
Strategies for Vaccination: Conventional Vaccine Approaches Versus New-Generation Strategies in Combination with Adjuvants

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Traditional Vaccines
3. Next-Generation Vaccines
3.1. Recombinant Protein Vaccines
3.2. Plasmid DNA Vaccines
3.3. Viral Vector Vaccines
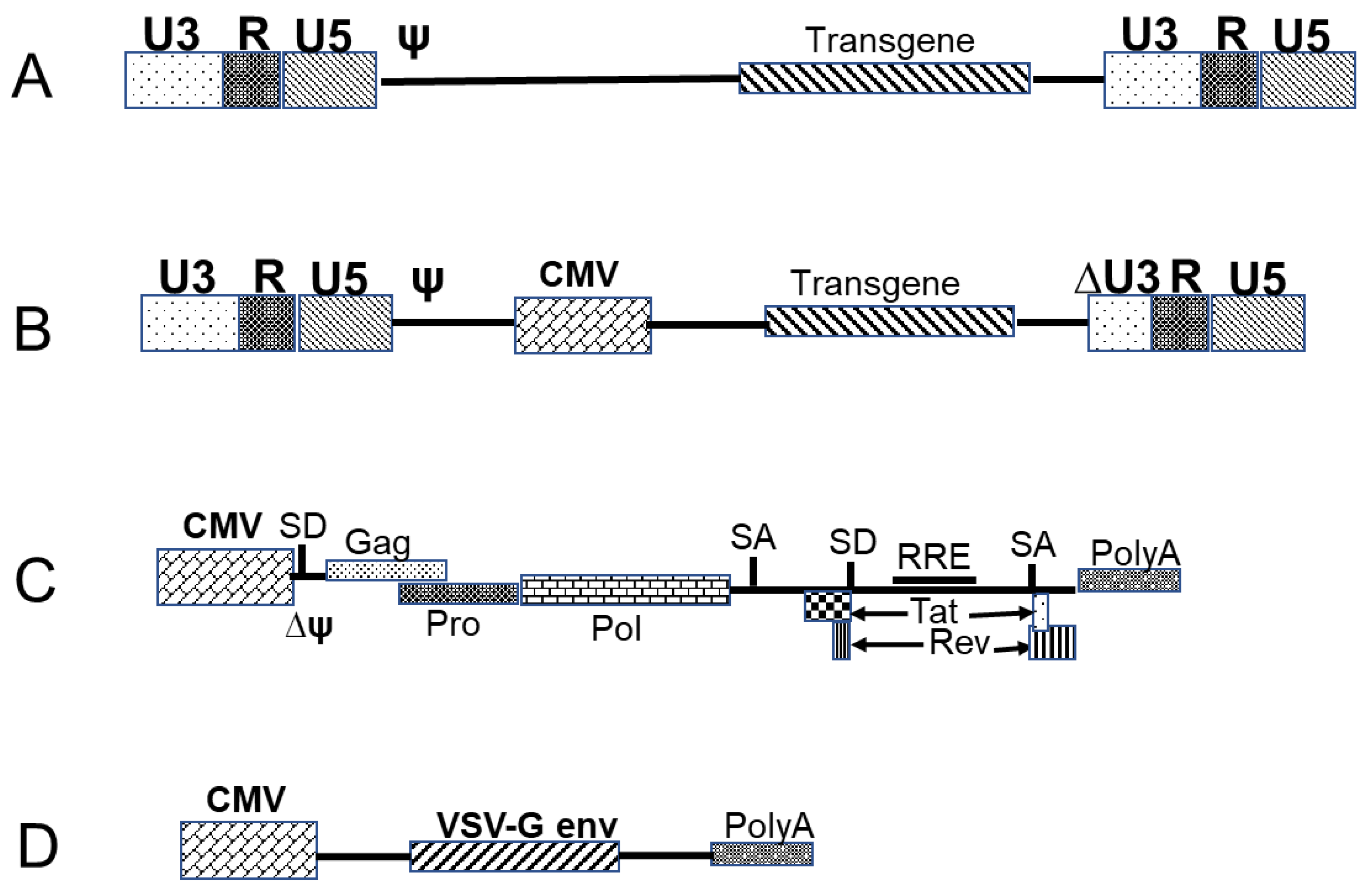
3.3.1. Retrovirus- and Lentivirus-Based Vectors
3.3.2. Adenovirus-Based Vectors
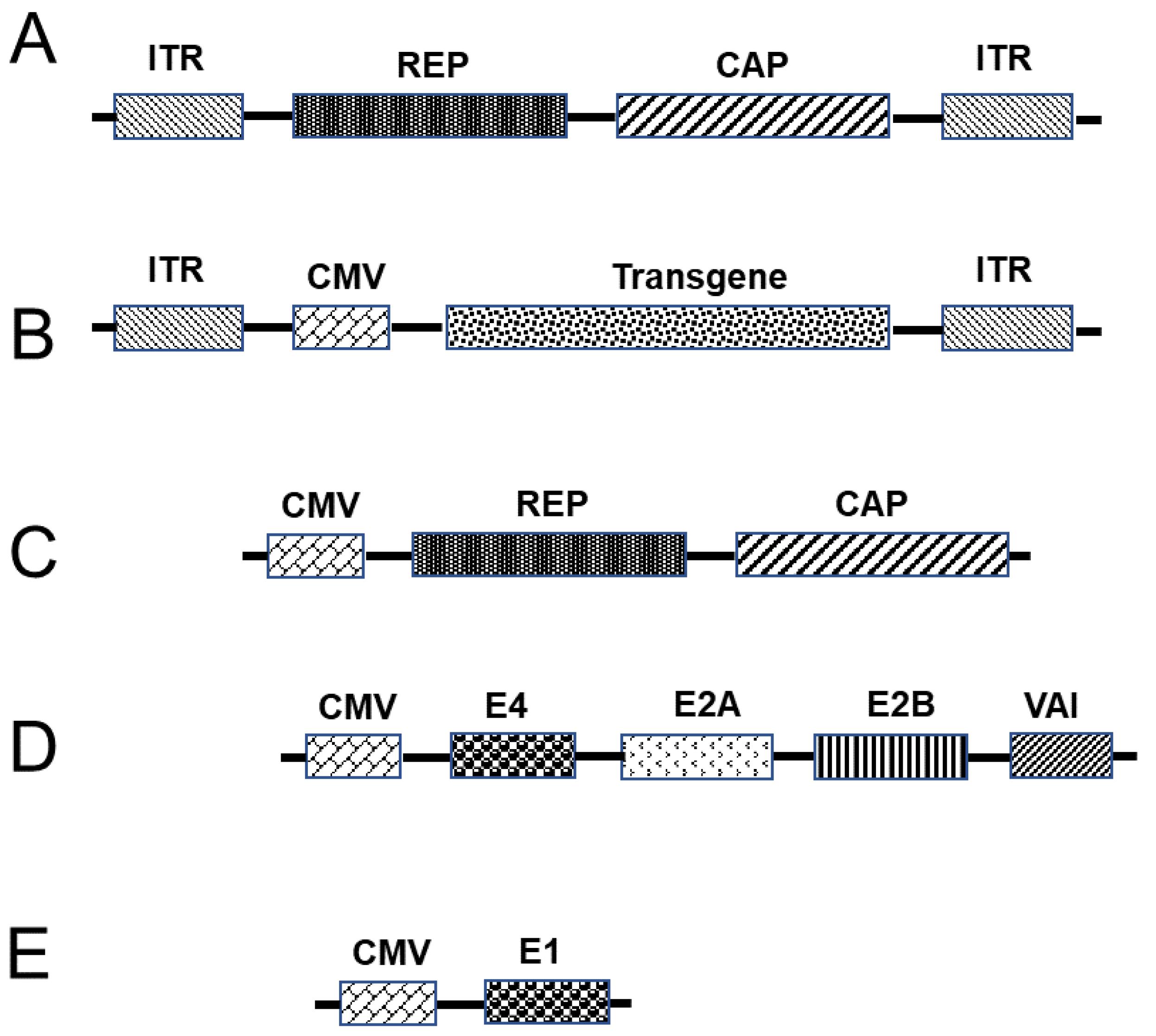
3.3.3. Adeno-Associated Virus Vectors as a Platform for Vaccination
3.4. RNA-Based Vaccines and Nanoparticle (NP) Formulations
3.4.1. RNA-Based Vaccines
3.4.2. NP Formulation for RNA Delivery
3.5. Vaccine Adjuvants
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Peng, Y.; Xu, H.; Cui, Z.; Williams, R.O., 3rd. The COVID-19 Vaccine Race: Challenges and Opportunities in Vaccine Formulation. AAPS PharmSciTech 2020, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Plett, P.C. Peter Plett and other discoverers of cowpox vaccination before Edward Jenner. Sudhoffs Arch 2006, 90, 219. [Google Scholar]
- Jenner, E. Jenner, on the Vaccine Inoculation. Med. Phys. J. 1800, 3, 502–503. [Google Scholar]
- Esparza, J.; Schrick, L.; Damaso, C.R.; Nitsche, A. Equination (inoculation of horsepox): An early alternative to vaccination (inoculation of cowpox) and the potential role of horsepox virus in the origin of the smallpox vaccine. Vaccine 2017, 35, 7222–7230. [Google Scholar] [CrossRef]
- Zepp, F. Principles of vaccine design-Lessons from nature. Vaccine 2010, 28 (Suppl 3), C14–C24. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C.; Janeway, C. Janeway’s Immunobiology; Garland science, Taylor & Francis Group LLC: New York, NY, USA, 2017. [Google Scholar]
- Offit, P.A. The Cutter Incident, 50 Years Later. N. Engl. J. Med. 2005, 352, 1411–1412. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Vartak, A.; Sucheck, S.J. Recent Advances in Subunit Vaccine Carriers. Vaccines 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingartl, H.; Czub, M.; Czub, S.; Neufeld, J.; Marszal, P.; Gren, J.; Smith, G.; Jones, S.; Proulx, R.; Deschambault, Y.; et al. Immunization with Modified Vaccinia Virus Ankara-Based Recombinant Vaccine against Severe Acute Respiratory Syndrome Is Associated with Enhanced Hepatitis in Ferrets. J. Virol. 2004, 78, 12672–12676. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhou, Y.; Liu, S.; Kou, Z.; Li, W.; Farzan, M.; Jiang, S. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 2004, 324, 773–781. [Google Scholar] [CrossRef]
- Du, L.; Zhao, G.; He, Y.; Guo, Y.; Zheng, B.J.; Jiang, S.; Zhou, Y. Receptor-binding domain of SARS-CoV spike protein induces long-term protective immunity in an animal model. Vaccine 2007, 25, 2832–2838. [Google Scholar] [CrossRef]
- Iyer, S.S.; Gangadhara, S.; Victor, B.; Shen, X.; Chen, X.; Nabi, R.; Kasturi, S.P.; Sabula, M.J.; Labranche, C.C.; Reddy, P.B.; et al. Virus-Like Particles Displaying Trimeric Simian Immunodeficiency Virus (SIV) Envelope gp160 Enhance the Breadth of DNA/Modified Vaccinia Virus Ankara SIV Vaccine-Induced Antibody Responses in Rhesus Macaques. J. Virol. 2016, 90, 8842–8854. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.; Cheng, H.; Dai, Z.; Bu, Z.; Sigal, L.J. Immunization with a single extracellular enveloped virus protein produced in bacteria provides partial protection from a lethal orthopoxvirus infection in a natural host. Virology 2006, 345, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, M.C.; Goenaga, J.; Wittek, R.; Rindisbacher, L. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology 1999, 254, 71–80. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.S.; MacLeod, M.K.; Kappler, J.W.; Marrack, P. Immune mechanisms of protection: Can adjuvants rise to the challenge? BMC Biol. 2010, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Kang, H.N.; Babiuk, L.A.; Liu, Q. Elicitation of strong immune responses by a DNA vaccine expressing a secreted form of hepatitis C virus envelope protein E2 in murine and porcine animal models. World J. Gastroenterol. 2006, 12, 7126–7135. [Google Scholar] [CrossRef] [PubMed]
