L.; Yang, C.; Hu, M.; Yang, T.; Zhang, J.; Hu, Q.; Zhang, Z. Transformable Nanoparticle-Enabled Synergistic Elicitation and Promotion of Immunogenic Cell Death for Triple-Negative Breast Cancer Immunotherapy. Adv. Funct. Mater. 2019, 29, 1905213. [Google Scholar] [CrossRef]
- Shashni, B.; Nagasaki, Y.J.B. Nitroxide radical-containing nanoparticles attenuate tumorigenic potential of triple negative breast cancer. Biomaterials 2018, 178, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, Q.; Lin, G.; Shi, Y.; Gu, Z.; Ding, T. Combination of using prodrug-modified cationic liposome nanocomplexes and a potentiating strategy via targeted co-delivery of gemcitabine and docetaxel for CD44-overexpressed triple negative breast cancer therapy. Acta Biomater. 2017, 62, 257–272. [Google Scholar] [CrossRef]
- Hu, G.; Chun, X.; Wang, Y.; He, Q.; Gao, H. Peptide mediated active targeting and intelligent particle size reduction-mediated enhanced penetrating of fabricated nanoparticles for triple-negative breast cancer treatment. Oncotarget 2015, 6, 41258. [Google Scholar] [CrossRef] [Green Version]
- Alimoradi, H.; Greish, K.; Barzegar-Fallah, A.; Alshaibani, L.; Pittalà, V. Nitric oxide-releasing nanoparticles improve doxorubicin anticancer activity. Int. J. Nanomed. 2018, 13, 7771. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xie, Y.; Li, J.; Peng, Z.-H.; Sheinin, Y.; Zhou, J.; Oupický, D. Tumor-penetrating nanoparticles for enhanced anticancer activity of combined photodynamic and hypoxia-activated therapy. ACS Nano 2017, 11, 2227–2238. [Google Scholar] [CrossRef]
- Saravanakumar, G.; Kim, J.; Kim, W.J. Reactive-Oxygen-Species-Responsive Drug Delivery Systems: Promises and Challenges. Adv. Sci. 2017, 4, 1600124. [Google Scholar] [CrossRef]
- Duwa, R.; Emami, F.; Lee, S.; Jeong, J.-H.; Yook, S. Polymeric and lipid-based drug delivery systems for treatment of glioblastoma multiforme. J. Ind. Eng. Chem. 2019, 79, 261–273. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami, F.; Banstola, A.; Vatanara, A.; Lee, S.; Kim, J.O.; Jeong, J.-H.; Yook, S. Doxorubicin and anti-PD-L1 antibody conjugated gold nanoparticles for colorectal cancer photochemotherapy. Mol. Pharm. 2019, 16, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Ruan, S.; Cao, X.; Cun, X.; Hu, G.; Zhou, Y.; Zhang, Y.; Lu, L.; He, Q.; Gao, H. Matrix metalloproteinase-sensitive size-shrinkable nanoparticles for deep tumor penetration and pH triggered doxorubicin release. Biomaterials 2015, 60, 100–110. [Google Scholar] [CrossRef]
- Miller, M.R.; Megson, I.L. Recent developments in nitric oxide donor drugs. Br. J. Pharmacol. 2007, 151, 305–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.-b.; Yazdi, S.J.M.; Lee, J.-H. Development of Wearable Temperature Sensor Based on Peltier Thermoelectric Device to Change Human Body Temperature. Sens. Mater. 2020, 32, 2959–2970. [Google Scholar] [CrossRef]
- Oliveira, A.M.; Oliveira, P.C.; Santos, A.M.; Zanin, M.H.A.; Ré, M.I. Synthesis and characterization of thermo-responsive particles of poly (hydroxybutirate-co-hydroxyvalerate)-b-poly (N-isopropylacrylamide). Braz. J. Phys. 2009, 39, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Moreno, P.; de Vicente, J.; Nardecchia, S.; Marchal, J.A.; Boulaiz, H. Thermo-Sensitive Nanomaterials: Recent Advance in Synthesis and Biomedical Applications. Nanomaterials 2018, 8, 935. [Google Scholar] [CrossRef] [Green Version]
- Truffi, M.; Mazzucchelli, S.; Bonizzi, A.; Sorrentino, L.; Allevi, R.; Vanna, R.; Morasso, C.; Corsi, F. Nano-Strategies to Target Breast Cancer-Associated Fibroblasts: Rearranging the Tumor Microenvironment to Achieve Antitumor Efficacy. Int. J. Mol. Sci. 2019, 20, 1263. [Google Scholar] [CrossRef] [Green Version]