- Wiest-Ladenburger, U.; Fortnagel, A.; Richter, W.; Reimann, J.; Boehm, B.O. DNA vaccination with glutamic acid decarboxylase (GAD) generates a strong humoral immune response in BALB/c, C57BL/6, and in diabetes-prone NOD mice. Horm. Metab. Res. 1998, 30, 605–609. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, N.Z.; Zhang, F.K.; Wang, M.; Hu, L.Y.; Zhu, X.Q. Immune response and protective effect against chronic Toxoplasma gondii infection induced by vaccination with a DNA vaccine encoding profilin. BMC Infect. Dis. 2018, 18, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobernik, D.; Bros, M. DNA Vaccines-How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.R.F.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.N.; Walker, S.N.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Kraynyak, K.A.; Patel, A.; Maslow, J.N.; Morrow, M.P.; Sylvester, A.J.; Knoblock, D.; Gillespie, E.; Amante, D.; Racine, T. Intradermal SynCon® Ebola GP DNA vaccine is temperature stable and safely demonstrates cellular and humoral immunogenicity advantages in healthy volunteers. J. Infect. Dis. 2019, 220, 400–410. [Google Scholar] [CrossRef]
- Yang, Z.-y.; Kong, W.-p.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.-d. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect. Dis. 2019, 19, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Muthumani, K.; Falzarano, D.; Reuschel, E.L.; Tingey, C.; Flingai, S.; Villarreal, D.O.; Wise, M.; Patel, A.; Izmirly, A.; Aljuaid, A. A synthetic consensus anti–spike protein DNA vaccine induces protective immunity against Middle East respiratory syndrome coronavirus in nonhuman primates. Sci. Transl. Med. 2015, 7, 301ra132. [Google Scholar] [CrossRef] [Green Version]
- Tebas, P.; Roberts, C.C.; Muthumani, K.; Reuschel, E.L.; Kudchodkar, S.B.; Zaidi, F.I.; White, S.; Khan, A.S.; Racine, T.; Choi, H.; et al. Safety and Immunogenicity of an Anti-Zika Virus DNA Vaccine—Preliminary Report. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.P.; Rabbitts, T.H. Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2004, 350, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Z.; Tian, H.; Qi, M.; Zhai, Z.; Li, S.; Li, R.; Zhang, H.; Wang, W.; Fu, S.; et al. Biodistribution and safety assessment of bladder cancer specific recombinant oncolytic adenovirus in subcutaneous xenografts tumor model in nude mice. Curr. Gene Ther. 2012, 12, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samulski, R.J.; Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.L.; Marconi, P.; Argnani, R.; Manservigi, R. HSV-1-derived recombinant and amplicon vectors for gene transfer and gene therapy. Curr. Gene Ther. 2005, 5, 445–458. [Google Scholar] [CrossRef]
- Ady, J.W.; Johnsen, C.; Mojica, K.; Heffner, J.; Love, D.; Pugalenthi, A.; Belin, L.J.; Chen, N.G.; Yu, Y.A.; Szalay, A.A.; et al. Oncolytic gene therapy with recombinant vaccinia strain GLV-2b372 efficiently kills hepatocellular carcinoma. Surgery 2015, 158, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Cohn, L.; Delamarre, L. Dendritic cell-targeted vaccines. Front. Immunol. 2014, 5, 255. [Google Scholar] [CrossRef]
- Schambach, A.; Morgan, M. Retroviral Vectors for Cancer Gene Therapy. Recent Results Cancer Res. 2016, 209, 17–35. [Google Scholar] [CrossRef]
- Cone, R.D.; Mulligan, R.C. High-efficiency gene transfer into mammalian cells: Generation of helper-free recombinant retrovirus with broad mammalian host range. Proc. Natl. Acad. Sci. USA 1984, 81, 6349–6353. [Google Scholar] [CrossRef] [Green Version]