- Goh, C.Y.; Wyse, C.; Ho, M.; O’Beirne, E.; Howard, J.; Lindsay, S.; Kelly, P.; Higgins, M.; McCann, A. Exosomes in Triple Negative Breast Cancer: Garbage Disposals or Trojan Horses? Cancer Lett. 2020, 73, 90–97. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticles | Stimuli | Carrier Type | Bioactive Compound | Ligand | Target | TNBC Cell Line | Outcome | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Multi stimuli NP | MMP-2 GSH Acidic pH | 2-(Nap)-FFKPt-2TPA-ADD-PLGVRGGGG(2-NPs) 2-(Nap)-FFKPt-2TPA-ADD-GGGPLGVRG-WKYMVm-mPEG1000(3-NPs) | Pt ADDWKYMVm | WKYMVm | FPR-1 | MDA-MB-231 4T1 MCF7ADR orthotopic model | Highest cell death in all three cell lines by both NPs No body weight loss in both NPs treated mice Well tolerable tumor inhibitory effects Prolonged tumor retention 93.1% tumor shrinkage in 4T1 model 2.7- fold increase of overall survival | [73] | |
| ROS-responsive NP | ROS scavenging Acidic pH | RNPN pH-sensitive | MeO-PEG-b-PMNT | TEMPO | - | ROS | MDA-MB-231 xenograft model | 61% MDA-MB-231 cell viability treated with RNP0 98% MDA-MB-231 cell viability treated with RNPN Considerable anti-migratory effect on MDA-MB-231 cell Higher invading inhibition potential for RNPN rather than RNP0 Significant anti-tumor effect and tumor weight decrease Important downregulation of MMP-2 and NF-κB in tumor Insignificant adverse effects on mouse body weight | [74] |
| RNP0pH-insensitive | MeO-PEG-b-PMOT | ||||||||
| pH-responsive NP | Acidic pH | DOTAP | DTX GEM | HA | CD44 | MDA-MB-231 | Highest induced apoptosis: 80 ± 5.12% Strongest anti-migration effects in MDA-MB-231 cell line by Combo NCs Almost 93 mm3 decrease in tumor volume in MDA-MB-231 tumor bearing mice Lack of considerable systemic toxicity in Combo NCs treatment | [75] | |
| Enzyme-responsive shrinkable NP | MMP2 enzyme NIR | G-AuNPs | DOX | RRGD | Extracellular matrix | 4T1 cells bearing mice | Improved tumor targeting Deep tumor penetration (75.5%) Enhanced tumor accumulation Acidic environment dependent drug release No considerable pulmonary metastasis Displaying the high tumor growth inhibition | [7] | |
| GNP with drug loaded DGL | DOX | Angiopep-2 | LRP1 | 4T1 cells bearing mice | Higher cellular uptake due to efficient targeting Considerable tumor accumulation Massive tumor cell apoptosis | [76] | |||
| NO-responsive NP | NO donor | SMA-tDodSNO and SMA | DOX | - | - | 4T1 cells bearing mice | A synergistic effect on cell survival with an IC50 of 1.79 ± 0.7 nM 87.4% of cell population in subG1 phase A drop in the alive cells’ percentage to 21.7 ± 3.9% Significant tumor growth inhibition | [77] | |
| Hypoxia-responsive NP | Hypoxia | Hybrid PLGA lipid NPs (DPPC, DSPE-PEG and DSPE-PEG-DBCO) | TPZ | iRGD | αυ-integrins neuropilin-1 | 4T1 cells bearing mice | Efficient targeting with almost 2-fold increase in comparison with non-targeted particles Significant cell cytotoxicity (IC50 in hypoxia: 3.7 μg/mL, IC50 in normoxia: 9.4 μg/mL) Possessing highest cellular uptake in spheroids High tumor penetration Strong tumor cell killing Successful metastasis inhibition | [78] | |
| Thermo-responsive NP | High temperature | poly ((2-(2-methoxyethoxy) ethyl methacrylate-co-oligo (ethylene glycol) methacrylate)-co-2-(dimethylamino) ethyl methacrylate-b-poly (d,l-lactic-coglycolide) and PDA as film coating | DOX PTX | siRNA | Survivin | MDA-MB-231 bearing mice | 80% tumor cell death Sensitized cancer cells to chemotherapy Non-significant adverse effects | [72] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keihan Shokooh, M.; Emami, F.; Jeong, J.-H.; Yook, S. Bio-Inspired and Smart Nanoparticles for Triple Negative Breast Cancer Microenvironment. Pharmaceutics 2021, 13, 287. https://doi.org/10.3390/pharmaceutics13020287
Keihan Shokooh M, Emami F, Jeong J-H, Yook S. Bio-Inspired and Smart Nanoparticles for Triple Negative Breast Cancer Microenvironment. Pharmaceutics. 2021; 13(2):287. https://doi.org/10.3390/pharmaceutics13020287
Chicago/Turabian StyleKeihan Shokooh, Mahsa, Fakhrossadat Emami, Jee-Heon Jeong, and Simmyung Yook. 2021. "Bio-Inspired and Smart Nanoparticles for Triple Negative Breast Cancer Microenvironment" Pharmaceutics 13, no. 2: 287. https://doi.org/10.3390/pharmaceutics13020287