- Yee, J.K.; Friedmann, T.; Burns, J.C. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994, 43, Pt A. 99–112. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.-P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Slavin, S.; Aker, M.; Ficara, F.; Deola, S.; Mortellaro, A.; Morecki, S.; Andolfi, G.; Tabucchi, A.; Carlucci, F. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 2002, 296, 2410–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.-L.; Fraser, C.C.; Cavazzana-Calvo, M. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; Di Serio, C. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knöss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [Green Version]
- Bouazzaoui, A.; Kreutz, M.; Eisert, V.; Dinauer, N.; Heinzelmann, A.; Hallenberger, S.; Strayle, J.; Walker, R.; Rubsamen-Waigmann, H.; Andreesen, R.; et al. Stimulated trans-acting factor of 50 kDa (Staf50) inhibits HIV-1 replication in human monocyte-derived macrophages. Virology 2006, 356, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Al-Allaf, F.A.; Abduljaleel, Z.; Taher, M.M.; Abdellatif, A.A.H.; Athar, M.; Bogari, N.M.; Al-Ahdal, M.N.; Al-Mohanna, F.; Al-Hassnan, Z.N.; Alzabeedi, K.H.Y.; et al. Molecular Dynamics Simulation Reveals Exposed Residues in the Ligand-Binding Domain of the Low-Density Lipoprotein Receptor that Interacts with Vesicular Stomatitis Virus-G Envelope. Viruses 2019, 11, 1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigna, E.; Naldini, L. Lentiviral vectors: Excellent tools for experimental gene transfer and promising candidates for gene therapy. J. Gene Med. 2000, 2, 308–316. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Lévy, C.; Verhoeyen, E.; Cosset, F.L. Surface engineering of lentiviral vectors for gene transfer into gene therapy target cells. Curr. Opin. Pharmacol. 2015, 24, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Loew, R.; Meyer, Y.; Kuehlcke, K.; Gama-Norton, L.; Wirth, D.; Hauser, H.; Stein, S.; Grez, M.; Thornhill, S.; Thrasher, A.; et al. A new PG13-based packaging cell line for stable production of clinical-grade self-inactivating γ-retroviral vectors using targeted integration. Gene Ther. 2010, 17, 272–280. [Google Scholar] [CrossRef]
- Luis, A. The Old and the New: Prospects for Non-Integrating Lentiviral Vector Technology. Viruses 2020, 12, 1103. [Google Scholar] [CrossRef]
- Sayedahmed, E.E.; Elkashif, A.; Alhashimi, M.; Sambhara, S.; Mittal, S.K. Adenoviral Vector-Based Vaccine Platforms for Developing the Next Generation of Influenza Vaccines. Vaccines 2020, 8, 574. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, L.; Ko, S.Y.; Kong, W.P.; Schmidt, S.D.; Gall, J.G.D.; Colloca, S.; Seder, R.A.; Mascola, J.R.; Nabel, G.J. Combination recombinant simian or chimpanzee adenoviral vectors for vaccine development. Vaccine 2015, 33, 7344–7351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Chi, Y.; Zhou, D. Development of Novel Vaccines Against Infectious Diseases Based on Chimpanzee Adenoviral Vector. Methods Mol. Biol. 2017, 1581, 3–13. [Google Scholar] [CrossRef]
- Liu, L. Fields Virology, 6th Edition. Clin. Infect. Dis. 2014, 59, 613. [Google Scholar] [CrossRef]
- Kovesdi, I.; Hedley, S.J. Adenoviral producer cells. Viruses 2010, 2, 1681–1703. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Alonso-Padilla, J.; Papp, T.; Kaján, G.L.; Benkő, M.; Havenga, M.; Lemckert, A.; Harrach, B.; Baker, A.H. Development of Novel Adenoviral Vectors to Overcome Challenges Observed With HAdV-5-based Constructs. Mol. Ther. 2016, 24, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbink, P.; Lemckert, A.A.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; et al. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 2007, 81, 4654–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledgerwood, J.E.; Costner, P.; Desai, N.; Holman, L.; Enama, M.E.; Yamshchikov, G.; Mulangu, S.; Hu, Z.; Andrews, C.A.; Sheets, R.A.; et al. A replication defective recombinant Ad5 vaccine expressing Ebola virus GP is safe and immunogenic in healthy adults. Vaccine 2010, 29, 304–313. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Patial, S.; McBride, A.; Godbehere, S.; Van Rooijen, N.; Parameswaran, N.; Amalfitano, A. Adenovirus Vector-Induced Innate Inflammatory Mediators, MAPK Signaling, As Well As Adaptive Immune Responses Are Dependent upon Both TLR2 and TLR9 In Vivo. J. Immunol. 2008, 181, 2134–2144. [Google Scholar] [CrossRef]
- Fejer, G.; Freudenberg, M.; Greber, U.F.; Gyory, I. Adenovirus-triggered innate signalling pathways. Eur. J. Microbiol. Immunol. (Bp) 2011, 1, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Huang, X.; Yang, Y. Innate Immune Response to Adenoviral Vectors Is Mediated by both Toll-Like Receptor-Dependent and -Independent Pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, B.; Jayandharan, G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, L.V.; Follett, E.A.; Burdon, M.G.; McGeoch, D.J. The DNA of a minute virus of mice. J. Gen. Virol. 1969, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.A.; Berns, K.I.; Hoggan, M.D.; Koczot, F.J. Evidence for a single-stranded adenovirus-associated virus genome: Formation of a DNA density hybrid on release of viral DNA. Proc. Natl. Acad. Sci. USA 1969, 64, 863–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.J.; Khoury, G.; Denhardt, D.T. Physical map and strand polarity of specific fragments of adenovirus-associated virus DNA produced by endonuclease R-EcoRI. J. Virol. 1975, 16, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Lusby, E.; Fife, K.H.; Berns, K.I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. J. Virol. 1980, 34, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Berns, K.I.; Pinkerton, T.C.; Thomas, G.F.; Hoggan, M.D. Detection of adeno-associated virus (AAV)-specific nucleotide sequences in DNA isolated from latently infected Detroit 6 cells. Virology 1975, 68, 556–560. [Google Scholar] [CrossRef]
- Cheung, A.K.; Hoggan, M.D.; Hauswirth, W.W.; Berns, K.I. Integration of the adeno-associated virus genome into cellular DNA in latently infected human Detroit 6 cells. J. Virol. 1980, 33, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Kotin, R.M.; Berns, K.I. Organization of adeno-associated virus DNA in latently infected Detroit 6 cells. Virology 1989, 170, 460–467. [Google Scholar] [CrossRef]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.J.; Khoury, G.; Rose, J.A. Adenovirus-associated virus multiplication. IX. Extent of transcription of the viral genome in vivo. J. Virol. 1972, 10, 1118–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauswirth, W.W.; Berns, K.I. Origin and termination of adeno-associated virus DNA replication. Virology 1977, 78, 488–499. [Google Scholar] [CrossRef]
- Marcus, C.J.; Laughlin, C.A.; Carter, B.J. Adeno-associated virus RNA transcription in vivo. Eur. J. Biochem. 1981, 121, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.W.; Carter, B.J. Assembly of adeno-associated virus. Virology 1980, 102, 71–82. [Google Scholar] [CrossRef]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughlin, C.A.; Tratschin, J.D.; Coon, H.; Carter, B.J. Cloning of infectious adeno-associated virus genomes in bacterial plasmids. Gene 1983, 23, 65–73. [Google Scholar] [CrossRef]
- Srivastava, A.; Lusby, E.W.; Berns, K.I. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 1983, 45, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Fields, B.N.; Knipe, D.M.; Howley, P.M.; Griffin, D.E. Fields’ Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Sonntag, F.; Schmidt, K.; Kleinschmidt, J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, F.; Köther, K.; Schmidt, K.; Weghofer, M.; Raupp, C.; Nieto, K.; Kuck, A.; Gerlach, B.; Böttcher, B.; Müller, O.J.; et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011, 85, 12686–12697. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Warrington, K.H., Jr.; Hearing, P.; Hughes, J.; Muzyczka, N. Adenovirus-facilitated nuclear translocation of adeno-associated virus type 2. J. Virol. 2002, 76, 11505–11517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolson, S.C.; Samulski, R.J. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J. Virol. 2014, 88, 4132–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelich, J.M.; Ma, J.; Dong, B.; Wang, Q.; Chin, M.; Magura, C.M.; Xiao, W.; Yang, W. Super-resolution imaging of nuclear import of adeno-associated virus in live cells. Mol. Ther. Methods Clin. Dev. 2015, 2, 15047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zeng, X.; Fan, Z.; Li, C.; McCown, T.; Samulski, R.J.; Xiao, X. Adeno-associated virus of a single-polarity DNA genome is capable of transduction in vivo. Mol. Ther. 2008, 16, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhou, X.; Li, Y.; Qing, K.; Xiao, X.; Samulski, R.J.; Srivastava, A. Single-polarity recombinant adeno-associated virus 2 vector-mediated transgene expression in vitro and in vivo: Mechanism of transduction. Mol. Ther. 2008, 16, 290–295. [Google Scholar] [CrossRef]
- Duan, D.; Yan, Z.; Yue, Y.; Engelhardt, J.F. Structural analysis of adeno-associated virus transduction circular intermediates. Virology 1999, 261, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Marie-Claude, G.; Anna, S. Helper Functions Required for Wild Type and Recombinant Adeno- Associated Virus Growth. Curr. Gene Ther. 2005, 5, 265–271. [Google Scholar] [CrossRef]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol. 2011, 18, 1586–1588. [Google Scholar] [CrossRef] [Green Version]
- Erles, K.; Sebökovà, P.; Schlehofer, J.R. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J. Med. Virol. 1999, 59, 406–411. [Google Scholar] [CrossRef]
- Li, C.; Narkbunnam, N.; Samulski, R.J.; Asokan, A.; Hu, G.; Jacobson, L.J.; Manco-Johnson, M.J.; Monahan, P.E. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012, 19, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, K.; Salvetti, A. AAV Vectors Vaccines Against Infectious Diseases. Front. Immunol. 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Linden, R.M. Adeno-associated virus biology. Methods Mol. Biol. 2011, 807, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Grieger, J.C.; Samulski, R.J. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol. 2012, 507, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.F. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008, 15, 840–848. [Google Scholar] [CrossRef]
- Liu, Y.L.; Wagner, K.; Robinson, N.; Sabatino, D.; Margaritis, P.; Xiao, W.; Herzog, R.W. Optimized production of high-titer recombinant adeno-associated virus in roller bottles. Biotechniques 2003, 34, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Robert, M.A.; Chahal, P.S.; Audy, A.; Kamen, A.; Gilbert, R.; Gaillet, B. Manufacturing of recombinant adeno-associated viruses using mammalian expression platforms. Biotechnol. J. 2017, 12, 1600193. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Gaudet, D.; Méthot, J.; Déry, S.; Brisson, D.; Essiembre, C.; Tremblay, G.; Tremblay, K.; de Wal, J.; Twisk, J.; van den Bulk, N.; et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: An open-label trial. Gene Ther. 2013, 20, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef]
- Büning, H. Gene therapy enters the pharma market: The short story of a long journey. EMBO Mol. Med. 2013, 5, 1–3. [Google Scholar] [CrossRef]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.X.; Wang, Y.; Blake, S.; Yu, M.; Mei, L.; Wang, H.; Shi, J. RNA Nanotechnology-Mediated Cancer Immunotherapy. Theranostics 2020, 10, 281–299. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramps, T.; Probst, J. Messenger RNA-based vaccines: Progress, challenges, applications. Wiley Interdiscip Rev. RNA 2013, 4, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Yanez Arteta, M.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef] [Green Version]
- Martens, T.F.; Remaut, K.; Demeester, J.; De Smedt, S.C.; Braeckmans, K. Intracellular delivery of nanomaterials: How to catch endosomal escape in the act. Nano Today 2014, 9, 344–364. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, X.; Dong, Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip Rev. Nanomed. Nanobiotechnol. 2019, 11, e1530. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, D.H.; Berglund, P. Amplifying RNA Vaccine Development. N. Engl. J. Med. 2020, 382, 2469–2471. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Lu, H.; Fan, L.; Yan, S.; Hu, K. Efficient Delivery of Therapeutic siRNA with Nanoparticles Induces Apoptosis in Prostate Cancer Cells. J. Nanomater. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Wang, M.; Ma, Y.; Xia, W.; Gu, H. A mesoporous silica nanoparticle--PEI--fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials 2013, 34, 1391–1401. [Google Scholar] [CrossRef]
- Gunther, M.; Lipka, J.; Malek, A.; Gutsch, D.; Kreyling, W.; Aigner, A. Polyethylenimines for RNAi-mediated gene targeting in vivo and siRNA delivery to the lung. Eur. J. Pharm. Biopharm. 2011, 77, 438–449. [Google Scholar] [CrossRef]
- Dimitriadis, G.J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 1978, 274, 923–924. [Google Scholar] [CrossRef]
- Grabbe, S.; Haas, H.; Diken, M.; Kranz, L.M.; Langguth, P.; Sahin, U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: Case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine (Lond.) 2016, 11, 2723–2734. [Google Scholar] [CrossRef]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, W.; Nguyen, G.N.; Zhang, C.; Zeng, C.; Yan, J.; Du, S.; Hou, X.; Li, W.; Jiang, J.; et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci. Adv. 2020, 6, eabc2315. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jarvis, R.; Zhu, K.; Glass, Z.; Ogurlu, R.; Gao, P.; Li, P.; Chen, J.; Yu, Y.; Yang, Y.; et al. Protein and mRNA Delivery Enabled by Cholesteryl-Based Biodegradable Lipidoid Nanoparticles. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle-Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Vet. Pathol. 2018, 55, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; DuRoss, A.; Sun, C.; Murphy-Benenato, K.E.; Mihai, C.; Almarsson, O.; Sahay, G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stoter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of therapeutic proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 252. [Google Scholar] [CrossRef]
- Siewert, C.D.; Haas, H.; Cornet, V.; Nogueira, S.S.; Nawroth, T.; Uebbing, L.; Ziller, A.; Al-Gousous, J.; Radulescu, A.; Schroer, M.A.; et al. Hybrid Biopolymer and Lipid Nanoparticles with Improved Transfection Efficacy for mRNA. Cells 2020, 9, 2034. [Google Scholar] [CrossRef]
- Wang, Y.; Su, H.H.; Yang, Y.; Hu, Y.; Zhang, L.; Blancafort, P.; Huang, L. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther. 2013, 21, 358–367. [Google Scholar] [CrossRef]
- Palama, I.E.; Cortese, B.; D’Amone, S.; Gigli, G. mRNA delivery using non-viral PCL nanoparticles. Biomater. Sci. 2015, 3, 144–151. [Google Scholar] [CrossRef]
- Luo, Y.H.; Chang, L.W.; Lin, P. Metal-Based Nanoparticles and the Immune System: Activation, Inflammation, and Potential Applications. Biomed Res. Int. 2015, 2015, 143720. [Google Scholar] [CrossRef]
- Chan, K.P.; Chao, S.H.; Kah, J.C.Y. Universal mRNA Translation Enhancement with Gold Nanoparticles Conjugated to Oligonucleotides with a Poly(T) Sequence. ACS Appl. Mater. Interfaces 2018, 10, 5203–5212. [Google Scholar] [CrossRef]
- Yeom, J.H.; Ryou, S.M.; Won, M.; Park, M.; Bae, J.; Lee, K. Inhibition of Xenograft tumor growth by gold nanoparticle-DNA oligonucleotide conjugates-assisted delivery of BAX mRNA. PLoS ONE 2013, 8, e75369. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Aguado, I.; Rodriguez-Castejon, J.; Vicente-Pascual, M.; Rodriguez-Gascon, A.; Solinis, M.A.; Del Pozo-Rodriguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdellatif, A.A.H.; Tawfeek, H.M. Development and evaluation of fluorescent gold nanoparticles. Drug Dev. Ind. Pharm. 2018, 44, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, A.A.; Zayed, G.; El-Bakry, A.; Zaky, A.; Saleem, I.Y.; Tawfeek, H.M. Novel gold nanoparticles coated with somatostatin as a potential delivery system for targeting somatostatin receptors. Drug Dev. Ind. Pharm. 2016, 42, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B. Vaccine Adjuvants; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Flach, T.L.; Ng, G.; Hari, A.; Desrosiers, M.D.; Zhang, P.; Ward, S.M.; Seamone, M.E.; Vilaysane, A.; Mucsi, A.D.; Fong, Y. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat. Med. 2011, 17, 479. [Google Scholar] [CrossRef]
- Quandt, D.; Rothe, K.; Baerwald, C.; Rossol, M. GPRC6A mediates Alum-induced Nlrp3 inflammasome activation but limits Th2 type antibody responses. Sci. Rep. 2015, 5, 16719. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Soullié, T.; van Nimwegen, M.; Willart, M.A.; Muskens, F.; Jung, S.; Hoogsteden, H.C.; Hammad, H.; Lambrecht, B.N. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 2008, 205, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Igietseme, J.U.; Eko, F.O.; He, Q.; Black, C.M. Antibody regulation of Tcell immunity: Implications for vaccine strategies against intracellular pathogens. Expert Rev. Vaccines 2004, 3, 23–34. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.D.; Mullen, G.E.; Pierce, M.; Martin, L.B.; Miura, K.; Fay, M.P.; Long, C.A.; Shaffer, D.; Saul, A.; Miller, L.H. A Phase 1 study of the blood-stage malaria vaccine candidate AMA1-C1/Alhydrogel® with CPG 7909, using two different formulations and dosing intervals. Vaccine 2009, 27, 4104–4109. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Aldayel, A.M.; Cui, Z. Aluminum hydroxide nanoparticles show a stronger vaccine adjuvant activity than traditional aluminum hydroxide microparticles. J. Control. Release 2014, 173, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wang, Q.; Li, L.; Zeng, Q.; Li, H.; Gong, T.; Zhang, Z.; Sun, X. Turning the old adjuvant from gel to nanoparticles to amplify CD8+ T cell responses. Adv. Sci. 2018, 5, 1700426. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Ott, G.S.; Nest, G.V.; Rappuoli, R.; Giudice, G.D. The history of MF59(®) adjuvant: A phoenix that arose from the ashes. Expert Rev. Vaccines 2013, 12, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Caillet, C.; Piras, F.; Bernard, M.-C.; de Montfort, A.; Boudet, F.; Vogel, F.R.; Hoffenbach, A.; Moste, C.; Kusters, I. AF03-adjuvanted and non-adjuvanted pandemic influenza A (H1N1) 2009 vaccines induce strong antibody responses in seasonal influenza vaccine-primed and unprimed mice. Vaccine 2010, 28, 3076–3079. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Huynh, C.; O’Hara, M.K.; Onu, A. Technology transfer of oil-in-water emulsion adjuvant manufacturing for pandemic influenza vaccine production in Romania. Vaccine 2013, 31, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Aucouturier, J.; Dupuis, L.; Deville, S.; Ascarateil, S.; Ganne, V. Montanide ISA 720 and 51: A new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev. Vaccines 2002, 1, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Ascarateil, S.; Puget, A.; Gaucheron, J.; Koziol, M.-E. Sustained release of actives with Montanide™ ISA 51 VG and Montanide™ ISA 720 VG, two adjuvants dedicated to human therapeutic vaccines. J. Immunol. Ther. Cancer 2015, 3, P429. [Google Scholar] [CrossRef] [Green Version]
- Ellebedy, A.H.; Lupfer, C.; Ghoneim, H.E.; DeBeauchamp, J.; Kanneganti, T.-D.; Webby, R.J. Inflammasome-independent role of the apoptosis-associated speck-like protein containing CARD (ASC) in the adjuvant effect of MF59. Proc. Natl. Acad. Sci. USA 2011, 108, 2927–2932. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Wu, J.; Du, Y.; Miao, C.; Su, Z.; Ma, G. Bridging Systemic Immunity with Gastrointestinal Immune Responses via Oil-in-Polymer Capsules. Adv. Mater. 2018, 30, 1801067. [Google Scholar] [CrossRef]
- Qureshi, N.; Mascagni, P.; Ribi, E.; Takayama, K. Monophosphoryl lipid A obtained from lipopolysaccharides of Salmonella minnesota R595. Purification of the dimethyl derivative by high performance liquid chromatography and complete structural determination. J. Biol. Chem. 1985, 260, 5271–5278. [Google Scholar] [CrossRef]
- Kensil, C.R.; Patel, U.; Lennick, M.; Marciani, D. Separation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortex. J. Immunol. 1991, 146, 431–437. [Google Scholar]
- Detienne, S.; Welsby, I.; Collignon, C.; Wouters, S.; Coccia, M.; Delhaye, S.; Van Maele, L.; Thomas, S.; Swertvaegher, M.; Detavernier, A.; et al. Central Role of CD169+ Lymph Node Resident Macrophages in the Adjuvanticity of the QS-21 Component of AS01. Sci. Rep. 2016, 6, 39475. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Dreno, B.; Thompson, J.F.; Smithers, B.M.; Santinami, M.; Jouary, T.; Gutzmer, R.; Levchenko, E.; Rutkowski, P.; Grob, J.-J.; Korovin, S. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 916–929. [Google Scholar] [CrossRef] [Green Version]
- Jahrsdörfer, B.; Weiner, G.J. CpG oligodeoxynucleotides as immunotherapy in cancer. Update Cancer Ther. 2008, 3, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirota, H.; Klinman, D. CpG Oligodeoxynucleotides as adjuvants for clinical use. In Immunopotentiators in Modern Vaccines; Elsevier: Amsterdam, The Netherlands, 2017; pp. 163–198. [Google Scholar]
- Wang, Z.-B.; Shan, P.; Li, S.-Z.; Zhou, Y.; Deng, X.; Li, J.-L.; Zhang, Y.; Gao, J.-S.; Xu, J. The mechanism of action of acid-soluble chitosan as an adjuvant in the formulation of nasally administered vaccine against HBV. RSC Adv. 2016, 6, 96785–96797. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Qiu, Y.-Y.; Zhao, Y.-H.; Liu, X.-T.; Liu, M.; Yu, A.-L. Immunogenicity of oral vaccination with Lactococcus lactis derived vaccine candidate antigen (UreB) of Helicobacter pylori fused with the human interleukin 2 as adjuvant. Mol. Cell. Probes 2014, 28, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Krupka, M.; Zachova, K.; Cahlikova, R.; Vrbkova, J.; Novak, Z.; Sebela, M.; Weigl, E.; Raska, M. Endotoxin-minimized HIV-1 p24 fused to murine hsp70 activates dendritic cells, facilitates endocytosis and p24-specific Th1 response in mice. Immunol. Lett. 2015, 166, 36–44. [Google Scholar] [CrossRef]
- Taylor, D.N.; Treanor, J.J.; Sheldon, E.A.; Johnson, C.; Umlauf, S.; Song, L.; Kavita, U.; Liu, G.; Tussey, L.; Ozer, K. Development of VAX128, a recombinant hemagglutinin (HA) influenza-flagellin fusion vaccine with improved safety and immune response. Vaccine 2012, 30, 5761–5769. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouazzaoui, A.; Abdellatif, A.A.H.; Al-Allaf, F.A.; Bogari, N.M.; Al-Dehlawi, S.; Qari, S.H. Strategies for Vaccination: Conventional Vaccine Approaches Versus New-Generation Strategies in Combination with Adjuvants. Pharmaceutics 2021, 13, 140. https://doi.org/10.3390/pharmaceutics13020140
Bouazzaoui A, Abdellatif AAH, Al-Allaf FA, Bogari NM, Al-Dehlawi S, Qari SH. Strategies for Vaccination: Conventional Vaccine Approaches Versus New-Generation Strategies in Combination with Adjuvants. Pharmaceutics. 2021; 13(2):140. https://doi.org/10.3390/pharmaceutics13020140
Chicago/Turabian StyleBouazzaoui, Abdellatif, Ahmed A. H. Abdellatif, Faisal A. Al-Allaf, Neda M. Bogari, Saied Al-Dehlawi, and Sameer H. Qari. 2021. "Strategies for Vaccination: Conventional Vaccine Approaches Versus New-Generation Strategies in Combination with Adjuvants" Pharmaceutics 13, no. 2: 140. https://doi.org/10.3390/pharmaceutics13020140